
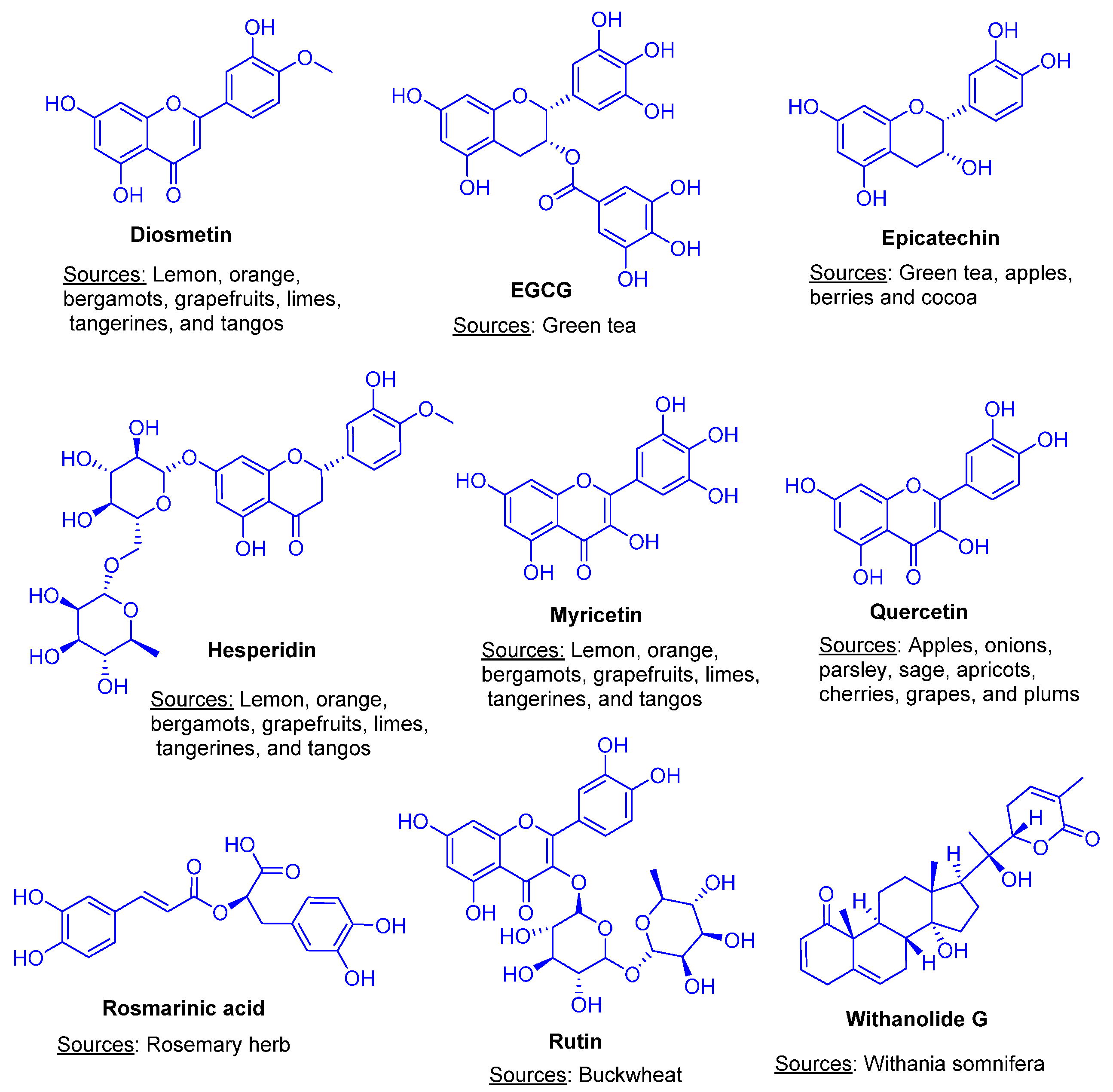
Computational Screening of Plant-Derived Natural Products against SARS-CoV-2 Variants

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. ADMET and Drug Likeness Study
2.2. Ligand Preparation
2.3. Protein Preparation and Grid Generation
2.4. Molecular Docking
2.5. Molecular Dynamic Simulation
2.6. DFT Calculation
3. Results
3.1. Evaluation and Analysis of ADMET and Drug Likeness Properties
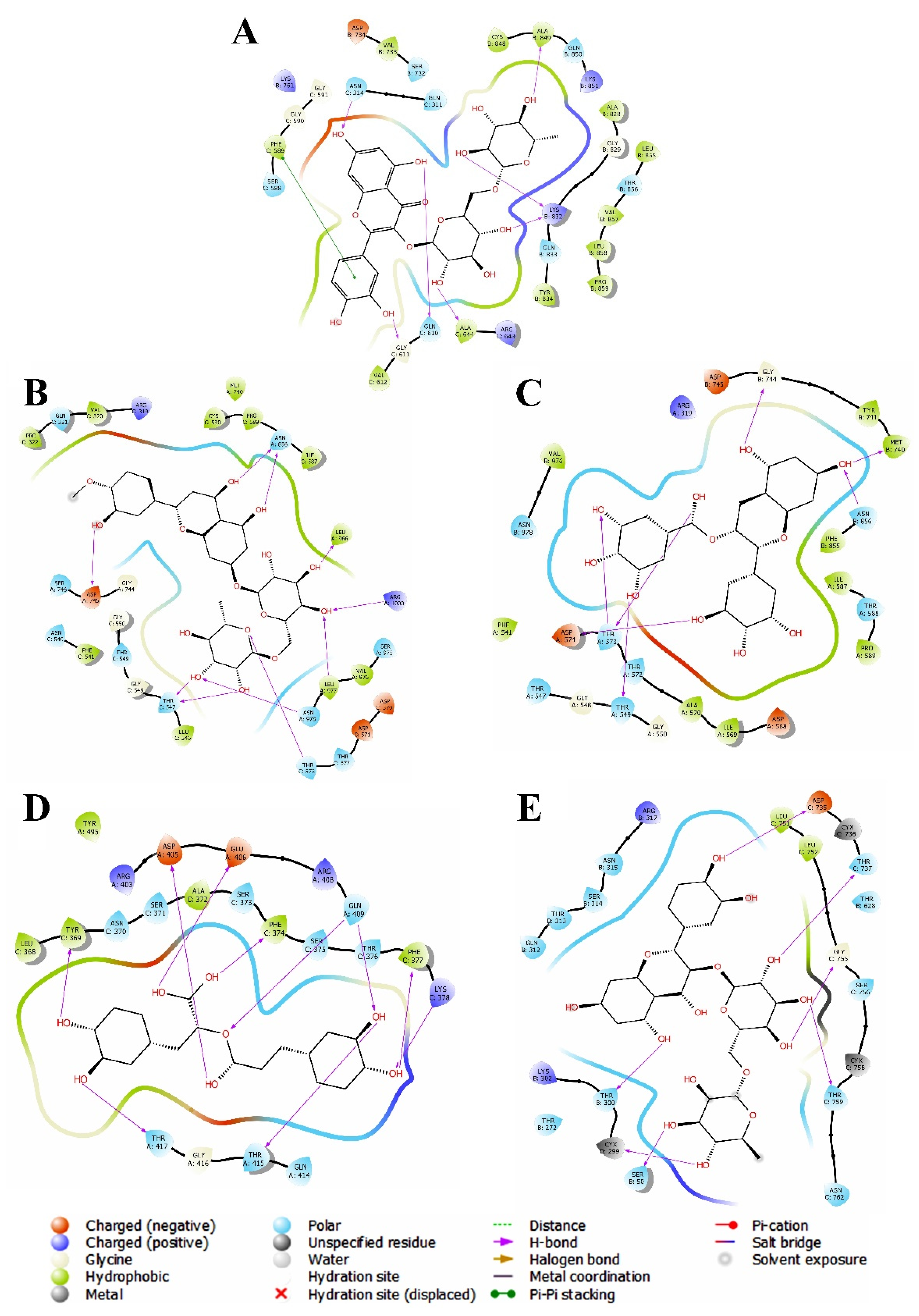
3.2. Evaluation and Analysis of Molecular Docking Analysis
3.2.1. Molecular Docking against Omicron Variant
3.2.2. Molecular Docking against Delta Variant
3.2.3. Molecular Docking against Alpha Variant
3.2.4. Molecular Docking against Beta Variant
3.2.5. Molecular Docking against Gamma Variant
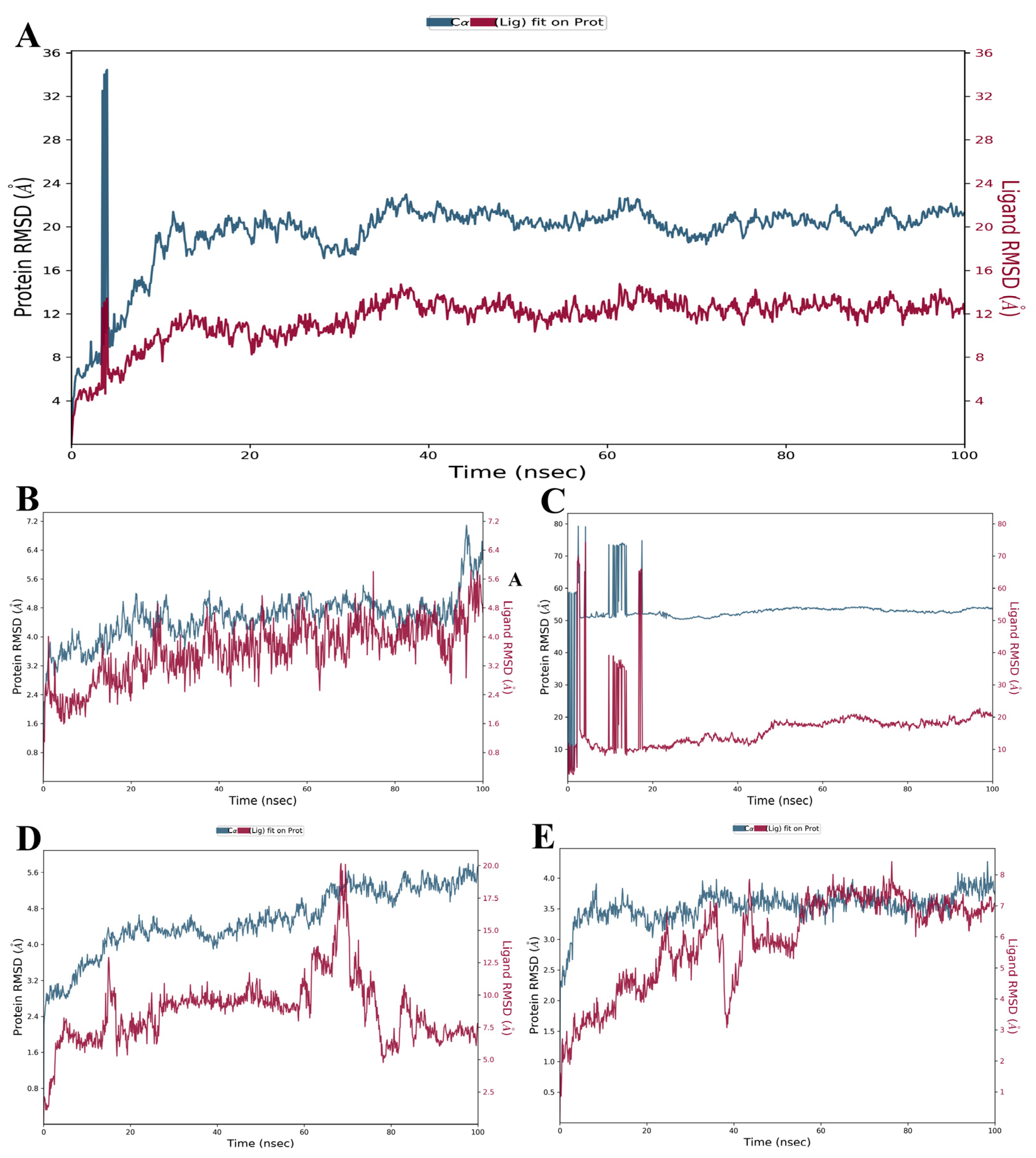
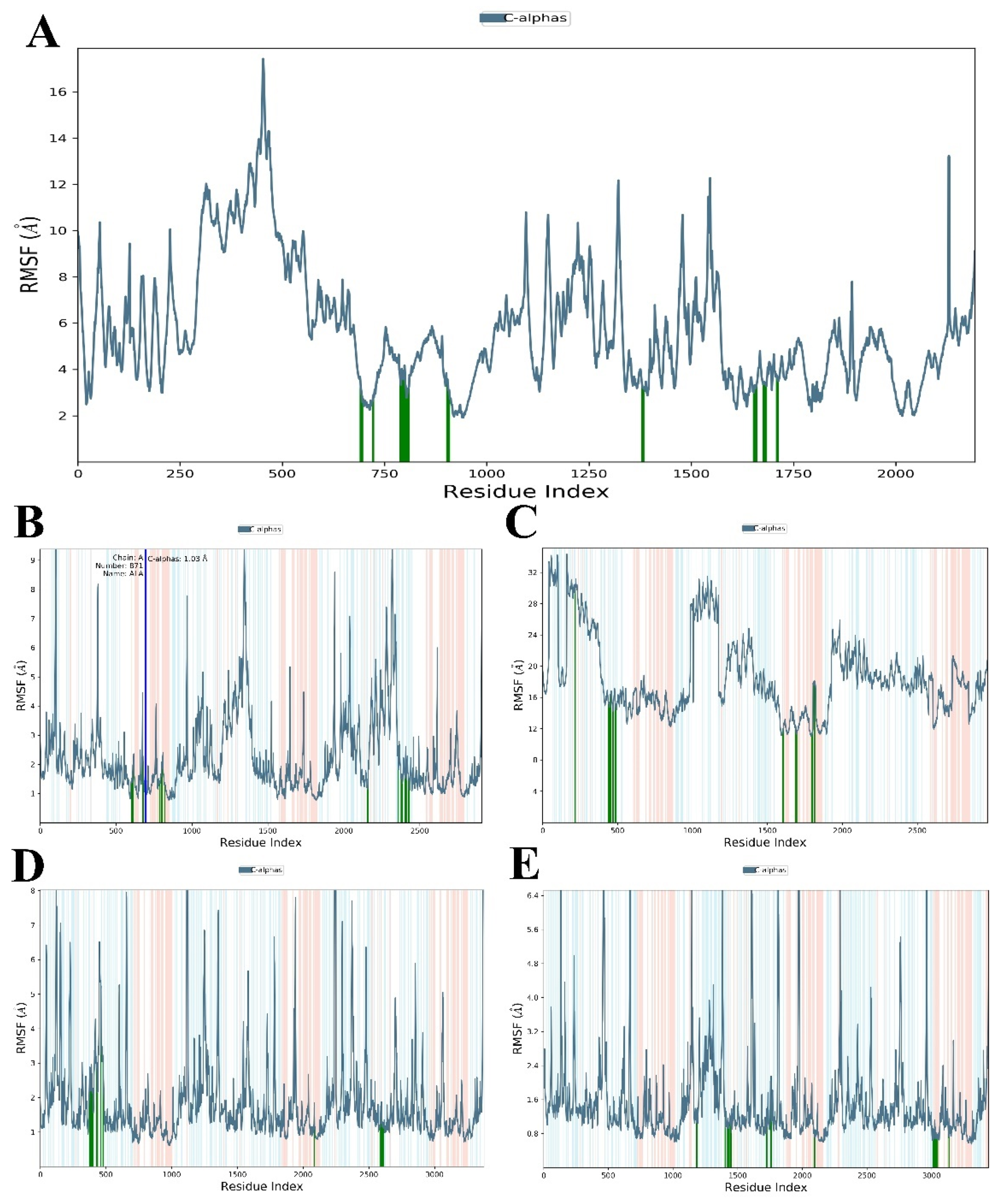
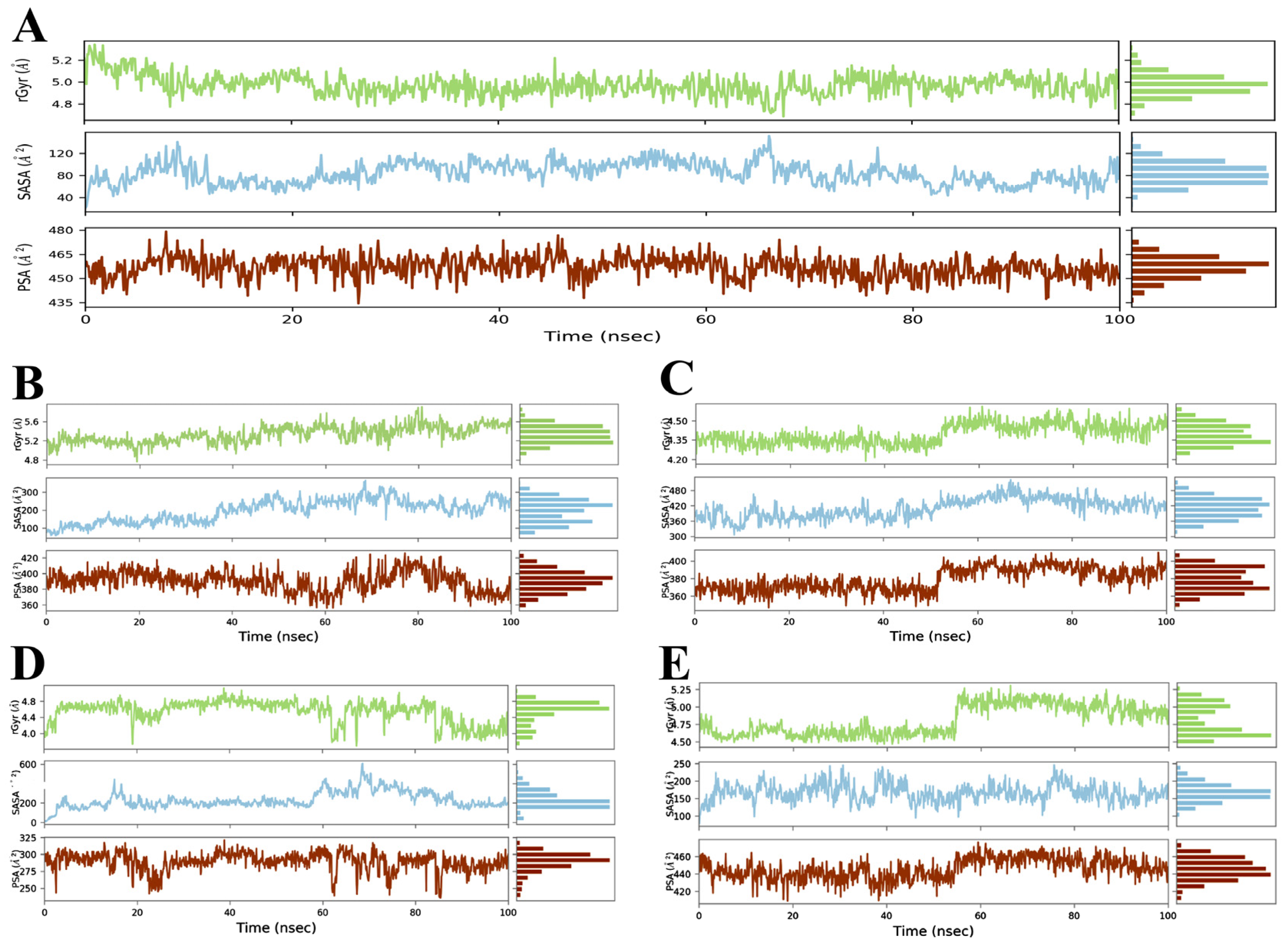
3.3. Evaluation and Analysis of Stability in Complexes through MD Simulation
3.3.1. RMSD and RMSF Analysis
3.3.2. Radius of Gyration (Rg) Analysis
3.3.3. Solvent Accessible Surface Area (SASA) Analysis
3.3.4. Polar Surface Area (PSA) Analysis
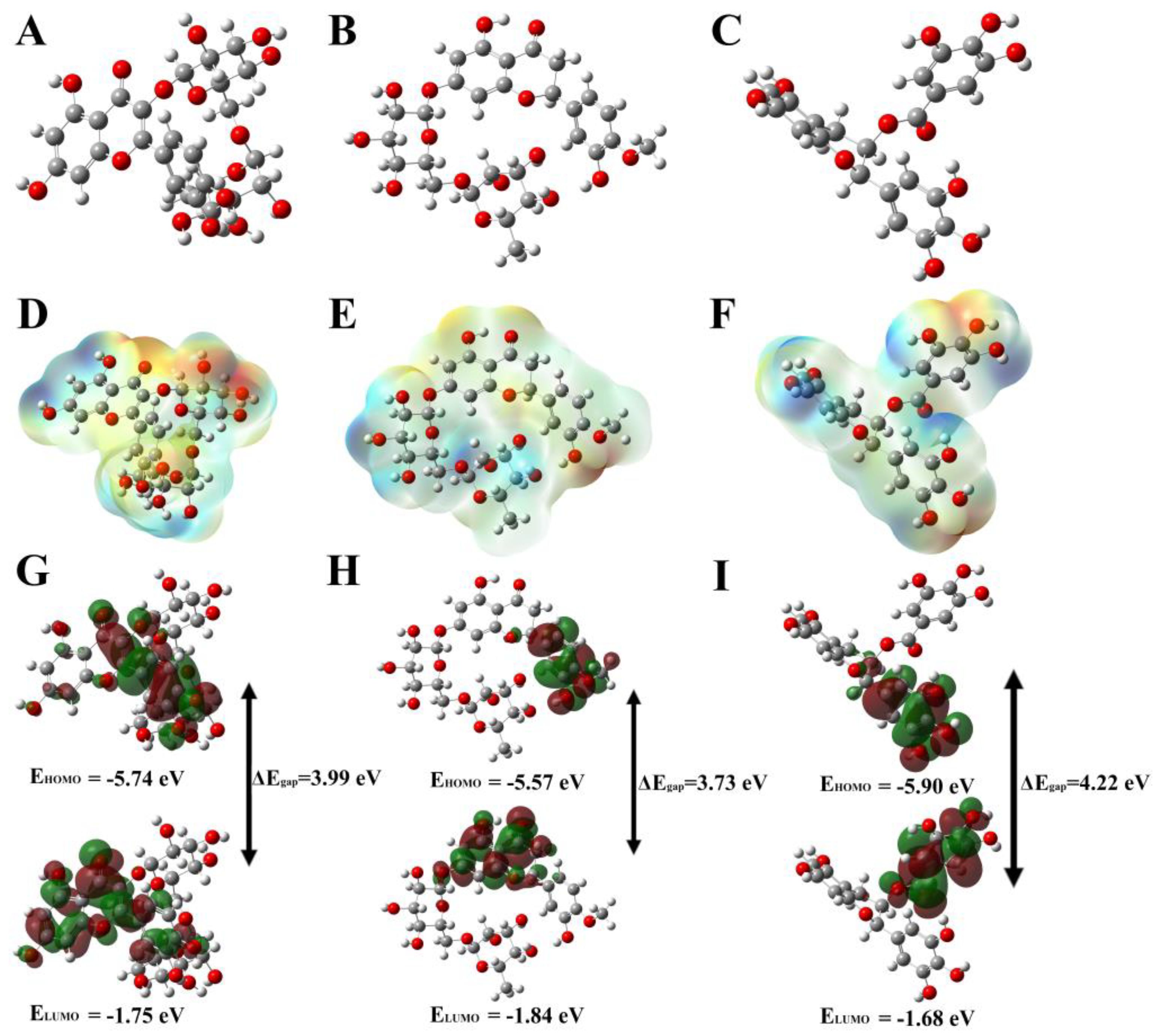
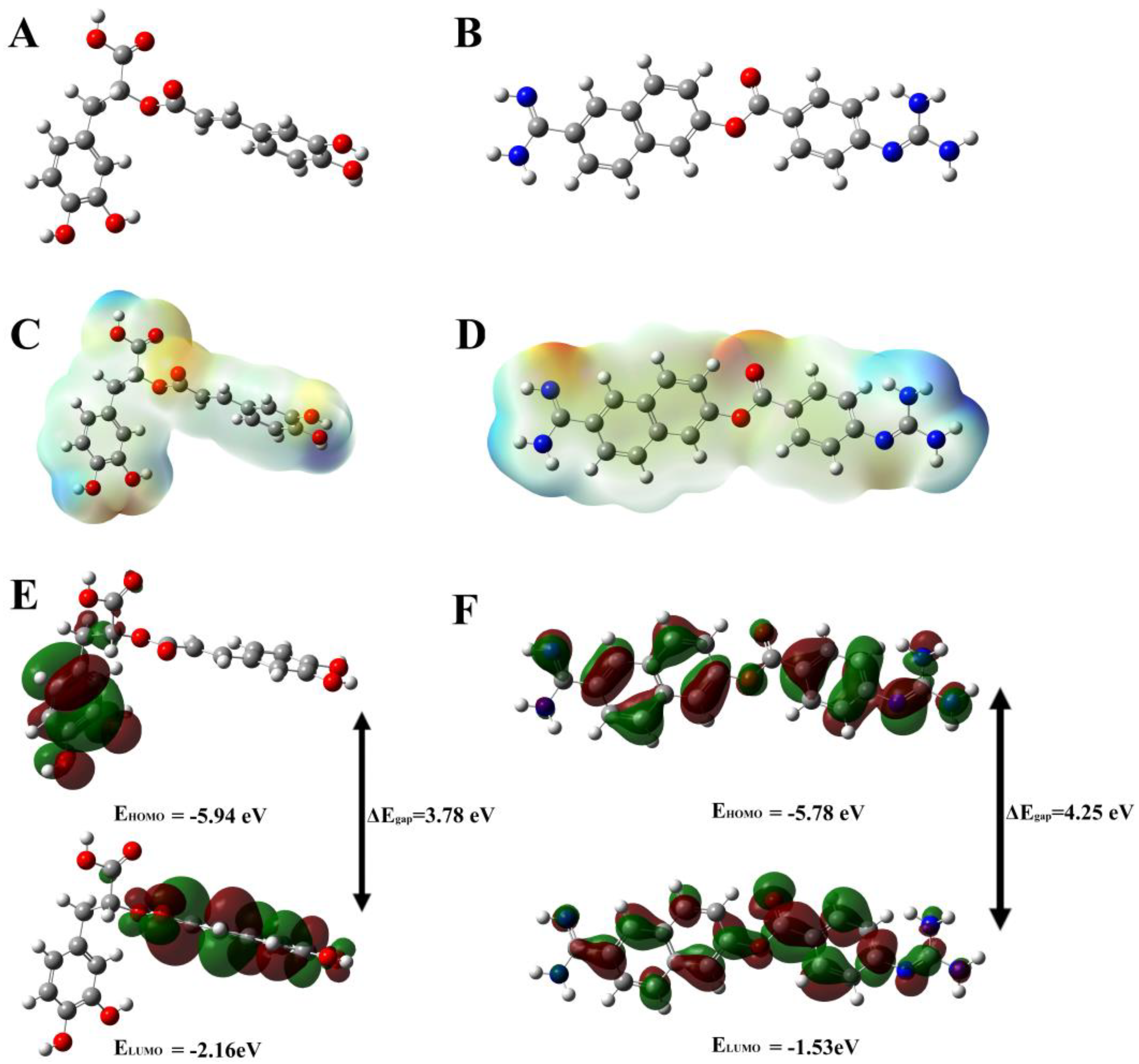
3.4. Evaluation and Analysis of Density-Function Theory (DFT) Method
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A Brief Summary and Comparison of Severe Acute Respiratory Infections Caused by Three Highly Pathogenic Human Coronaviruses. Respir. Res. 2020, 21, 224. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 Genome, Structure, Evolution, Pathogenesis and Therapies: Structural Genomics Approach. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Ahamad, S.; Branch, S.; Harrelson, S.; Hussain, K.; Saquib, M.; Khan, S. Primed for global coronavirus pandemic: Emerging research and clinical outcome. Eur. J. Med. Chem. 2021, 209, 112862. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular Interaction and Inhibition of SARS-CoV-2 Binding to the ACE2 Receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef] [PubMed]
- Hakmaoui, A.; Khan, F.; Liacini, A.; Kaur, A.; Berka, Y.; Machraoui, S.; Soualhine, H.; Berka, N.; Rais, H.; Admou, B. Relevant SARS-CoV-2 Genome Variation through Six Months of Worldwide Monitoring. BioMed Res. Int. 2021, 2021, 5553173. [Google Scholar] [CrossRef]
- Karunamoorthi, K.; Jegajeevanram, K.; Vijayalakshmi, J.; Mengistie, E. Traditional Medicinal Plants: A Source of Phytotherapeutic Modality in Resource-Constrained Health Care Settings. J. Evid. Based Complement. Altern. Med. 2013, 18, 67–74. [Google Scholar] [CrossRef]
- Siddiqui, A.; Iram, F. Role of Natural Products in Drug Discovery Process. Int. J. Drug Dev. Res. 2014, 6, 172–204. [Google Scholar]
- Levy, E.; Delvin, E.; Marcil, V.; Spahis, S. Can Phytotherapy with Polyphenols Serve as a Powerful Approach for the Prevention and Therapy Tool of Novel Coronavirus Disease 2019 (COVID-19)? Am. J. Physiol. Endocrinol. Metab. 2020, 319, E689–E708. [Google Scholar] [CrossRef]
- Khan, M.F.; Khan, M.A.; Khan, Z.A.; Ahamad, T.; Ansari, W.A. In-Silico Study to Identify Dietary Molecules as Potential SARS-CoV-2 Agents. Lett. Drug Des. Discov. 2020, 18, 562–573. [Google Scholar] [CrossRef]
- Ansari, W.A.; Ahamad, T.; Khan, M.A.; Khan, Z.A.; Khan, M.F. Exploration of Luteolin as Potential Anti-COVID-19 Agent: Molecular Docking, Molecular Dynamic Simulation, ADMET and DFT Analysis. Lett. Drug Des. Discov. 2021, 19, 741–756. [Google Scholar] [CrossRef]
- Agrawal, P.K.; Agrawal, C.; Blunden, G. Rutin: A Potential Antiviral for Repurposing as a SARS-CoV-2 Main Protease (M pro) Inhibitor. Nat. Prod. Commun. 2021, 16, 1934578X21991723. [Google Scholar] [CrossRef]
- Borse, S.; Joshi, M.; Saggam, A.; Bhat, V.; Walia, S.; Marathe, A.; Sagar, S.; Chavan-Gautam, P.; Girme, A.; Hingorani, L.; et al. Ayurveda Botanicals in COVID-19 Management: An in Silico Multi-Target Approach. PLoS ONE 2021, 16, e0248479. [Google Scholar] [CrossRef]
- Khan, J.; Sakib, S.A.; Mahmud, S.; Khan, Z.; Islam, M.N.; Sakib, M.A.; bin Emran, T.; Simal-Gandara, J. Identification of Potential Phytochemicals from Citrus Limon against Main Protease of SARS-CoV-2: Molecular Docking, Molecular Dynamic Simulations and Quantum Computations. J. Biomol. Struct. Dyn. 2021, 1–12. [Google Scholar] [CrossRef]
- Pokharkar, O.; Lakshmanan, H.; Zyryanov, G.; Tsurkan, M. In Silico Evaluation of Antifungal Compounds from Marine Sponges against COVID-19-Associated Mucormycosis. Mar. Drugs 2022, 20, 215. [Google Scholar] [CrossRef] [PubMed]
- Basha, S.; Tripathi, D.; Koora, S.; Satyanarayana, K.; Jayaraman, S. Molecular docking analysis of potential compounds from an Indian medicinal soup “kabsura kudineer” extract with IL-6. Biomed. Inf. 2021, 17, 568–572. [Google Scholar]
- Sharma, V.; Sharma, P.C.; Kumar, V. In silico molecular docking analysis of natural pyridoacridines as anticancer agents. Adv. Chem. 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Goodford, P. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1984, 27, 557. [Google Scholar] [CrossRef] [PubMed]
- Ansari, W.; Rizvi, F.; Khan, M.; Khan, Z.; Khan, M. Computational Study Reveals the Inhibitory Effects of Chemical Constituents from Azadirachta indica (Indian Neem) Against Delta and Omicron Variants of SARS-CoV-2. Coronaviruses 2022, 3, e270822208065. [Google Scholar]
- Sheikh, S.; Hassan, F.; Khan, M.; Ahamad, T.; Ansari, W.; Akhter, Y.; Khafagy, E.; Khan, A.; Nasibullah, M. Drug Repurposing to Discover Novel Anti-Inflammatory Agents Inhibiting JAK3/STAT Signaling. Russ. J. Bioorg. Chem. 2022, 48, 958–975. [Google Scholar] [CrossRef]
- Khan, M.; Ansari, W.; Ahamad, T.; Khan, M.; Khan, Z.; Sarfraz, A.; Khan, M. Bioactive components of different nasal spray solutions may defeat SARS-CoV-2: Repurposing and in silico studies. J. Mol. Model. 2022, 28, 212. [Google Scholar] [CrossRef] [PubMed]
- Saini, M.; Sangwan, R.; Khan, M.; Kumar, A.; Verma, R.; Jain, S. Specioside (SS) & verminoside (VS) (Iridoid glycosides): Isolation, characterization and comparable quantum chemical studies using density functional theory (DFT). Heliyon 2019, 5, e1118. [Google Scholar]
- Thangavel, N.; Al Bratty, M.; Al Hazmi, H.; Najmi, A.; Alaqi, R. Molecular Docking, and Molecular Dynamics Aided Virtual Search of OliveNetTM Directory for Secoiridoids to Combat SARS-CoV-2 Infection and Associated Hyperinflammatory Responses. Front. Mol. Biosci. 2021, 7, 627767. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Yadav, A.; Sarkar, P. Molecular docking and ADMET study of bioactive compounds of Glycyrrhiza glabra against main protease of SARS-CoV-2. Mater. Today Proc. 2022, 49, 2999–3007. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef]
- Gurung, A.; Ali, M.; Elshikh, M.; Aref, I.; Amina, M.; Lee, J. An in silico approach unveils the potential of antiviral compounds in preclinical and clinical trials as SARS-CoV-2 omicron inhibitors. Saudi J. Biol. Sci. 2022, 29, 103297. [Google Scholar] [CrossRef]
- Muhammad, S.; Fatima, N. In Silico Analysis and Molecular Docking Studies of Potential Angiotensin-Converting Enzyme Inhibitor Using Quercetin Glycosides. Pharmacogn. Mag. 2015, 11, 123. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, A.; Jamil, K. Drug Targets for Cell Cycle Dysregulators in Leukemogenesis: In Silico Docking Studies. PLoS ONE 2014, 9, e86310. [Google Scholar] [CrossRef] [PubMed]
- Sixto-López, Y.; Correa-Basurto, J.; Bello, M.; Landeros-Rivera, B.; Garzón-Tiznado, J.A.; Montaño, S. Structural Insights into SARS-CoV-2 Spike Protein and Its Natural Mutants Found in Mexican Population. Sci. Rep. 2021, 11, 4659. [Google Scholar] [CrossRef] [PubMed]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the HACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.; Mostafa, A.; Al-Karmalawy, A.A.; Zidan, A.; Abulkhair, H.S.; Mahmoud, S.H.; Shehata, M.; Elhefnawi, M.M.; Ali, M.A. Telaprevir Is a Potential Drug for Repurposing against SARS-CoV-2: Computational and in Vitro Studies. Heliyon 2021, 7, e07962. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; Saha, A.; Osman, S.M.; Alasmary, F.A.; Almutairi, T.M.; Islam, M.A. Structure-Based Identification of SARS-CoV-2 Main Protease Inhibitors from Anti-Viral Specific Chemical Libraries: An Exhaustive Computational Screening Approach. Mol. Divers. 2021, 25, 1979–1997. [Google Scholar] [CrossRef]
- Dhankhar, P.; Dalal, V.; Kotra, D.; Kumar, P. In-silico approach to identify novel potent inhibitors against GraR of S. aureus. Front. Biosci. 2020, 25, 1337–1360. [Google Scholar]
- Hassan, M.; Shahzadi, S.; Seo, S.Y.; Alashwal, H.; Zaki, N.; Moustafa, A.A. Molecular Docking and Dynamic Simulation of AZD3293 and Solanezumab Effects against BACE1 to Treat Alzheimer’s Disease. Front. Comput. Neurosci. 2018, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Grover, S.; Tyagi, C.; Goyal, S.; Jamal, S.; Singh, A.; Grover, A. Hydrophobic Interactions Are a Key to MDM2 Inhibition by Polyphenols as Revealed by Molecular Dynamics Simulations and MM/PBSA Free Energy Calculations. PLoS ONE 2016, 11, e0149014. [Google Scholar] [CrossRef]
- Abdalla, M.; Mohapatra, R.K.; Sarangi, A.K.; Mohapatra, P.K.; Eltayb, W.A.; Alam, M.; El-Arabey, A.A.; Azam, M.; Al-Resayes, S.I.; Seidel, V.; et al. In Silico Studies on Phytochemicals to Combat the Emerging COVID-19 Infection. J. Saudi Chem. Soc. 2021, 25, 101367. [Google Scholar] [CrossRef]
- Waqas Alam, M.; Farhan, M.; Souayeh, B.; Aamir, M.; Khan, M.S. Synthesis, Crystal Structure, Density Functional Theory (DFT) Calculations and Molecular Orbital Calculations of 4-Bromoanilinium Perchlorate Single Crystal. Crystals 2021, 11, 1070. [Google Scholar] [CrossRef]
- Arivazhagan, M.; Kumar, J. Molecular structure, vibrational spectral assignments, HOMO–LUMO, MESP, Mulliken analysis and thermodynamic properties of 2,6-xylenol and 2,5-dimethyl cyclohexanol based on DFT calculation. Spectrochim. Acta A 2015, 137, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Sanches, P.R.S.; Charlie-Silva, I.; Braz, H.L.B.; Bittar, C.; Freitas Calmon, M.; Rahal, P.; Cilli, E.M. Recent Advances in SARS-CoV-2 Spike Protein and RBD Mutations Comparison between New Variants Alpha (B.1.1.7, United Kingdom), Beta (B.1.351, South Africa), Gamma (P.1, Brazil) and Delta (B.1.617.2, India). J. Virus Erad. 2021, 7, 100054. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, S.; Ciccozzi, M.; Zella, D.; Bianchi, M.; Benedetti, F.; Benvenuto, D.; Broccolo, F.; Cauda, R.; Caruso, A.; Angeletti, S.; et al. SARS-CoV-2 B.1.617 Indian Variants: Are Electrostatic Potential Changes Responsible for a Higher Transmission Rate? J. Med. Virol. 2021, 93, 6551–6556. [Google Scholar] [CrossRef]
- Adhikari, B.; Marasini, B.; Rayamajhee, B.; Bhattarai, B.; Lamichhane, G.; Khadayat, K.; Adhikari, A.; Khanal, S.; Parajuli, N. Potential roles of medicinal plants for the treatment of viral diseases focusing on COVID-19: A review. Phytother. Res. 2021, 35, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Santana, F.P.R.; Pinheiro, N.M.; Mernak, M.I.B.; Righetti, R.F.; Martins, M.A.; Lago, J.H.G.; Lopes, F.D.T.Q.D.S.; Tibério, I.F.L.C.; Prado, C.M. Evidences of Herbal Medicine-Derived Natural Products Effects in Inflammatory Lung Diseases. Mediat. Inflamm. 2016, 2016, 2348968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montenegro-Landívar, M.; Tapia-Quirós, P.; Vecino, X.; Reig, M.; Valderrama, C.; Grandos, M.; Luis Cortina, J.; Saurina, J. Polyphenols and their potential role to fight viral diseases: An overview. Sci. Total Environ. 2021, 801, 149719. [Google Scholar] [CrossRef] [PubMed]
- Chojnacka, K.; Skrzypczak, D.; Izydorczyk, G.; Mikula, K.; Szopa, D.; Witek-Krowiak, A. Antiviral Properties of Polyphenols from Plants. Foods 2021, 10, 2277. [Google Scholar] [CrossRef]
- Chen, Y.; Kirchmair, J. Cheminformatics in Natural Product-based Drug Discovery. Mol. Inform. 2022, 39, 2000171. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.; Amaro, R.; Andrade, C.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.; Merz, K.; et al. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121. [Google Scholar] [CrossRef] [PubMed]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.; Gimeno, A.; Cereto-Massgué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste make waste: A critical review of docking –based virtual screening in drug repurposing for SARS-CoV-2 main protease (Mpro) inhibition. Med. Res. Rev. 2021, 42, 744–769. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bodnar, B.H.; Meng, F.; Khan, A.I.; Wang, X.; Saribas, S.; Wang, T.; Lohani, S.C.; Wang, P.; Wei, Z.; et al. Epigallocatechin gallate from green tea effectively blocks infection of SARS-CoV-2 and new variants by inhibiting spike binding to ACE2 receptor. Cell Biosci. 2021, 11, 168. [Google Scholar] [CrossRef]
- Haggag, Y.A.; El-Ashmawy, N.E.; Okasha, K.M. Is Hesperidin Essential for Prophylaxis and Treatment of COVID-19 Infection? Med. Hypotheses 2020, 144, 109957. [Google Scholar] [CrossRef]
- Cheng, F.; Huynh, T.; Yang, C.; Hu, D.; Shen, Y.; Tu, C.; Wu, Y.; Tang, C.; Huang, W.; Chen, Y.; et al. Hesperidin Is a Potential Inhibitor against SARS-CoV-2 Infection. Nutrients 2021, 13, 2800. [Google Scholar] [CrossRef]
- Ferreira, J.; Fadl, S.; Villanueva, A.; Rabeh, W. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef]
- Xiao, T.; Cui, M.; Zheng, C.; Wang, M.; Sun, R.; Gao, D.; Bao, J.; Ren, S.; Yang, B.; Lin, J.; et al. Myricetin Inhibits SARS-CoV-2 Viral Replication by Targeting Mpro and Ameliorates Pulmonary Inflammation. Front. Pharmacol. 2021, 12, 669642. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Singh, P.; Sharma, S.; Singh, T.P.; Ethayathulla, A.S.; Kaur, P. Identification of Bioactive Molecule from Withania Somnifera (Ashwagandha) as SARS-CoV-2 Main Protease Inhibitor. J. Biomol. Struct. Dyn. 2020, 39, 5668–5681. [Google Scholar] [CrossRef] [PubMed]
- Straughn, A.R.; Kakar, S.S. Withaferin A: A Potential Therapeutic Agent against COVID-19 Infection. J. Ovarian Res. 2020, 14, 126. [Google Scholar] [CrossRef]
- Elebeedy, D.; Elkhatib, W.F.; Kandeil, A.; Ghanem, A.; Kutkat, O.; Alnajjar, R.; Saleh, M.A.; Abd El Maksoud, A.I.; Badawy, I.; Al-Karmalawy, A.A. Anti-SARS-CoV-2 Activities of Tanshinone IIA, Carnosic Acid, Rosmarinic Acid, Salvianolic Acid, Baicalein, and Glycyrrhetinic Acid between Computational Andin Vitroinsights. RSC Adv. 2021, 11, 29267–29286. [Google Scholar] [CrossRef]
- Oselusi, S.O.; Christoffels, A.; Egieyeh, S.A. Cheminformatic Characterization of Natural Antimicrobial Products for the Development of New Lead Compounds. Molecules 2021, 26, 3970. [Google Scholar] [CrossRef]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-Score-a Comprehensive Scoring Function for Evaluation of Chemical Drug-Likeness. Med. Chem. Commun. 2019, 10, 148–157. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | MW | HBA | HBD | TPSA | MLogP | LogS | GI | BBB | Ro5 |
|---|---|---|---|---|---|---|---|---|---|
| Epicatechin | 290.27 | 6 | 5 | 110.38 | 0.24 | −2.22 | High | No | Yes |
| EGCG | 458.37 | 11 | 8 | 197.37 | −0.44 | −3.56 | Low | No | No |
| Rutin | 610.52 | 16 | 10 | 269.43 | −3.89 | −3.30 | Low | No | No |
| Quercetin | 302.24 | 7 | 5 | 131.36 | −0.56 | −3.16 | High | No | Yes |
| Myricetin | 318.24 | 8 | 6 | 151.59 | −1.08 | −3.01 | Low | No | Yes |
| Diosmetin | 300.26 | 6 | 3 | 100.13 | 0.22 | −4.06 | High | No | Yes |
| Rosmarinic acid | 360.31 | 8 | 5 | 144.52 | 0.90 | −3.44 | Low | No | Yes |
| Hesperidin | 610.56 | 15 | 8 | 234.29 | −3.04 | −3.28 | Low | No | No |
| Withanolide G | 454.60 | 5 | 2 | 83.83 | 3.48 | −4.71 | High | No | Yes |
| Nafamostat | 347.37 | 4 | 4 | 140.57 | 2.96 | −3.40 | Low | No | Yes |
| Ligand | Mutagenic | Tumorigenic | Irritant | Reproductive Effect |
|---|---|---|---|---|
| Epicatechin | No | No | No | No |
| EGCG | No | No | No | No |
| Rutin | No | No | No | No |
| Quercetin | Yes | Yes | No | No |
| Myricetin | Yes | No | No | No |
| Diosmetin | No | No | No | No |
| Rosmarinic acid | No | No | No | No |
| Hesperidin | No | No | No | No |
| Withanolide G | No | No | No | No |
| Nafamostat | No | No | No | No |
| PubChem | Smiles | Compounds | Docking Score | ||||
|---|---|---|---|---|---|---|---|
| ID | Omicron | Delta | Alpha | Beta | Gamma | ||
| 5280805 |  | Rutin | −10.224 | −8.160 | −8.589 | −8.095 | −5.891 |
| 10621 |  | Hesperidin | −9.029 | −7.873 | −8.993 | −7.559 | −4.055 |
| 107905 |  | ECG | −8.202 | −7.454 | −7.765 | −7.429 | −6.615 |
| 65064 |  | EGCG | −7.700 | −7.993 | −7.851 | −8.369 | −7.532 |
| 21679023 |  | Withanolide G | −5.817 | −6.159 | −8.766 | −6.590 | −4.443 |
| 5281792 |  | Rosmarinic acid | −6.739 | −6.629 | −8.761 | −7.187 | −9.235 |
| 5281612 |  | Diosmetin | −7.659 | −5.867 | −6.173 | −8.200 | −6.439 |
| 5281672 |  | Myricetin | −6.877 | −6.521 | −6.916 | −8.102 | −5.968 |
| 72276 |  | Epicatechin | −7.596 | −7.736 | −7.799 | −7.367 | −8.896 |
| 5280343 |  | Quercetin | −6.092 | −6.795 | −6.825 | −6.844 | −8.021 |
| 4413 |  | Nafamostat | −5.324 | −5.665 | −5.340 | −4.260 | −5.325 |
| Properties | Omicron Variant | Delta Variant | Gamma Variant | Beta Variant | Alpha Variant |
|---|---|---|---|---|---|
| RMSD value (Å) (Protein) | 2.48–34.46 | 2.19–4.27 | 2.21–5.80 | 2.75–79.25 | 2.00–7.08 |
| RMSD value (Å) (Ligand) | 1.29–14.75 | 0.85–8.42 | 1.09–20.16 | 2.16–74.14 | 1.01–5.84 |
| RMSF value (Å) (Protein) | 1.91–17.43 | 0.05–9.24 | 0.61–9.72 | 11.05–34.33 | 0.254–9.75 |
| Rg (Å) | 4.68–5.34 | 4.46–5.30 | 3.68–5.12 | 4.18–4.60 | 4.76–5.90 |
| SASA (Å2) | 20.85–152.19 | 78.64–246.68 | 10.77–605.76 | 306.64–522.23 | 59.43–365.01 |
| PSA (Å2) | 434.30–479.31 | 408.97–476.18 | 235.79–321.38 | 346.76–409.92 | 356.01–426.20 |
| Compounds | HOMO | LUMO | * ΔEgap |
|---|---|---|---|
| Rutin | −5.74 | −1.75 | 3.99 |
| Hesperidin | −5.57 | −1.84 | 3.73 |
| EGCG | −5.90 | −1.68 | 4.22 |
| Rosmarinic acid | −5.94 | −2.16 | 3.78 |
| Nafamostat | −5.78 | −1.53 | 4.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansari, W.A.; Khan, M.A.; Rizvi, F.; Ali, K.; Hussain, M.K.; Saquib, M.; Khan, M.F. Computational Screening of Plant-Derived Natural Products against SARS-CoV-2 Variants. Future Pharmacol. 2022, 2, 558-578. https://doi.org/10.3390/futurepharmacol2040034
Ansari WA, Khan MA, Rizvi F, Ali K, Hussain MK, Saquib M, Khan MF. Computational Screening of Plant-Derived Natural Products against SARS-CoV-2 Variants. Future Pharmacology. 2022; 2(4):558-578. https://doi.org/10.3390/futurepharmacol2040034
Chicago/Turabian StyleAnsari, Waseem Ahmad, Mohd Aamish Khan, Fahmina Rizvi, Kajim Ali, Mohd Kamil Hussain, Mohammad Saquib, and Mohammad Faheem Khan. 2022. "Computational Screening of Plant-Derived Natural Products against SARS-CoV-2 Variants" Future Pharmacology 2, no. 4: 558-578. https://doi.org/10.3390/futurepharmacol2040034