
PNPT1 Spectrum Disorders: An Underrecognized and Complex Group of Neurometabolic Disorders

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| ADCA | Autosomal Dominant Cerebellar Ataxias |
| AMP | Association for Molecular Pathology |
| ARSACS | Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay |
| CAPOS | Cerebellar Ataxia, Areflexia, Pes Cavus, Optic Atrophy, and Sensorineural Hearing Loss |
| HSP | Hereditary Spastic Paraplegia |
| ICARS | International Cooperative Ataxia Rating Scale |
| IEM | Inborn Errors of Metabolism |
| MRI | Magnetic Resonance Imaging |
| mtRNA | Mitochondrial RNA |
| NGS | Next-Generation Sequencing |
| PNPase | Polynucleotide phosphorylase |
| SARA | Scale for the Assessment and Rating of Ataxia |
| SCA | Spinocerebellar Ataxia |
| SCA7 | Spinocerebellar Ataxia type 7 |
| SCA25 | Spinocerebellar Ataxia type 25 |
| SPAX | Spastic ataxias |
| SPG | Spastic Paraplegia |
| SPOAN | Spastic Paraplegia, Optic Atrophy, and Neuropathy |
| SPRS | Spastic Paraplegia Rating Scale |
| SWI | Susceptibility weighted imaging |
| WES | Whole-Exome Sequencing |
References
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Beaudin, M.; Matilla-Dueñas, A.; Soong, B.W.; Pedroso, J.L.; Barsottini, O.G.; Mitoma, H.; Tsuji, S.; Schmahmann, J.D.; Manto, M.; Rouleau, G.A.; et al. The classification of Autosomal Recessive Cerebellar Ataxias: A Consensus Statement from the Society for Research on the Cerebellum and Ataxias Task Force. Cerebellum 2019, 18, 1098–1125. [Google Scholar] [CrossRef]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Primers 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.V.S.; Pinto, W.B.V.R.; Batistella, G.N.R.; Bortholin, T.; Oliveira, A.S.B. Hereditary Spastic Paraplegia: Clinical and genetic hallmarks. Cerebellum 2017, 16, 525–551. [Google Scholar] [CrossRef] [PubMed]
- Freitas, J.L.; Rezende Filho, F.M.; Sallum, J.M.F.; França, M.C., Jr.; Pedroso, J.L.; Barsottini, O.G.P. Ophthalmological changes in hereditary spastic paraplegia and other genetic diseases with spastic paraplegia. J. Neurol. Sci. 2020, 409, 116620. [Google Scholar] [CrossRef]
- Panwala, T.F.; Garcia-Santibanez, R.; Vizcarra, J.A.; Garcia, A.G.; Verma, S. Childhood-onset Hereditary Spastic Paraplegia (HSP): A case series and review of literature. Pediatr. Neurol. 2022, 130, 7–13. [Google Scholar] [CrossRef]
- Bot, S.T.; Willemsen, M.A.A.P.; Vermeer, S.; Kremer, H.P.H.; van de Warrenburg, B.P.C. Reviewing the genetic causes of spastic-ataxias. Neurology 2012, 79, 1507–1514. [Google Scholar] [CrossRef]
- Pedroso, J.L.; Vale, T.C.; França Junior, M.C.; Kauffman, M.A.; Teive, H.; Barsottini, O.G.P.; Munhoz, R.P. A diagnostic approach to Spastic ataxia syndromes. Cerebellum 2022, 21, 1073–1084. [Google Scholar] [CrossRef]
- Ortigoza-Escobar, J.D. A proposed diagnostic algorithm for Inborn Errors of Metabolism presenting with movement disorders. Front. Neurol. 2020, 11, 582160. [Google Scholar] [CrossRef]
- Silver, G.; Mercimek-Andrews, S. Inherited metabolic disorders presenting with Ataxia. Int. J. Mol. Sci. 2020, 21, 5519. [Google Scholar] [CrossRef]
- Lopriore, P.; Ricciarini, V.; Siciliano, G.; Mancuso, M.; Montano, V. Mitochondrial ataxias: Molecular classification and clinical heterogeneity. Neurol. Int. 2022, 14, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Kaminiów, K.; Rygula, I.; Paprocka, J. Ataxia in Neurometabolic Disorders. Metabolites 2023, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Braga-Neto, P.; Godeiro-Junior, C.; Dutra, L.A.; Pedroso, J.L.; Barsottini, O.G.P. Translation and validation into Brazilian version of the Scale of the Assessment and Rating of Ataxia (SARA). Arq. Neuropsiquiatr. 2010, 68, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Maggi, F.A.; Braga-Neto, P.; Chien, H.F.; Gama, M.T.D.; Rezende Filho, F.M.; Saraiva-Pereira, M.L.; Jardim, L.B.; Voos, M.C.; Pedroso, J.L.; Barsottini, O.G.P. Cross-cultural adaptation and validation of the International Cooperative Ataxia Rating Scale (ICARS) to Brazilian Portuguese. Arq. Neuropsiquiatr. 2018, 76, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Servelhere, K.R.; Faber, I.; Coan, A.C.; França Junior, M. Translation and validation into Brazilian Portuguese of the Spastic Paraplegia Rating Scale (SPRS). Arq. Neuropsiquiatr. 2016, 74, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Baralle, M. Splicing in action: Assessing disease causing sequence changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Lima, F.D.; Faber, I.; Servelhere, K.R.; Bittar, M.F.R.; Martinez, A.R.M.; Piovesana, L.G.; Martins, M.P.; Martins, C.R.; Benaglia, T.; Carvalho, B.d.S.; et al. Randomized trial of Botulinum Toxin Type A in Hereditary Spastic Paraplegia–The SPASTOX trial. Mov. Disord. 2021, 36, 1654–1663. [Google Scholar] [CrossRef]
- Vedrenne, V.; Gowher, A.; De Lonlay, P.; Nitschke, P.; Serre, V.; Boddaert, N.; Altuzarra, C.; Mager-Heckel, A.-M.; Chretien, F.; Entelis, N.; et al. Mutation in PNPT1, which encodes a Polyribonucleotide Nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am. J. Hum. Genet. 2012, 91, 912–918. [Google Scholar] [CrossRef]
- Wang, G.; Chen, H.W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE regulates RNA import into mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef]
- Liu, X.; Fu, R.; Pan, Y.; Meza-Sosa, K.F.; Zhang, Z.; Lieberman, J. PNPT1 release from mitochondria during apoptosis triggers decay of Poly(A) RNAs. Cell 2018, 174, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Eaton, A.; Bernier, F.P.; Goedhart, C.; Caluseriu, O.; Lamont, R.E.; Boycott, K.M.; Parboosingh, J.S.; Innes, A.M. Is PNPT1-related hearing loss ever non-syndromic? Whole exome sequencing of adult siblings expands the natural history of PNPT1-related disorders. Am. J. Med. Genet. A 2018, 176, 2487–2493. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.W.; Rainey, R.N.; Balatoni, C.E.; Dawson, D.W.; Troke, J.J.; Wasiak, S.; Hong, J.S.; McBride, H.M.; Koehler, C.M.; Teitell, M.A.; et al. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol. Cell. Biol. 2006, 26, 8475–8487. [Google Scholar] [CrossRef] [PubMed]
- Barbier, M.; Bahlo, M.; Pennisi, A.; Jacoupy, M.; Tankard, R.M.; Ewenczyk, C.; Davies, K.C.; Lino-Coulon, P.; Colace, C.; Rafehi, H.; et al. Heterozygous PNPT1 variants cause Spinocerebellar ataxia type 25. Ann. Neurol. 2022, 92, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, A.; Rötig, A.; Roux, C.J.; Lévy, R.; Henneke, M.; Gärtner, J.; Kisa, P.T.; Sarioglu, F.C.; Yiş, U.; Konczal, L.L.; et al. Heterogeneity of PNPT1 neuroimaging: Mitochondriopathy, interferonopathy or both? J. Med. Genet. 2022, 59, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.G.; Li, W.; Sowden, M.; Chávez, C.L.; Berk, B.C. Pnpt1 mediates NLRP3 inflammasome activation by MAVS and metabolic reprogramming in macrophages. Cell. Mol. Immunol. 2023, 20, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Von Ameln, S.; Wang, G.; Boulouiz, R.; Rutherford, M.A.; Smith, G.M.; Li, Y.; Pogoda, H.-M.; Nürnberg, G.; Stiller, B.; Volk, A.E.; et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am. J. Hum. Genet. 2012, 91, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rötig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef]
- Stevanin, G.; Bouslam, N.; Thobois, S.; Azzedine, H.; Ravaux, L.; Boland, A.; Schalling, M.; Broussolle, E.; Dürr, A.; Brice, A. Spinocerebellar ataxia with sensory neuropathy (SCA25) maps to chromosome 2p. Ann. Neurol. 2004, 55, 97–104. [Google Scholar] [CrossRef]
- Stevanin, G.; Broussolle, E.; Streichenberger, N.; Kopp, N.; Brice, A.; Durr, A. Spinocerebellar ataxia with sensory neuropathy (SCA25). Cerebellum 2005, 4, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Tranchant, C.; Anheim, M. Movement disorders in mitochondrial diseases. Rev. Neurol. 2016, 172, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Alodaib, A.; Sobreira, N.; Gold, W.A.; Riley, L.G.; Van Bergen, N.J.; Wilson, M.J.; Bennetts, B.; Thorburn, D.R.; Boehm, C.; Christodoulou, J. Whole-exome sequencing identifies novel variants in PNPT1 causing oxidative phosphorylation defects and severe multisystem disease. Eur. J. Hum. Genet. 2017, 25, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Souissi, A.; Said, M.B.; Ayed, I.B.; Elloumi, I.; Bouzid, A.; Mosrati, M.A.; Hasnaoui, M.; Belcadhi, M.; Idriss, N.; Kamoun, H.; et al. Novel pathogenic mutations and further evidence for clinical relevance of genes and variants causing hearing impairment in Tunisian population. J. Adv. Res. 2021, 31, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Vanniya, S.P.; Chandru, J.; Jeffrey, J.M.; Rabinowitz, T.; Brownstein, Z.; Krishnamoorthy, M.; Avraham, K.B.; Cheng, L.; Shomron, N.; Srisailapathy, C.R.S. PNPT1, MYO15A, PTPRQ, and SLC12A2-associated genetic and phenotypic heterogeneity among hearing impaired assortative mating families in Southern India. Ann. Hum. Genet. 2022, 86, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Arai-Ichinoi, N.; Kikuchi, A.; Matsuhashi, T.; Numata-Uematsu, Y.; Uematsu, M.; Fujii, Y.; Murayama, K.; Ohtake, A.; Abe, T.; et al. Novel biallelic mutation in the PNPT1 gene encoding a mitochondrial-RNA-import protein PNPase cause delayed myelination. Clin. Genet. 2018, 93, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, S.; Carroll, C.J.; Richter, U.; Euro, L.; Pohjanpelto, M.; Paetau, A.; Isohanni, P.; Suomalainen, A. Defective mitochondrial RNA processing due to PNPT1 variants causes Leigh syndrome. Hum. Mol. Genet. 2017, 26, 3352–3361. [Google Scholar] [CrossRef]
- Rius, R.; Van Bergen, N.J.; Compton, A.G.; Riley, L.G.; Kava, M.P.; Balasubramaniam, S.; Amor, D.J.; Fanjul-Fernandez, M.; Cowley, M.J.; Fahey, M.C.; et al. Clinical spectrum and functional consequences associated with bi-allelic pathogenic PNPT1 variants. J. Clin. Med. 2019, 8, 2020. [Google Scholar] [CrossRef]
- Bamborschke, D.; Kreutzer, M.; Koy, A.; Koerber, F.; Lucas, N.; Huenseler, C.; Herkenrath, P.; Lee-Kirsch, M.A.; Cirak, S. PNPT1 mutations may cause Aicardi-Goutières-syndrome. Brain Dev. 2021, 43, 320–324. [Google Scholar] [CrossRef]
- Arnoldi, A.; Tonelli, A.; Crippa, F.; Villani, G.; Pacelli, C.; Sironi, M.; Pozzoli, U.; D’Angelo, M.G.; Meola, G.; Martinuzzi, A.; et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum. Mutat. 2008, 29, 522–531. [Google Scholar] [CrossRef]
- Sáenz-Farret, M.; Lang, A.E.; Kalia, L.; Cunha, I.; Sousa, M.; Kuhlman, G.; Ganos, C.; Munhoz, R.P.; Fasano, A.; Piña-Avilés, C.E.; et al. Spastic paraplegia type 7 and movement disorders: Beyond the Spastic Paraplegia. Mov. Disord. Clin. Pract. 2022, 9, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, K.O.; Wigers, A.R.; Wedding, I.M.; Erichsen, A.K.; Barøy, T.; Søberg, K.; Jørstad, Ø.K. A novel homozygous variant in the SPG7 gene presenting with childhood optic nerve atrophy. Am. J. Ophthalmol. Case Rep. 2022, 26, 101400. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.V.S.; Bortholin, T.; Naylor, F.G.M.; Chieia, M.A.T.; Pinto, W.B.V.R.; Oliveira, A.S.B. Motor neuron disease in inherited neurometabolic disorders. Rev. Neurol. 2018, 174, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Macedo-Souza, L.I.; Kok, F.; Santos, S.; Amorim, S.C.; Starling, A.; Nishimura, A.; Lezirovitz, K.; Lino, A.M.M.; Zatz, M. Spastic paraplegia, optic atrophy, and neuropathy is linked to chromosome 11q13. Ann. Neurol. 2005, 57, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Melo, U.S.; Macedo-Souza, L.I.; Figueiredo, T.; Muotri, A.R.; Gleeson, J.G.; Coux, G.; Armas, P.; Calcaterra, N.B.; Kitajima, J.P.; Amorim, S.; et al. Overexpression of KLC2 due to a homozygous deletion in the non-coding region causes SPOAN syndrome. Hum. Mol. Genet. 2015, 24, 6877–6885. [Google Scholar] [CrossRef]
- Albuquerque, M.V.C.; Pedroso, J.L.; Braga Neto, P.; Barsottini, O.G.P. Phenotype variability and early onset ataxia symptoms in spinocerebellar ataxia type 7: Comparison and correlation with other spinocerebellar ataxias. Arq. Neuropsiquiatr. 2015, 73, 18–21. [Google Scholar] [CrossRef]
- Bah, M.G.; Rodriguez, D.; Cazeneuve, C.; Mochel, F.; Devos, D.; Suppiej, A.; Roubertie, A.; Meunier, I.; Gitiaux, C.; Curie, A.; et al. Deciphering the natural history of SCA7 in children. Eur. J. Neurol. 2020, 27, 2267–2276. [Google Scholar] [CrossRef]



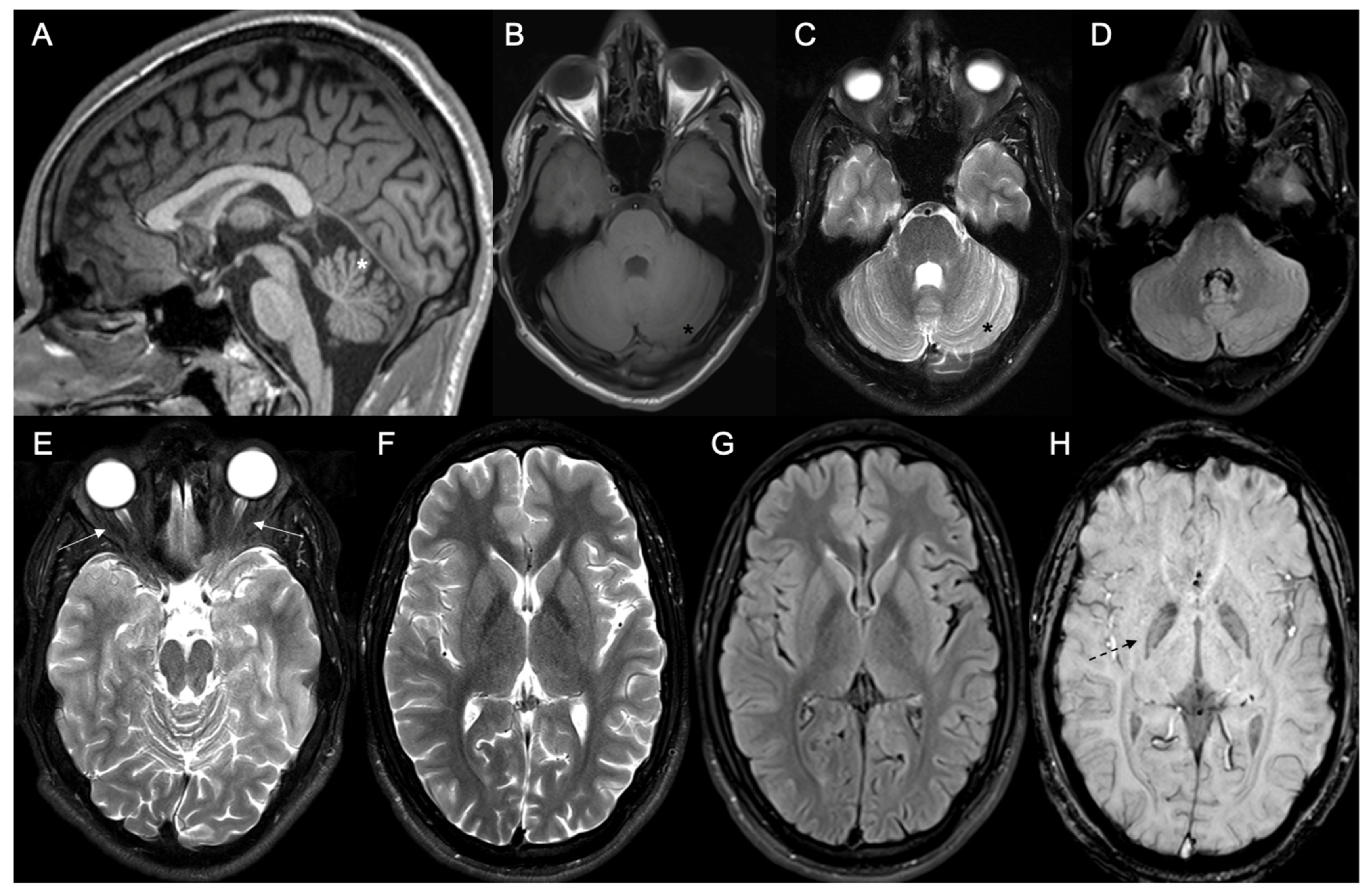{kind=link}
{kind=link}
| Pattern of Inheritance (Zygosity) | Phenotypes (#MIM) | Age at Onset/Clinical Course | Family History/PNPT1 Variants | Spasticity/Cerebellar Ataxia | Neuromuscular Involvement/Neurophysiological Features | Sensorineural Deafness/Neuro-Ophthalmological Disturbance | Other Movement Disorders (Parkinsonism, Chorea, Dystonia, Myoclonus) | Neuropsychiatric and Cognitive Involvement/Neuroimaging Findings | Skeletal Features/Gastrointestinal and Genitourinary Involvement |
|---|---|---|---|---|---|---|---|---|---|
| Autosomal recessive forms (compound heterozygous or homozygous variants) | Combined Oxidative Phosphorylation Deficiency type 13 (COXPD13) (MIM #614932) | Infancy onset; rapidly progressive and severe, then nonprogressive after | Consanguinity; positive history in recurrence/variants inside the catalytic core and active site of the protein—affects multimerization and destabilizes the mutated protein | ++/++ | Myopathy, axial hypotonia, dysphagia; sensory neuropathy; axonal and demyelinating sensory neuropathy; autonomic neuropathy | ++/optic atrophy, macular pigmentary changes; nystagmus, strabismus, cataracts; chorioretinal defects | Dystonia, choreoathetosis, dyskinesia; myoclonus | Neurological regression (more marked in epileptic encephalopathy forms); global developmental delay/Cerebellar atrophy, leukodystrophy (including cystic leukodystrophy and hypomyelination), thin corpus callosum; Leigh syndrome-like pattern (caudate head, putamen); optic tract atrophy; possible lactate peak on MR spectroscopy | Scoliosis/Gastroparesis; constipation; Gastroesophageal reflux |
| Differential diagnosis: Other primary respiratory chain complex defects (oxidative phosphorylation defects); organic acidurias (i.e., glutaric aciduria type 1); Leigh syndrome; infantile neuroaxonal dystrophy (PLA2G6); Neuronal Ceroid Lipofuscinosis; Sphingolipidosis | |||||||||
| Autosomal Recessive Deafness with or without adult-onset Neurodegeneration (DFNB70) (MIM #614934) | Congenital hearing loss, neurological adulthood; progressive | Consanguinity; positive history in recurrence/variants outside the catalytic core of the protein—hypofunctional protein and abnormal PNPase trimerization | +/++ | Late-onset dysphagia; late-onset myopathy; unremarkable neurophysiological studies | +++/Late-onset optic atrophy | Dystonia | Cognitive decline; mood disorders, obsessive compulsive disorder; psychotic features/normal, mild cortical cerebral or cerebellar atrophy | ---/late-onset urinary incontinence | |
| Differential diagnosis: Mitochondrial DNA disorders (MELAS syndrome, MT-TL1, MT-ND5); Mohr–Tranebjaerg syndrome; Usher syndrome spectrum; Riboflavin transporter defects; Wolfram and Wolfram-like syndromes; MEGDEL syndrome; Arts syndrome; Neuroacanthocytosis; Late-onset Friedreich ataxia | |||||||||
| Autosomal dominant form or sporadic cases (heterozygous variants) | Spinocerebellar Ataxia type 25 (SCA25) (MIM #608703) | Childhood or juvenile onset; progressive | Positive family history, despite some sporadic cases; incomplete penetrance/splicing and nonsense variants with premature stop codon | +++/+++ | Sensory neuropathy; absent tendon reflexes; axonal sensory neuropathy (ganglionopathy) | +/Strabismus; ophthalmoparesis; gaze-evoked nystagmus; slow saccadic pursuit; optic atrophy; late-onset diplopia | Facial tics; facial myokymia; dystonia; late-onset head tremor | Cognitive decline; intellectual disability (minor)/normal, cerebellar atrophy | Pes cavus, scoliosis/Gastroparesis; constipation; urinary urgency or incontinence |
| Differential diagnosis: SPG7; SPOAN syndrome; SCA7; SPG35, SPG79B, SPG11, SPG15; CAPOS syndrome; SPAX7, SPAX4; ARSACS; nuclear genome-associated mitocondrial disorders (MFN2, OPA1, OPA2, OPA10); mitochondrial DNA mutations | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sgobbi, P.; Farias, I.B.; Serrano, P.d.L.; Badia, B.d.M.L.; Oliveira, H.B.d.; Barbosa, A.S.; Pereira, C.A.; Moreira, V.d.F.; Chieia, M.A.T.; Barbosa, A.R.; et al. PNPT1 Spectrum Disorders: An Underrecognized and Complex Group of Neurometabolic Disorders. Muscles 2024, 3, 4-15. https://doi.org/10.3390/muscles3010002
Sgobbi P, Farias IB, Serrano PdL, Badia BdML, Oliveira HBd, Barbosa AS, Pereira CA, Moreira VdF, Chieia MAT, Barbosa AR, et al. PNPT1 Spectrum Disorders: An Underrecognized and Complex Group of Neurometabolic Disorders. Muscles. 2024; 3(1):4-15. https://doi.org/10.3390/muscles3010002
Chicago/Turabian StyleSgobbi, Paulo, Igor Braga Farias, Paulo de Lima Serrano, Bruno de Mattos Lombardi Badia, Hélvia Bertoldo de Oliveira, Alana Strucker Barbosa, Camila Alves Pereira, Vanessa de Freitas Moreira, Marco Antônio Troccoli Chieia, Adriel Rêgo Barbosa, and et al. 2024. "PNPT1 Spectrum Disorders: An Underrecognized and Complex Group of Neurometabolic Disorders" Muscles 3, no. 1: 4-15. https://doi.org/10.3390/muscles3010002