
Sex-Specific Associations between Prenatal Exposure to Di(2-ethylhexyl) Phthalate, Epigenetic Age Acceleration, and Susceptibility to Early Childhood Upper Respiratory Infections

 , , , and
, , , and
Abstract
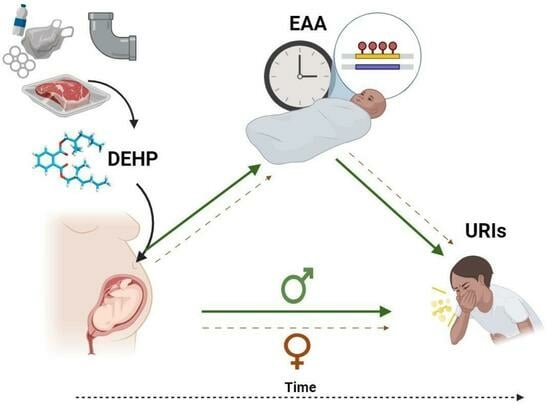
:
1. Introduction
2. Results
2.1. Sample Characteristics
2.2. Phthalate Exposure
2.3. Estimated Cell Type Proportions
2.4. Epigenetic Age Acceleration
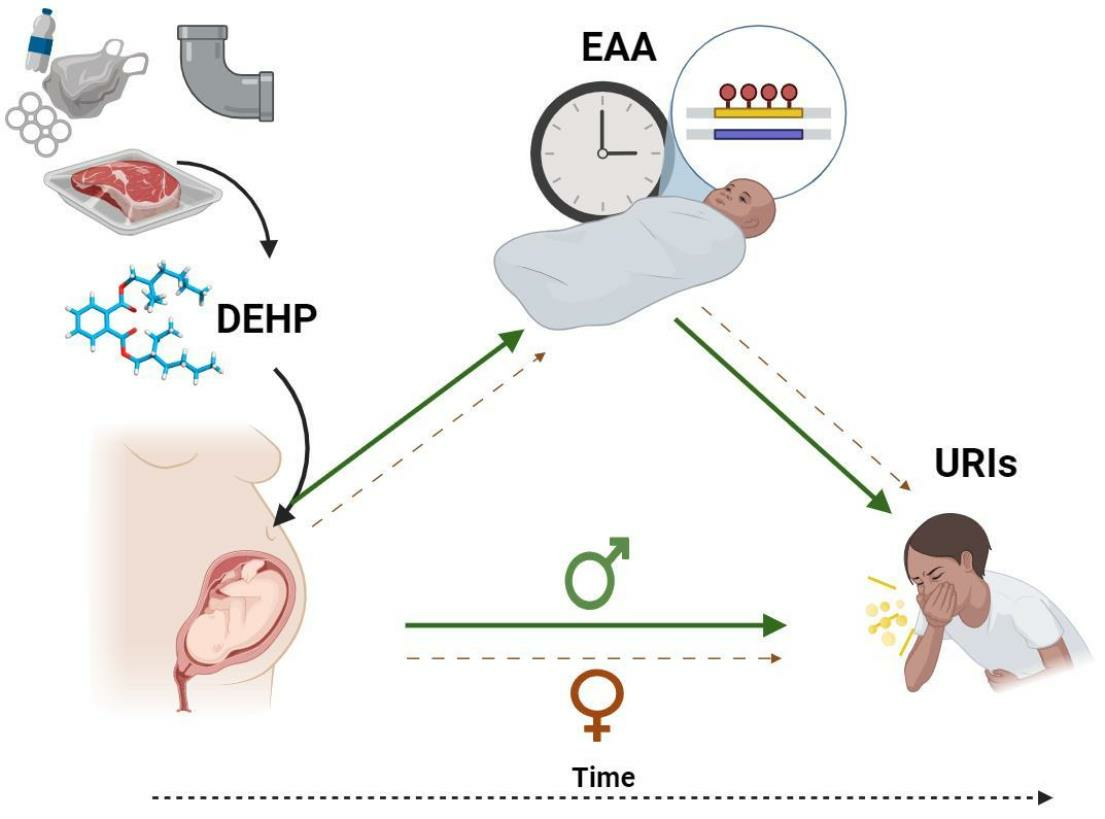
2.5. Overall Associations between Prenatal DEHP Exposure, Epigenetic Age, and Early Childhood URIs
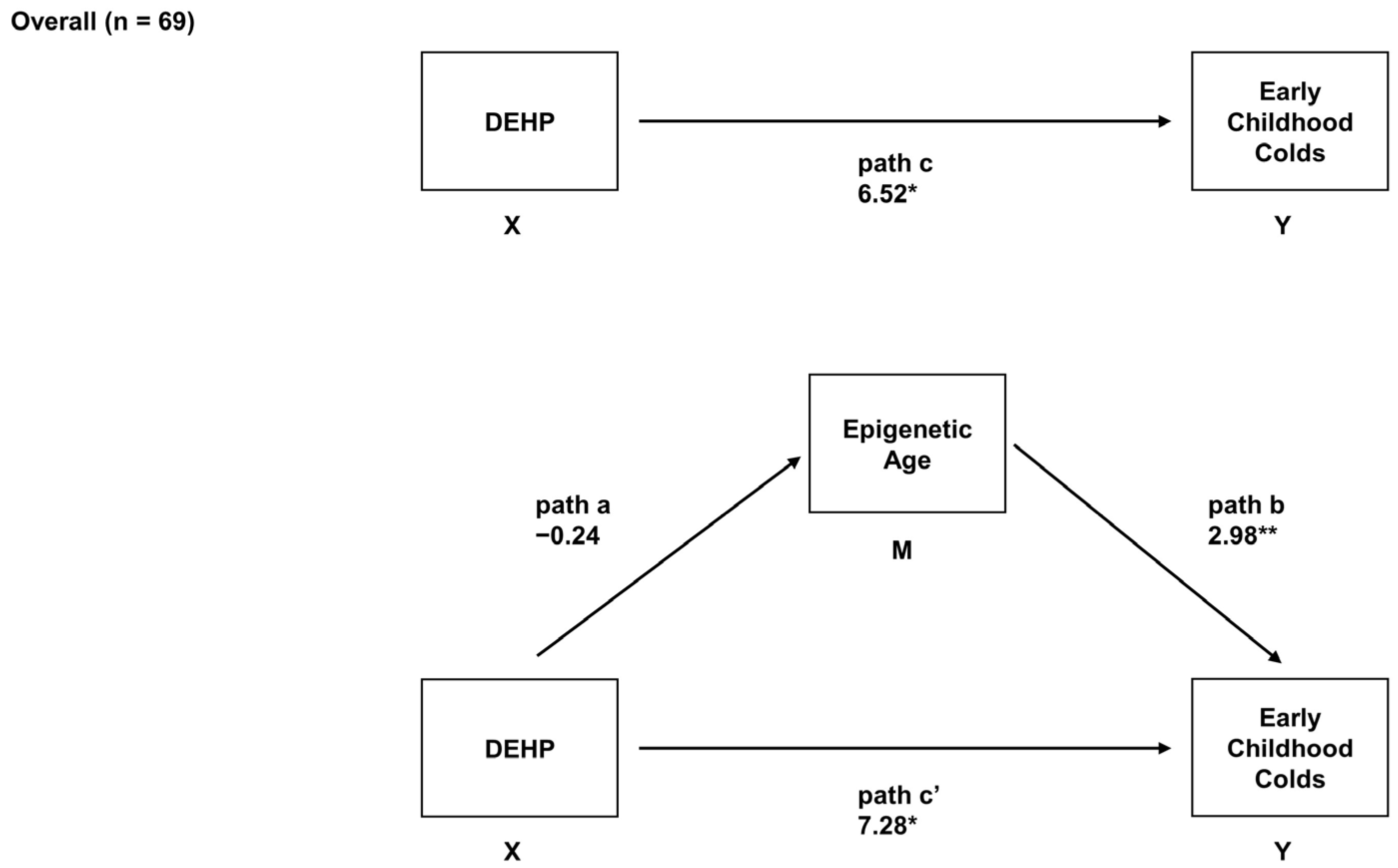
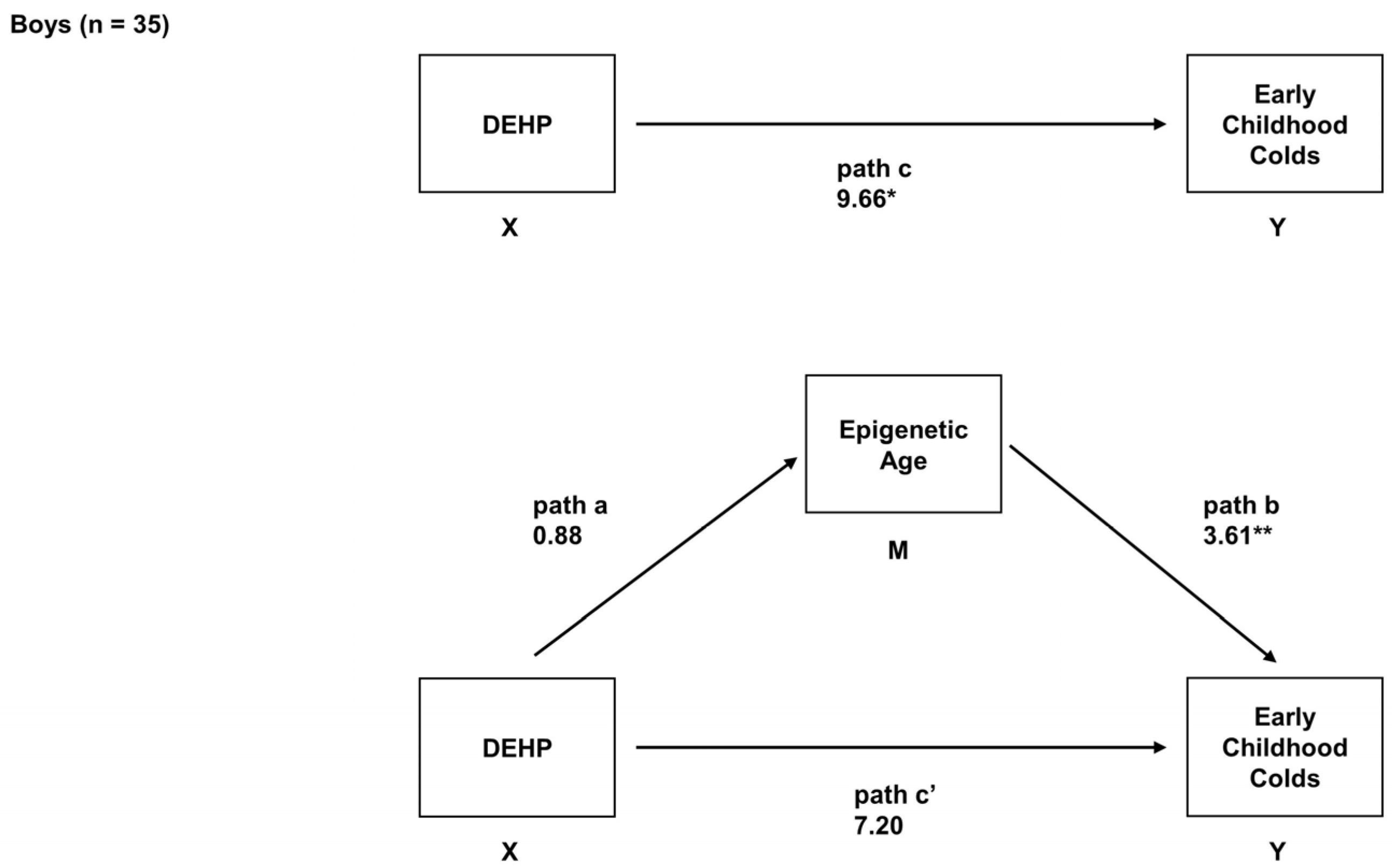
2.6. Sex-Specific Associations between Prenatal DEHP Exposure, Epigenetic Age, and Early Childhood URIs
2.7. Post Hoc and Sensitivity Analyses
3. Discussion
4. Materials and Methods
4.1. Participants and Procedure
4.2. Urinary DEHP Assessment
4.3. Children’s URIs
4.4. Epigenetic Age Calculation
4.4.1. Infant Blood Sample Collection
4.4.2. DNA Methylation Assay and Data Processing
4.4.3. Estimated Cell Type Proportions
4.4.4. Epigenetic Clock
4.5. Covariates
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worrall, G. Common Cold. Can. Fam. Physician 2011, 57, 1289–1290. [Google Scholar]
- Pappas, D.E. The Common Cold. Princ. Pract. Pediatr. Infect. Dis. 2018, 199–202.e1. [Google Scholar] [CrossRef]
- Canadian Paediatric Society Infectious Diseases and Immunization Committee Colds in Children. Paediatr. Child. Health 2005, 10, 493–495.
- Di Cicco, M.; D’Elios, S.; Peroni, D.G.; Comberiati, P. The Role of Atopy in Asthma Development and Persistence. Curr. Opin. Allergy Clin. Immunol. 2020, 20, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Jartti, T.; Bønnelykke, K.; Elenius, V.; Feleszko, W. Role of Viruses in Asthma. Semin. Immunopathol. 2020, 42, 61–74. [Google Scholar] [CrossRef]
- Ma, K.C.; Winn, A.; Moline, H.L.; Scobie, H.M.; Midgley, C.M.; Kirking, H.L.; Adjemian, J.; Hartnett, K.P.; Johns, D.; Jones, J.M.; et al. Increase in Acute Respiratory Illnesses Among Children and Adolescents Associated with Rhinoviruses and Enteroviruses, Including Enterovirus D68—United States, July–September 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 1265–1270. [Google Scholar] [CrossRef]
- Mohn, C.H.; Blix, H.S.; Halvorsen, J.A.; Nafstad, P.; Valberg, M.; Lagerløv, P. Incidence Trends of Atopic Dermatitis in Infancy and Early Childhood in a Nationwide Prescription Registry Study in Norway. JAMA Netw. Open 2018, 1, e184145. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Sekhon, S.; Sanchez, I.M.; Beck, K.M.; Bhutani, T. Recent Developments in Atopic Dermatitis. Pediatrics 2018, 142, e20181102. [Google Scholar] [CrossRef] [PubMed]
- Garner, R.; Kohen, D. Changes in the Prevalence of Asthma among Canadian Children. Health Rep. 2008, 19, 45–50. [Google Scholar]
- Gershon, A.S.; Guan, J.; Wang, C.; To, T. Trends in Asthma Prevalence and Incidence in Ontario, Canada, 1996–2005: A Population Study. Am. J. Epidemiol. 2010, 172, 728–736. [Google Scholar] [CrossRef]
- Gascon, M.; Casas, M.; Morales, E.; Valvi, D.; Ballesteros-Gómez, A.; Luque, N.; Rubio, S.; Monfort, N.; Ventura, R.; Martínez, D.; et al. Prenatal Exposure to Bisphenol A and Phthalates and Childhood Respiratory Tract Infections and Allergy. J. Allergy Clin. Immunol. 2015, 135, 370–378. [Google Scholar] [CrossRef]
- Sotir, M.; Yeatts, K.; Shy, C. Presence of Asthma Risk Factors and Environmental Exposures Related to Upper Respiratory Infection-Triggered Wheezing in Middle School-Age Children. Environ. Health Perspect. 2003, 111, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Kimber, I.; Dearman, R.J. An Assessment of the Ability of Phthalates to Influence Immune and Allergic Responses. Toxicology 2010, 271, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.; Miller, R. The Impact of Bisphenol A and Phthalates on Allergy, Asthma, and Immune Function: A Review of Latest Findings. Curr. Environ. Health Rep. 2015, 2, 379–387. [Google Scholar] [CrossRef]
- Kahn, L.G.; Philippat, C.; Nakayama, S.F.; Slama, R.; Trasande, L. Endocrine-Disrupting Chemicals: Implications for Human Health. Lancet Diabetes Endocrinol. 2020, 8, 703–718. [Google Scholar] [CrossRef]
- Koch, H.M.; Lorber, M.; Christensen, K.L.Y.; Pälmke, C.; Koslitz, S.; Brüning, T. Identifying Sources of Phthalate Exposure with Human Biomonitoring: Results of a 48h Fasting Study with Urine Collection and Personal Activity Patterns. Int. J. Hydrogen Environ. Health 2013, 216, 672–681. [Google Scholar] [CrossRef]
- Pacyga, D.C.; Sathyanarayana, S.; Strakovsky, R.S. Dietary Predictors of Phthalate and Bisphenol Exposures in Pregnant Women. Adv. Nutr. 2019, 10, 803–815. [Google Scholar] [CrossRef] [PubMed]
- McKeen, L.W. 3—Plastics Used in Medical Devices. In Handbook of Polymer Applications in Medicine and Medical Devices; Modjarrad, K., Ebnesajjad, S., Eds.; Plastics Design Library; William Andrew Publishing: Oxford, UK, 2014; pp. 21–53. ISBN 978-0-323-22805-3. [Google Scholar]
- Zarean, M.; Keikha, M.; Poursafa, P.; Khalighinejad, P.; Amin, M.; Kelishadi, R. A Systematic Review on the Adverse Health Effects of Di-2-Ethylhexyl Phthalate. Environ. Sci. Pollut. Res. 2016, 23, 24642–24693. [Google Scholar] [CrossRef]
- Guo, J.; Wu, M.; Gao, X.; Chen, J.; Li, S.; Chen, B.; Dong, R. Meconium Exposure to Phthalates, Sex and Thyroid Hormones, Birth Size and Pregnancy Outcomes in 251 Mother–Infant Pairs from Shanghai. Int. J. Environ. Res. Public. Health 2020, 17, 7711. [Google Scholar] [CrossRef]
- Katsikantami, I.; Tzatzarakis, M.N.; Alegakis, A.K.; Karzi, V.; Hatzidaki, E.; Stavroulaki, A.; Vakonaki, E.; Xezonaki, P.; Sifakis, S.; Rizos, A.K.; et al. Phthalate Metabolites Concentrations in Amniotic Fluid and Maternal Urine: Cumulative Exposure and Risk Assessment. Toxicol. Rep. 2020, 7, 529–538. [Google Scholar] [CrossRef]
- Li, L.-X.; Chen, L.; Meng, X.-Z.; Chen, B.-H.; Chen, S.-Q.; Zhao, Y.; Zhao, L.-F.; Liang, Y.; Zhang, Y.-H. Exposure Levels of Environmental Endocrine Disruptors in Mother-Newborn Pairs in China and Their Placental Transfer Characteristics. PLoS ONE 2013, 8, e62526. [Google Scholar] [CrossRef]
- Mose, T.; Mortensen, G.K.; Hedegaard, M.; Knudsen, L.E. Phthalate Monoesters in Perfusate from a Dual Placenta Perfusion System, the Placenta Tissue and Umbilical Cord Blood. Reprod. Toxicol. 2007, 23, 83–91. [Google Scholar] [CrossRef]
- Mose, T.; Knudsen, L.E.; Hedegaard, M.; Mortensen, G.K. Transplacental Transfer of Monomethyl Phthalate and Mono(2-Ethylhexyl) Phthalate in a Human Placenta Perfusion System. Int. J. Toxicol. 2007, 26, 221–229. [Google Scholar] [CrossRef]
- Shin, I.-S.; Lee, M.-Y.; Cho, E.-S.; Choi, E.; Son, H.-Y.; Lee, K.-Y. Effects of Maternal Exposure to Di(2-Ethylhexyl)Phthalate (DEHP) during Pregnancy on Susceptibility to Neonatal Asthma. Toxicol. Appl. Pharmacol. 2014, 274, 402–407. [Google Scholar] [CrossRef]
- Walker, C.; Ghazisaeidi, S.; Collet, B.; Boisvert, A.; Culty, M. In Utero Exposure to Low Doses of Genistein and Di-(2-Ethylhexyl) Phthalate (DEHP) Alters Innate Immune Cells in Neonatal and Adult Rat Testes. Andrology 2020, 8, 943–964. [Google Scholar] [CrossRef]
- Yen, P.-L.; Yang, C.-R.; Huang, M.-L.; Lin, T.-A.; Liao, V.H.-C. Chronic Exposure to Di(2-Ethylhexyl) Phthalate (DEHP) Weakens Innate Immunity and Leads to Immunosenescence in C. Elegans. Environ. Toxicol. Pharmacol. 2023, 98, 104071. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Han, B.; Qin, L.; Li, B.; You, H.; Yang, J.; Liu, D.; Wei, C.; Nanberg, E.; Bornehag, C.-G.; et al. Pulmonary Toxicity and Adjuvant Effect of Di-(2-Exylhexyl) Phthalate in Ovalbumin-Immunized BALB/c Mice. PLoS ONE 2012, 7, e39008. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.T.; Hansen, J.S.; Hansen, E.W.; Clausen, P.A.; Nielsen, G.D. Airway Inflammation and Adjuvant Effect after Repeated Airborne Exposures to Di-(2-Ethylhexyl)Phthalate and Ovalbumin in BALB/c Mice. Toxicology 2007, 235, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lyu, L.; Tao, Y.; Ju, H.; Chen, J. Health Risks of Phthalates: A Review of Immunotoxicity. Environ. Pollut. 2022, 313, 120173. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S. Targeting the Macrophage: Immune Cells May Be the Key to Phthalate-Induced Liver Toxicity. Environ. Health Perspect. 2022, 130, 034003. [Google Scholar] [CrossRef] [PubMed]
- Ashley-Martin, J.; Dodds, L.; Levy, A.R.; Platt, R.W.; Marshall, J.S.; Arbuckle, T.E. Prenatal Exposure to Phthalates, Bisphenol A and Perfluoroalkyl Substances and Cord Blood Levels of IgE, TSLP and IL-33. Environ. Res. 2015, 140, 360–368. [Google Scholar] [CrossRef]
- Tsai, C.-K.; Cheng, H.-H.; Hsu, T.-Y.; Wang, J.-Y.; Hung, C.-H.; Tsai, C.-C.; Lai, Y.-J.; Lin, Y.-J.; Huang, H.-C.; Chan, J.Y.H.; et al. Prenatal Exposure to Di-Ethyl Phthalate (DEP) Is Related to Increasing Neonatal IgE Levels and the Altering of the Immune Polarization of Helper-T Cells. Int. J. Environ. Res. Public. Health 2021, 18, 6364. [Google Scholar] [CrossRef]
- Eisner, A.; Gao, Y.; Collier, F.; Drummond, K.; Thomson, S.; Burgner, D.; Vuillermin, P.; Tang, M.L.; Mueller, J.; Symeonides, C.; et al. Cord Blood Immune Profile: Associations with Higher Prenatal Plastic Chemical Levels. Environ. Pollut. 2022, 315, 120332. [Google Scholar] [CrossRef]
- Jøhnk, C.; Høst, A.; Husby, S.; Schoeters, G.; Timmermann, C.A.G.; Kyhl, H.B.; Beck, I.H.; Andersson, A.-M.; Frederiksen, H.; Jensen, T.K. Maternal Phthalate Exposure and Asthma, Rhinitis and Eczema in 552 Children Aged 5 Years; a Prospective Cohort Study. Environ. Health 2020, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- James, K.M.; Peebles, R.S.; Hartert, T.V. Response to Infections in Patients with Asthma and Atopic Disease: An Epiphenomenon or Reflection of Host Susceptibility? J. Allergy Clin. Immunol. 2012, 130, 343–351. [Google Scholar] [CrossRef]
- Kansen, H.M.; Lebbink, M.A.; Mul, J.; van Erp, F.C.; van Engelen, M.; de Vries, E.; Prevaes, S.M.P.J.; Le, T.M.; van der Ent, C.K.; Verhagen, L.M. Risk Factors for Atopic Diseases and Recurrent Respiratory Tract Infections in Children. Pediatr. Pulmonol. 2020, 55, 3168–3179. [Google Scholar] [CrossRef]
- Winans, B.; Humble, M.C.; Lawrence, B.P. Environmental Toxicants and the Developing Immune System: A Missing Link in the Global Battle against Infectious Disease? Reprod. Toxicol. 2011, 31, 327–336. [Google Scholar] [CrossRef]
- Aristizabal, M.J.; Anreiter, I.; Halldorsdottir, T.; Odgers, C.L.; McDade, T.W.; Goldenberg, A.; Mostafavi, S.; Kobor, M.S.; Binder, E.B.; Sokolowski, M.B.; et al. Biological Embedding of Experience: A Primer on Epigenetics. Proc. Natl. Acad. Sci. USA 2020, 117, 23261–23269. [Google Scholar] [CrossRef]
- Benjamin, S.; Masai, E.; Kamimura, N.; Takahashi, K.; Anderson, R.C.; Faisal, P.A. Phthalates Impact Human Health: Epidemiological Evidences and Plausible Mechanism of Action. J. Hazard. Mater. 2017, 340, 360–383. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Haggerty, D.K.; Rappolee, D.A.; Ruden, D.M. Phthalate Exposure and Long-Term Epigenomic Consequences: A Review. Front. Genet. 2020, 11, 405. [Google Scholar] [CrossRef] [PubMed]
- Khodasevich, D.; Holland, N.; Hubbard, A.; Harley, K.; Deardorff, J.; Eskenazi, B.; Cardenas, A. Associations between Prenatal Phthalate Exposure and Childhood Epigenetic Age Acceleration. Environ. Res. 2023, 231, 116067. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Chandra, T. Epigenetic Age Prediction. Aging Cell 2021, 20, e13452. [Google Scholar] [CrossRef]
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Binder, A.M.; Horvath, S. Epigenetic Clocks. In Epigenetic Epidemiology; Michels, K.B., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 261–276. ISBN 978-3-030-94475-9. [Google Scholar]
- Bejaoui, Y.; Amanullah, F.H.; Saad, M.; Taleb, S.; Bradic, M.; Megarbane, A.; Hssain, A.A.; Khalil, C.A.; Hajj, N.E. Epigenetic Age Acceleration in Surviving versus Deceased COVID-19 Patients with Acute Respiratory Distress Syndrome Following Hospitalization. Clin. Epigenetics 2023, 15, 186. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Cardenas, A.; Rifas-Shiman, S.L.; Hivert, M.-F.; Gold, D.R.; Platts-Mills, T.A.; Lin, X.; Oken, E.; Avila, L.; Celedón, J.C.; et al. Epigenetic Age Acceleration Is Associated with Allergy and Asthma in Children in Project Viva. J. Allergy Clin. Immunol. 2019, 143, 2263–2270.e14. [Google Scholar] [CrossRef]
- Vasileva, D.; Greenwood, C.M.T.; Daley, D. A Review of the Epigenetic Clock: Emerging Biomarkers for Asthma and Allergic Disease. Genes 2023, 14, 1724. [Google Scholar] [CrossRef] [PubMed]
- Merrill, S.M.; Gladish, N.; Fu, M.P.; Moore, S.R.; Konwar, C.; Giesbrecht, G.F.; MacIssac, J.L.; Kobor, M.S.; Letourneau, N.L. Associations of Peripheral Blood DNA Methylation and Estimated Monocyte Proportion Differences during Infancy with Toddler Attachment Style. Attach. Hum. Dev. 2023, 25, 132–161. [Google Scholar] [CrossRef]
- Fang, F.; Zhou, L.; Perng, W.; Marsit, C.J.; Knight, A.K.; Cardenas, A.; Aung, M.T.; Hivert, M.-F.; Aris, I.M.; Goodrich, J.M.; et al. Evaluation of Pediatric Epigenetic Clocks across Multiple Tissues. Clin. Epigenetics 2023, 15, 142. [Google Scholar] [CrossRef]
- Heikkinen, T.; Järvinen, A. The Common Cold. Lancet 2003, 361, 51–59. [Google Scholar] [CrossRef]
- Hellgren, J.; Cervin, A.; Nordling, S.; Bergman, A.; Cardell, L.O. Allergic Rhinitis and the Common Cold—High Cost to Society. Allergy 2010, 65, 776–783. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.Q. Phthalate Exposure and Lung Disease: The Epidemiological Evidences, Plausible Mechanism and Advocacy of Interventions. Rev. Environ. Health 2022. [Google Scholar] [CrossRef]
- Zhang, Y.; Mustieles, V.; Sun, Y.; Oulhote, Y.; Wang, Y.-X.; Messerlian, C. Association between Serum Per- and Polyfluoroalkyl Substances Concentrations and Common Cold among Children and Adolescents in the United States. Environ. Int. 2022, 164, 107239. [Google Scholar] [CrossRef]
- Taneja, V. Sex Hormones Determine Immune Response. Front. Immunol. 2018, 9, 1931. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.A.; Sheikh, I.A. Endocrine Disruption: Structural Interactions of Androgen Receptor against Di(2-Ethylhexyl) Phthalate and Its Metabolites. Toxics 2020, 8, 115. [Google Scholar] [CrossRef]
- Borch, J.; Metzdorff, S.B.; Vinggaard, A.M.; Brokken, L.; Dalgaard, M. Mechanisms Underlying the Anti-Androgenic Effects of Diethylhexyl Phthalate in Fetal Rat Testis. Toxicology 2006, 223, 144–155. [Google Scholar] [CrossRef]
- Foster, P.M.D. Mode of Action: Impaired Fetal Leydig Cell Function—Effects on Male Reproductive Development Produced by Certain Phthalate Esters. Crit. Rev. Toxicol. 2005, 35, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Parks, L.G.; Ostby, J.S.; Lambright, C.R.; Abbott, B.D.; Klinefelter, G.R.; Barlow, N.J.; Gray, L.E. The Plasticizer Diethylhexyl Phthalate Induces Malformations by Decreasing Fetal Testosterone Synthesis during Sexual Differentiation in the Male Rat. Toxicol. Sci. 2000, 58, 339–349. [Google Scholar] [CrossRef]
- Sathyanarayana, S.; Barrett, E.; Butts, S.; Wang, C.; Swan, S.H. Phthalate Exposure and Reproductive Hormone Concentrations in Pregnancy. Reproduction 2014, 147, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-C.; Wang, S.-L.; Chang, Y.-C.; Huang, P.-C.; Cheng, J.-T.; Su, P.-H.; Liao, P.-C. Associations between Maternal Phthalate Exposure and Cord Sex Hormones in Human Infants. Chemosphere 2011, 83, 1192–1199. [Google Scholar] [CrossRef]
- Guerra-Silveira, F.; Abad-Franch, F. Sex Bias in Infectious Disease Epidemiology: Patterns and Processes. PLoS ONE 2013, 8, e62390. [Google Scholar] [CrossRef]
- Ait Bamai, Y.; Miyashita, C.; Araki, A.; Nakajima, T.; Sasaki, S.; Kishi, R. Effects of Prenatal Di(2-Ethylhexyl) Phthalate Exposure on Childhood Allergies and Infectious Diseases: The Hokkaido Study on Environment and Children’s Health. Sci. Total Environ. 2018, 618, 1408–1415. [Google Scholar] [CrossRef]
- Huang, Y.; Garcia, J.M.; Shu, W.; Rong, H.; Zhang, L.; Wang, Y.; Tan, Y.; Lin, H.; Zeng, H.; Chen, J. Peroxisome Proliferator Activated Receptor Gamma in Human Placenta May Mediate the Adverse Effects of Phthalates Exposure in Pregnancy. Reprod. Toxicol. 2018, 75, 121–126. [Google Scholar] [CrossRef]
- Schlezinger, J.J.; Howard, G.J.; Hurst, C.H.; Emberley, J.K.; Waxman, D.J.; Webster, T.; Sherr, D.H. Environmental and Endogenous Peroxisome Proliferator-Activated Receptor γ Agonists Induce Bone Marrow B Cell Growth Arrest and Apoptosis: Interactions between Mono(2-Ethylhexyl)Phthalate, 9-Cis-Retinoic Acid, and 15-Deoxy-Δ12,14-Prostaglandin J21. J. Immunol. 2004, 173, 3165–3177. [Google Scholar] [CrossRef]
- Xu, M.; Li, Y.; Wang, X.; Zhang, Q.; Wang, L.; Zhang, X.; Cui, W.; Han, X.; Ma, N.; Li, H.; et al. Role of Hepatocyte- and Macrophage-Specific PPARγ in Hepatotoxicity Induced by Diethylhexyl Phthalate in Mice. Environ. Health Perspect. 2022, 130, 017005. [Google Scholar] [CrossRef]
- Palacios-Arreola, M.I.; Morales-Montor, J.; Cazares-Martinez, C.J.; Gomez-Arroyo, S.; Nava-Castro, K.E. Environmental Pollutants: An Immunoendocrine Perspective on Phthalates. Front. Biosci.-Landmark 2020, 26, 401–430. [Google Scholar] [CrossRef]
- Shepherd, R.; Cheung, A.S.; Pang, K.; Saffery, R.; Novakovic, B. Sexual Dimorphism in Innate Immunity: The Role of Sex Hormones and Epigenetics. Front. Immunol. 2020, 11, 604000. [Google Scholar] [CrossRef]
- AbdelHamid, S.G.; Refaat, A.A.; Benjamin, A.M.; Elmawardy, L.A.; Elgendy, L.A.; Manolly, M.M.; Elmaksoud, N.A.; Sherif, N.; Hamdy, N.M. Deciphering Epigenetic(s) Role in Modulating Susceptibility to and Severity of COVID-19 Infection and/or Outcome: A Systematic Rapid Review. Environ. Sci. Pollut. Res. 2021, 28, 54209–54221. [Google Scholar] [CrossRef] [PubMed]
- Crimi, E.; Benincasa, G.; Figueroa-Marrero, N.; Galdiero, M.; Napoli, C. Epigenetic Susceptibility to Severe Respiratory Viral Infections and Its Therapeutic Implications: A Narrative Review. Br. J. Anaesth. 2020, 125, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, W.-H. Epigenetic Clocks in the Pediatric Population: When and Why They Tick? Chin. Med. J. 2021, 134, 2901–2910. [Google Scholar] [CrossRef] [PubMed]
- Mansell, T.; Saffery, R. The End of the Beginning: Epigenetic Variation in Utero as a Mediator of Later Human Health and Disease. Epigenomics 2017, 9, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Faul, J.D.; Kim, J.K.; Levine, M.E.; Thyagarajan, B.; Weir, D.R.; Crimmins, E.M. Epigenetic-Based Age Acceleration in a Representative Sample of Older Americans: Associations with Aging-Related Morbidity and Mortality. Proc. Natl. Acad. Sci. USA 2023, 120, e2215840120. [Google Scholar] [CrossRef] [PubMed]
- Yusupov, N.; Dieckmann, L.; Erhart, M.; Sauer, S.; Rex-Haffner, M.; Kopf-Beck, J.; Brückl, T.M.; Czamara, D.; Binder, E.B. Transdiagnostic Evaluation of Epigenetic Age Acceleration and Burden of Psychiatric Disorders. Neuropsychopharmacology 2023, 48, 1409–1417. [Google Scholar] [CrossRef]
- Roubinov, D.; Meaney, M.J.; Boyce, W.T. Change of Pace: How Developmental Tempo Varies to Accommodate Failed Provision of Early Needs. Neurosci. Biobehav. Rev. 2021, 131, 120–134. [Google Scholar] [CrossRef]
- England-Mason, G.; Merrill, S.M.; Gladish, N.; Moore, S.R.; Giesbrecht, G.F.; Letourneau, N.; MacIsaac, J.L.; MacDonald, A.M.; Kinniburgh, D.W.; Ponsonby, A.-L.; et al. Prenatal Exposure to Phthalates and Peripheral Blood and Buccal Epithelial DNA Methylation in Infants: An Epigenome-Wide Association Study. Environ. Int. 2022, 163, 107183. [Google Scholar] [CrossRef]
- Engelbrecht, H.-R.; Merrill, S.M.; Gladish, N.; MacIsaac, J.L.; Lin, D.T.S.; Ecker, S.; Chrysohoou, C.A.; Pes, G.M.; Kobor, M.S.; Rehkopf, D.H. Sex Differences in Epigenetic Age in Mediterranean High Longevity Regions. Front. Aging 2022, 3, 1007098. [Google Scholar] [CrossRef] [PubMed]
- Kankaanpää, A.; Tolvanen, A.; Saikkonen, P.; Heikkinen, A.; Laakkonen, E.K.; Kaprio, J.; Ollikainen, M.; Sillanpää, E. Do Epigenetic Clocks Provide Explanations for Sex Differences in Life Span? A Cross-Sectional Twin Study. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 1898–1906. [Google Scholar] [CrossRef]
- Haugen, A.C.; Schug, T.T.; Collman, G.; Heindel, J.J. Evolution of DOHaD: The Impact of Environmental Health Sciences. J. Dev. Orig. Health Dis. 2015, 6, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.Q.V.; Miyake, K. Neurodevelopmental Disorders and Environmental Toxicants: Epigenetics as an Underlying Mechanism. Int. J. Genom. 2017, 2017, 7526592. [Google Scholar] [CrossRef]
- Bulka, C.M.; Enggasser, A.E.; Fry, R.C. Epigenetics at the Intersection of COVID-19 Risk and Environmental Chemical Exposures. Curr. Environ. Health Rep. 2022, 9, 477–489. [Google Scholar] [CrossRef]
- Laffont, S.; Guéry, J.-C. Deconstructing the Sex Bias in Allergy and Autoimmunity: From Sex Hormones and Beyond. Adv. Immunol. 2019, 142, 35–64. [Google Scholar] [CrossRef]
- Zazara, D.E.; Arck, P.C. Developmental Origin and Sex-Specific Risk for Infections and Immune Diseases Later in Life. Semin. Immunopathol. 2019, 41, 137–151. [Google Scholar] [CrossRef]
- Dietert, R.R. Developmental Immunotoxicity, Perinatal Programming, and Noncommunicable Diseases: Focus on Human Studies. Adv. Med. 2014, 2014, 867805. [Google Scholar] [CrossRef]
- Lucaccioni, L.; Trevisani, V.; Passini, E.; Righi, B.; Plessi, C.; Predieri, B.; Iughetti, L. Perinatal Exposure to Phthalates: From Endocrine to Neurodevelopment Effects. Int. J. Mol. Sci. 2021, 22, 4063. [Google Scholar] [CrossRef] [PubMed]
- Main, K.M.; Mortensen, G.K.; Kaleva, M.M.; Boisen, K.A.; Damgaard, I.N.; Chellakooty, M.; Schmidt, I.M.; Suomi, A.-M.; Virtanen, H.E.; Petersen, J.H.; et al. Human Breast Milk Contamination with Phthalates and Alterations of Endogenous Reproductive Hormones in Infants Three Months of Age. Environ. Health Perspect. 2006, 114, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Haro, M.Y.; Camacho-Pacheco, R.T.; Brito-Pérez, Y.; Mancilla-Herrera, I. The Hormonal Physiology of Immune Components in Breast Milk and Their Impact on the Infant Immune Response. Mol. Cell. Endocrinol. 2023, 572, 111956. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, B.J.; Giesbrecht, G.F.; Leung, B.M.Y.; Field, C.J.; Dewey, D.; Bell, R.C.; Manca, D.P.; O’Beirne, M.; Johnston, D.W.; Pop, V.J.; et al. The Alberta Pregnancy Outcomes and Nutrition (APrON) Cohort Study: Rationale and Methods. Matern. Child. Nutr. 2014, 10, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, N.; Aghajafari, F.; Bell, R.C.; Deane, A.J.; Dewey, D.; Field, C.; Giesbrecht, G.; Kaplan, B.; Leung, B.; Ntanda, H. The Alberta Pregnancy Outcomes and Nutrition (APrON) Longitudinal Study: Cohort Profile and Key Findings from the First Three Years. BMJ Open 2022, 12, e047503. [Google Scholar] [CrossRef] [PubMed]
- England-Mason, G.; Grohs, M.N.; Reynolds, J.E.; MacDonald, A.; Kinniburgh, D.; Liu, J.; Martin, J.W.; Lebel, C.; Dewey, D.; APrON Study Team. White Matter Microstructure Mediates the Association between Prenatal Exposure to Phthalates and Behavior Problems in Preschool Children. Environ. Res. 2020, 182, 109093. [Google Scholar] [CrossRef] [PubMed]
- England-Mason, G.; Martin, J.W.; MacDonald, A.; Kinniburgh, D.; Giesbrecht, G.F.; Letourneau, N.; Dewey, D. Similar Names, Different Results: Consistency of the Associations between Prenatal Exposure to Phthalates and Parent-Ratings of Behavior Problems in Preschool Children. Environ. Int. 2020, 142, 105892. [Google Scholar] [CrossRef]
- Hornung, R.W.; Reed, L.D. Estimation of Average Concentration in the Presence of Nondetectable Values. Appl. Occup. Environ. Hyg. 1990, 5, 46–51. [Google Scholar] [CrossRef]
- Messerlian, C.; Wylie, B.J.; Minguez-Alarcon, L.; Williams, P.L.; Ford, J.B.; Souter, I.C.; Calafat, A.M.; Hauser, R. Urinary Concentrations of Phthalate Metabolites in Relation to Pregnancy Loss among Women Conceiving with Medically Assisted Reproduction. Epidemiology 2016, 27, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, N.L.; Kozyrskyj, A.L.; Cosic, N.; Ntanda, H.N.; Anis, L.; Hart, M.J.; Campbell, T.S.; Giesbrecht, G.F. Maternal Sensitivity and Social Support Protect against Childhood Atopic Dermatitis. Allergy Asthma Clin. Immunol. 2017, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High Density DNA Methylation Array with Single CpG Site Resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA Methylation Arrays as Surrogate Measures of Cell Mixture Distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Gervin, K.; Salas, L.A.; Bakulski, K.M.; Van Zelm, M.C.; Koestler, D.C.; Wiencke, J.K.; Duijts, L.; Moll, H.A.; Kelsey, K.T.; Kobor, M.S.; et al. Systematic Evaluation and Validation of Reference and Library Selection Methods for Deconvolution of Cord Blood DNA Methylation Data. Clin. Epigenetics 2019, 11, 125. [Google Scholar] [CrossRef]
- Koestler, D.C.; Jones, M.J.; Usset, J.; Christensen, B.C.; Butler, R.A.; Kobor, M.S.; Wiencke, J.K.; Kelsey, K.T. Improving Cell Mixture Deconvolution by Identifying Optimal DNA Methylation Libraries (IDOL). BMC Bioinform. 2016, 17, 120. [Google Scholar] [CrossRef]
- Salas, L.A.; Zhang, Z.; Koestler, D.C.; Butler, R.A.; Hansen, H.M.; Molinaro, A.M.; Wiencke, J.K.; Kelsey, K.T.; Christensen, B.C. Enhanced Cell Deconvolution of Peripheral Blood Using DNA Methylation for High-Resolution Immune Profiling. Nat. Commun. 2022, 13, 761. [Google Scholar] [CrossRef]
- Merrill, S.M.; Moore, S.R.; Gladish, N.; Giesbrecht, G.F.; Dewey, D.; Konwar, C.; MacIssac, J.L.; Kobor, M.S.; Letourneau, N.L. Paternal Adverse Childhood Experiences: Associations with Infant DNA Methylation. Dev. Psychobiol. 2021, 63, e22174. [Google Scholar] [CrossRef]
- Hermansen, M.C. Nucleated Red Blood Cells in the Fetus and Newborn. Arch. Dis. Child. Fetal Neonatal Ed. 2001, 84, F211–F215. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA Methylation Aging Clocks: Challenges and Recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef]
- Moore, S.R.; McEwen, L.M.; Quirt, J.; Morin, A.; Mah, S.M.; Barr, R.G.; Boyce, W.T.; Kobor, M.S. Epigenetic Correlates of Neonatal Contact in Humans. Dev. Psychopathol. 2017, 29, 1517–1538. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic Clock for Skin and Blood Cells Applied to Hutchinson Gilford Progeria Syndrome and Ex Vivo Studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Impinen, A.; Nygaard, U.C.; Lødrup Carlsen, K.C.; Mowinckel, P.; Carlsen, K.H.; Haug, L.S.; Granum, B. Prenatal Exposure to Perfluoralkyl Substances (PFASs) Associated with Respiratory Tract Infections but Not Allergy- and Asthma-Related Health Outcomes in Childhood. Environ. Res. 2018, 160, 518–523. [Google Scholar] [CrossRef]
- Lourenço, V.M.; Pires, A.M.; Kirst, M. Robust Linear Regression Methods in Association Studies. Bioinformatics 2011, 27, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Venables; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Greenland, S.; Daniel, R.; Pearce, N. Outcome Modelling Strategies in Epidemiology: Traditional Methods and Basic Alternatives. Int. J. Epidemiol. 2016, 45, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Zu, J.; Yuan, K.-H. Local Influence and Robust Procedures for Mediation Analysis. Multivar. Behav. Res. 2010, 45, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.F. Introduction to Mediation, Moderation, and Conditional Process Analysis: A Regression-Based Approach; Guilford Publications: New York, NY, USA, 2022; ISBN 978-1-4625-4903-0. [Google Scholar]
- MacKinnon, D.P.; Fairchild, A.J.; Fritz, M.S. Mediation Analysis. Annu. Rev. Psychol. 2007, 58, 593. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Fritz, M.S.; Mackinnon, D.P. Required Sample Size to Detect the Mediated Effect. Psychol. Sci. 2007, 18, 233–239. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n (%) | Mean (SD) | |
|---|---|---|
| Maternal Characteristics | ||
| Age (years) | - | 31.24 (4.01) |
| White | 64 (92.75%) | - |
| Married/Cohabiting | 69 (100.00%) | - |
| Household Income > CAD 70k 1 | 56 (81.16%) | - |
| Child Characteristics | ||
| Birthweight (g) | - | 3479.42 (505.91) |
| Gestational age at birth (weeks) | - | 39.49 (1.40) |
| Sex (Female) | 34 (49.28%) | - |
| Age at blood draw (weeks) | - | 12.52 (0.96) |
| Number of colds | - | 2.54 (1.51) |
| Metabolite | % > LOD | Minimum | Maximum | GM | 25th Percentile | 50th Percentile | 75th Percentile |
|---|---|---|---|---|---|---|---|
| MEHP 1 | 100% | 0.712 | 31.0 | 3.52 | 2.26 | 3.12 | 4.95 |
| MEHHP 1 | 100% | 2.32 | 33.5 | 10.6 | 7.05 | 9.81 | 16.4 |
| MEOHP 1 | 100% | 2.58 | 25.7 | 9.13 | 6.53 | 8.83 | 13.0 |
| MECPP 1 | 100% | 6.41 | 62.4 | 16.6 | 11.9 | 16.0 | 23.3 |
| DEHP 2 | - | 0.0222 | 0.285 | 0.0814 | 0.0534 | 0.0755 | 0.126 |
| IEAA | EAA (Skin and Blood Clock) | |
|---|---|---|
| B (95% CI) | B (95% CI) | |
| Overall Sample (n = 69) | ||
| Total Effect (path c) 1 | - | - |
| Path a | −0.34 (−1.30, 0.61) | −0.03 (−0.58, 0.51) |
| Path b | 2.98 † (1.67, 4.28) | 4.15 † (1.38, 6.92) |
| Direct effect (path c’) | 7.81 † (2.54, 13.07) | 7.11 † (1.63, 12.58) |
| Indirect effect (path ab) | −1.79 (−5.41, 1.26) | −0.07 (−2.34, 2.35) |
| Girls (n = 34) | ||
| Total Effect (path c) 1 | - | - |
| Path a | −2.40 † (−3.54, −1.27) | −0.80 † (−1.59, −0.02) |
| Path b | 1.04 (−1.88, 3.96) | 3.47 (−0.79, 7.73) |
| Direct effect (path c’) | −3.47 (−15.03, 8.09) | −2.24 (−11.86, 7.39) |
| Indirect effect (path ab) | −2.11 (−8.56, 5.87) | −3.90 † (−10.56, −0.23) |
| Boys (n = 35) | ||
| Total Effect (path c) 1 | - | - |
| Path a | 0.85 (−0.54, 2.25) | 0.50 (−0.16, 1.17) |
| Path b | 3.70 † (1.74, 5.66) | 4.55 † (0.28, 8.81) |
| Direct effect (path c’) | 7.29 (−0.12, 14.70) | 8.38 (−0.11, 16.87) |
| Indirect effect (path ab) | 3.27 (−0.78, 8.58) | 3.15 † (0.04, 9.52) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merrill, S.M.; Letourneau, N.; Giesbrecht, G.F.; Edwards, K.; MacIsaac, J.L.; Martin, J.W.; MacDonald, A.M.; Kinniburgh, D.W.; Kobor, M.S.; Dewey, D.; et al. Sex-Specific Associations between Prenatal Exposure to Di(2-ethylhexyl) Phthalate, Epigenetic Age Acceleration, and Susceptibility to Early Childhood Upper Respiratory Infections. Epigenomes 2024, 8, 3. https://doi.org/10.3390/epigenomes8010003
Merrill SM, Letourneau N, Giesbrecht GF, Edwards K, MacIsaac JL, Martin JW, MacDonald AM, Kinniburgh DW, Kobor MS, Dewey D, et al. Sex-Specific Associations between Prenatal Exposure to Di(2-ethylhexyl) Phthalate, Epigenetic Age Acceleration, and Susceptibility to Early Childhood Upper Respiratory Infections. Epigenomes. 2024; 8(1):3. https://doi.org/10.3390/epigenomes8010003
Chicago/Turabian StyleMerrill, Sarah M., Nicole Letourneau, Gerald F. Giesbrecht, Karlie Edwards, Julia L. MacIsaac, Jonathan W. Martin, Amy M. MacDonald, David W. Kinniburgh, Michael S. Kobor, Deborah Dewey, and et al. 2024. "Sex-Specific Associations between Prenatal Exposure to Di(2-ethylhexyl) Phthalate, Epigenetic Age Acceleration, and Susceptibility to Early Childhood Upper Respiratory Infections" Epigenomes 8, no. 1: 3. https://doi.org/10.3390/epigenomes8010003