
The Thirty-Fifth Anniversary of K+ Channels in O2 Sensing: What We Know and What We Don’t Know

Abstract
:1. Physiological Oxygen Sensing: The Carotid Body as an O2 Sensor
2. Oxygen-Sensitive K+ Channels in the Carotid Body
2.1. KV Channels and Oxygen Sensitivity in the Carotid Body
2.2. BKCa Channels and Oxygen Sensitivity in the Carotid Body
2.3. K2P Channels and Oxygen Sensitivity in the Carotid Body
3. What We Know and What We Do Not about the Oxygen Sensors in Chemoreceptor Cells
4. Coupling Mechanisms between the Sensor and the K+ Channels
5. Concluding Remarks on O2 Sensing in the Carotid Body
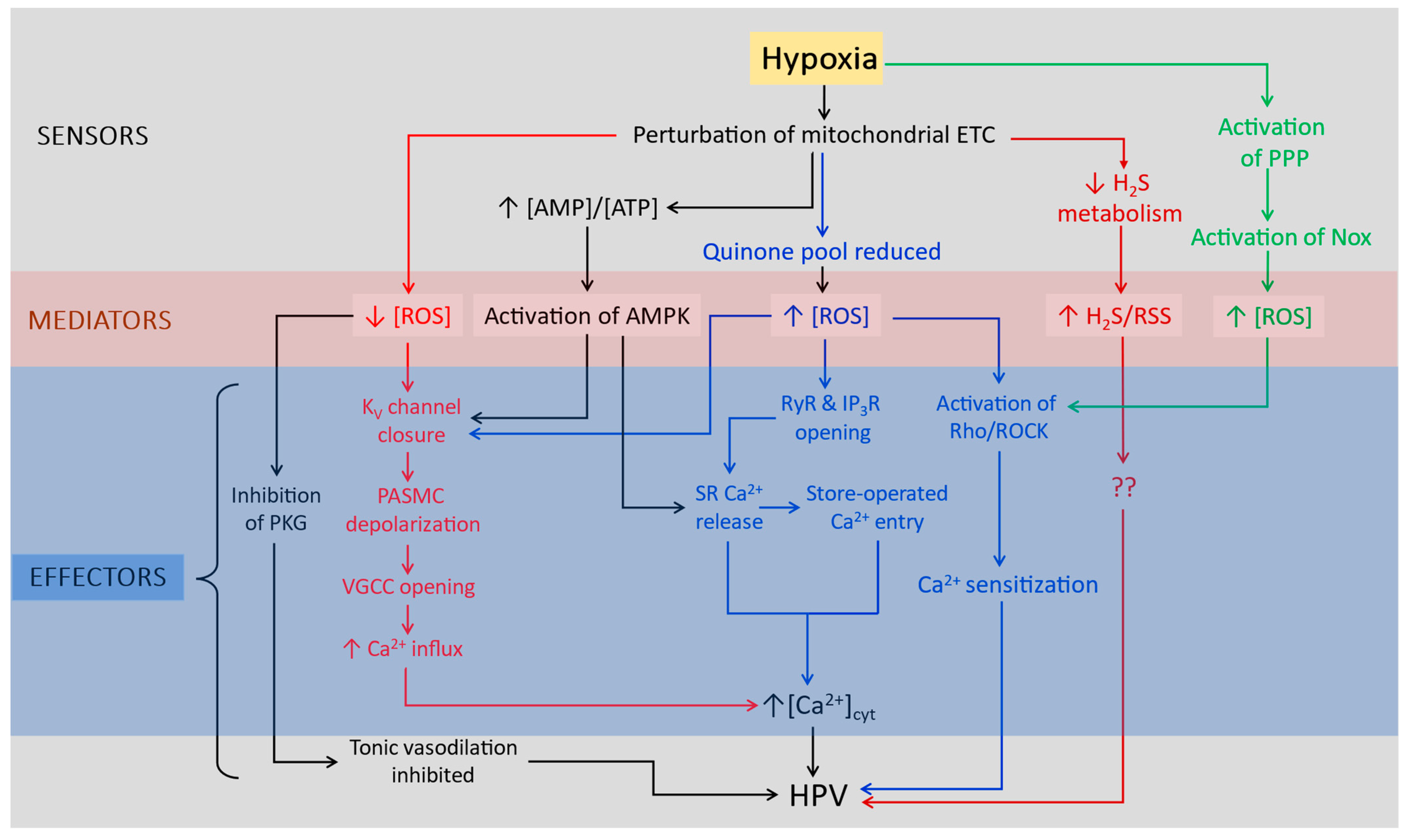
6. O2-Sensitive K+ Channels in the Pulmonary Vasculature
KV Channels and O2 Sensing in PASMC
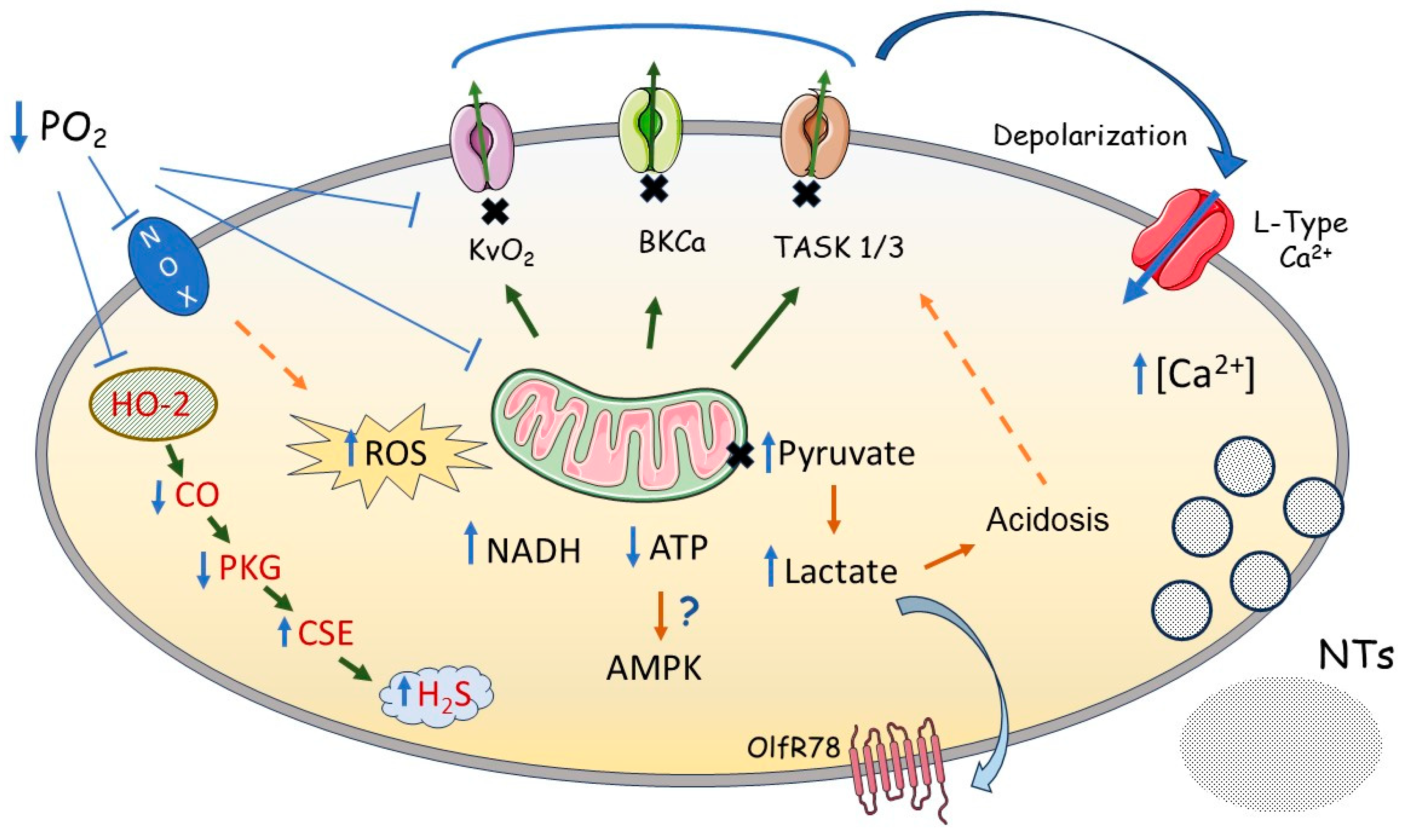
7. How Does PO2 Regulate K+ Channels in PASMC?
7.1. Evidence for KV Channel Regulation by Cytoplasmic Redox State
7.2. Evidence for TASK-1 Channel Regulation by Cytoplasmic Redox State
7.3. Other Factors Regulating PASMC K+ Currents during Hypoxia
8. Concluding Remarks: O2 Sensing in CBCC versus PASMCs
Author Contributions
Funding
Conflicts of Interest
References
- Lopez-Barneo, J.; Lopez-Lopez, J.R.; Urena, J.; Gonzalez, C. Chemotransduction in the carotid body: K+ current modulated by PO2 in type I chemoreceptor cells. Science 1988, 241, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Almaraz, L.; Obeso, A.; Rigual, R. Carotid body chemoreceptors: From natural stimuli to sensory discharges. Physiol. Rev. 1994, 74, 829–898. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.E.; Marshall, C.; Frasch, F.; Hanson, C.W. Role of hypoxic pulmonary vasoconstriction in pulmonary gas exchange and blood flow distribution. 1. Physiologic concepts. Intensive Care Med. 1994, 20, 291–297. [Google Scholar] [CrossRef]
- Peers, C.; Kemp, P.J. Acute oxygen sensing: Diverse but convergent mechanisms in airway and arterial chemoreceptors. Respir. Res. 2001, 2, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Seidler, F.J. Adrenomedullary catecholamine release in the fetus and newborn: Secretory mechanisms and their role in stress and survival. J. Dev. Physiol. 1988, 10, 1–16. [Google Scholar]
- Pflüger, E. Ueber die ursacheder athembewegungen, sowie der dyspnoë und apnoë. Arch. Gesamte Physiol. Menschen Tiere 1868, 1, 61–106. [Google Scholar] [CrossRef]
- Boycott, A.E.; Haldane, J.S. The effects of low atmospheric pressures on respiration. J. Physiol. 1908, 37, 355–377. [Google Scholar] [CrossRef]
- De Castro, F. Sur la structure et l’innervation du sinus carotidien de l’homme et des mammifères: Nouveaux faits sur l’innervation et la fonction du glomus caroticum. Trab. Lab. Invest. Biol. Univ. Madrid 1928, 25, 330–380. [Google Scholar]
- Gonzalez, C.; Conde, S.V.; Gallego-Martín, T.; Olea, E.; Gonzalez-Obeso, E.; Ramirez, M.; Yubero, S.; Agapito, M.T.; Gomez-Nino, A.; Obeso, A.; et al. Fernando de Castro and the discovery of the arterial chemoreceptors. Front. Neuroanat. 2014, 8, 25. [Google Scholar] [CrossRef]
- Anichkov, S.V.; Belen’kii, M.L. Pharmacology of the Carotid Body Chemoreceptors; McMillan: New York, NY, USA, 1963. [Google Scholar]
- Mills, E.; Jöbsis, F.F. Simultaneous measurement of cytochrome a3 reduction and chemoreceptor afferent activity in the carotid body. Nature 1970, 225, 1147–1149. [Google Scholar] [CrossRef]
- Mills, E.; Jöbsis, F.F. Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. J. Neurophysiol. 1972, 35, 405–428. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R.; Biscoe, T.J. Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J. Physiol. 1992, 450, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R.; Biscoe, T.J. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J. Physiol. 1992, 450, 33–61. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Rumsey, W.L.; Wilson, D.F.; Iturriaga, R. Contribution of in vivo microvascular PO2 in the cat carotid body chemotransduction. J. Appl. Physiol. (1985) 1993, 75, 1035–1043. [Google Scholar] [CrossRef]
- Buckler, K.J.; Turner, P.J. Oxygen sensitivity of mitochondrial function in rat arterial chemoreceptor cells. J. Physiol. 2013, 591, 3549–3563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chien, M.S.; Kaleem, S.; Matsunami, H. Single cell transcriptome analysis of mouse carotid body glomus cells. J. Physiol. 2016, 594, 4225–4251. [Google Scholar] [CrossRef]
- Gao, L.; Bonilla-Henao, V.; García-Flores, P.; Arias-Mayenco, I.; Ortega-Sáenz, P.; López-Barneo, J. Gene expression analyses reveal metabolic specifications in acute O2-sensing chemoreceptor cells. J. Physiol. 2017, 595, 6091–6120. [Google Scholar] [CrossRef]
- Moreno-Domínguez, A.; Ortega-Sáenz, P.; Gao, L.; Colinas, O.; García-Flores, P.; Bonilla-Henao, V.; Aragonés, J.; Hüttemann, M.; Grossman, L.I.; Weissmann, N.; et al. Acute O2 sensing through HIF2α-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci. Signal. 2020, 13, eaay9452. [Google Scholar] [CrossRef]
- Almaraz, L.; Gonzalez, C.; Obeso, A. Effects of high potassium on the release of [3H] dopamine from the cat carotid body in vitro. J. Physiol. 1986, 379, 293–307. [Google Scholar] [CrossRef]
- Rocher, A.; Obeso, A.; Herreros, B.; Gonzalez, C. Activation of the release of dopamine in the carotid body by veratridine. Evidence for the presence of voltage-dependent Na+ channels in type I cells. Neurosci. Lett. 1988, 94, 274–278. [Google Scholar] [CrossRef]
- Lopez-Lopez, J.R.; Gonzalez, C.; Urena, J.; Lopez-Barneo, J. Low pO2 selectively inhibits K channel activity in chemoreceptor cells of the mammalian carotid body. J. Gen. Physiol. 1989, 93, 1001–1015. [Google Scholar] [CrossRef]
- Peers, C. Hypoxic suppression of K+ currents in type I carotid body cells: Selective effect on the Ca2+-activated K+ current. Neurosci. Lett. 1990, 119, 253–256. [Google Scholar] [CrossRef]
- Buckler, K.J. A novel oxygen-sensitive potassium current in rat carotid body type I cells. J. Physiol. 1997, 498, 649–662. [Google Scholar] [CrossRef]
- Buckler, K.J. Background leak K+ currents and oxygen sensing in carotid body type 1 cells. Respir. Physiol. 1999, 115, 179–187. [Google Scholar] [CrossRef]
- Rocher, A.; Obeso, A.; Cachero, M.T.; Herreros, B.; Gonzalez, C. Participation of Na+ channels in the response of carotid body chemoreceptor cells to hypoxia. Am. J. Physiol. 1994, 267, C738–C744. [Google Scholar] [CrossRef]
- Obeso, A.; Rocher, A.; Fidone, S.; Gonzalez, C. The role of dihydropyridine-sensitive Ca2+ channels in stimulus-evoked catecholamine release from chemoreceptor cells of the carotid body. Neuroscience 1992, 47, 463–472. [Google Scholar] [CrossRef]
- Buckler, K.J.; Vaughan-Jones, R.D. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J. Physiol. 1994, 476, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Stea, A.; Nurse, C.A. Whole-cell and perforated-patch recordings from O2-sensitive rat carotid body cells grown in short- and long-term culture. Pflügers Arch. 1991, 418, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Ureña, J.; Fernández-Chacón, R.; Benot, A.R.; Alvarez de Toledo, G.A.; López-Barneo, J. Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc. Natl. Acad. Sci. USA 1994, 91, 10208–10211. [Google Scholar] [CrossRef] [PubMed]
- Rocher, A.; Geijo-Barrientos, E.; Caceres, A.I.; Rigual, R.; Gonzalez, C.; Almaraz, L. Role of voltage-dependent calcium channels in stimulus-secretion coupling in rabbit carotid body chemoreceptor cells. J. Physiol. 2005, 562, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Youngson, C.; Nurse, C.; Yeger, H.; Cutz, E. Oxygen sensing in airway chemoreceptors. Nature 1993, 365, 153–155. [Google Scholar] [CrossRef]
- Weir, E.K.; Archer, S.L. The mechanism of acute hypoxic pulmonary vasoconstriction: The tale of two channels. FASEB J. 1995, 9, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Peers, C.; Buckler, K.J. Transduction of chemostimuli by the type I carotid body cell. J. Membr. Biol. 1995, 144, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, M.T.; Lopez-Lopez, J.R. Are Kv channels the essence of O2 sensing? Circ. Res. 2000, 86, 490–491. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Honore, E. Molecular physiology of oxygen-sensitive potassium channels. Eur. Respir. J. 2001, 8, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Coppock, E.A.; Martens, J.R.; Tamkun, M.M. Molecular basis of hypoxia-induced pulmonary vasoconstriction: Role of voltage-gated K+ channels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L1–L12. [Google Scholar] [CrossRef] [PubMed]
- Kemp, P.J. Detecting acute changes in oxygen: Will the real sensor please stand up? Exp. Physiol. 2006, 91, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Peers, C.; Wyatt, C.N.; Evans, A.M. Mechanisms for acute oxygen sensing in the carotid body. Respir. Physiol. Neurobiol. 2010, 174, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Jan, L.Y.; Jan, Y.N. Cloned potassium channels from eukaryotes and procaryotes. Annu. Rev. Neurosci. 1997, 20, 91–123. [Google Scholar] [CrossRef]
- Coetzee, W.A.; Amarillo, Y.; Chiu, J.; Chow, A.; Lau, D.; McCormack, T.; Moreno, H.; Nadal, M.S.; Ozaita, A.; Pountney, D.; et al. Molecular diversity of K+ channels. Ann. N. Y. Acad. Sci. 1999, 868, 233–285. [Google Scholar] [CrossRef]
- Lotshaw, D.P. Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem. Biophys. 2007, 47, 209–256. [Google Scholar] [CrossRef]
- Gutman, G.A.; Chandy, K.G.; Adelman, J.P.; Aiyar, J.; Bayliss, D.A.; Clapham, D.E.; Covarriubias, M.; Desir, G.V.; Furuichi, K.; Ganetzky, B.; et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: Potassium channels. Pharmacol. Rev. 2003, 55, 583–586. [Google Scholar] [CrossRef]
- Bocksteins, E.; Snyders, D.J. Electrically silent Kv subunits: Their molecular and functional characteristics. Physiology 2012, 27, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.R.; Kwak, Y.G.; Tamkun, M.M. Modulation of Kv channel α/β subunit interactions. Trends Cardiovasc. Med. 1999, 9, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Biggin, P.C.; Roosild, T.; Choe, S. Potassium channel structure: Domain by domain. Curr. Opin. Struct. Biol. 2000, 10, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, M.T.; López-López, J.R.; González, C. Kvβ1.2 subunit coexpression in HEK293 cells confers O2 sensitivity to kv4.2 but not to Shaker channels. J. Gen. Physiol. 1999, 113, 897–907. [Google Scholar]
- Coppock, E.A.; Tamkun, M.M. Differential expression of KV channel α- and β-subunits in the bovine pulmonary arterial circulation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L1350–L1360. [Google Scholar] [CrossRef] [PubMed]
- Abbott, G.W. Kv Channel Ancillary Subunits: Where Do We Go from Here? Physiology 2022, 37, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Ganfornina, M.D.; López-Barneo, J. Gating of O2-sensitive K+ channels of arterial chemoreceptor cells and kinetic modifications induced by low PO2. J. Gen. Physiol. 1992, 100, 427–455. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, J.R.; Gonzalez, C. Time course of K+ current inhibition by low oxygen in chemoreceptor cells of adult rabbit carotid body: Effects of carbon monoxide. FEBS Lett. 1992, 299, 251–254. [Google Scholar] [CrossRef]
- Perez-Garcia, M.T.; Lopez-Lopez, J.R.; Riesco, A.M.; Hoppe, U.; Gonzalez, C.; Marban, E.; Johns, D.C. Supression of transient outward K+ currents in chemoreceptor cells of the rabbit carotid body by viral gene transfer of inducible dominant negative KV4.3 constructs. J. Neurosci. 2000, 20, 5689–5695. [Google Scholar] [CrossRef]
- López-López, J.R.; Pérez-García, M.T.; Sanz-Alfayate, G.; Obeso, A.; Gonzalez, C. Functional identification of Kvα subunits contributing to the O2-sensitive K+ current in rabbit CB chemoreceptor cells. Adv. Exp. Med. Biol. 2003, 536, 33–39. [Google Scholar]
- Sanchez, D.; López-López, J.R.; Pérez-García, M.T.; Sanz-Alfayate, G.; Obeso, A.; Ganfornina, M.D.; Gonzalez, C. Molecular identification of KVα subunits that contribute to the oxygen-sensitive K+ current of chemoreceptor cells of the rabbit carotid body. J. Physiol. 2002, 542, 369–382. [Google Scholar] [CrossRef]
- Pérez-García, M.T.; Colinas, O.; Miguel-Velado, E.; Moreno-Domínguez, A.; López-López, J.R. Characterization of the Kv channels of mouse carotid body chemoreceptor cells and their role in oxygen sensing. J. Physiol. 2004, 557, 457–471. [Google Scholar] [CrossRef] [PubMed]
- López-López, J.R.; Pérez-García, M.T. Oxygen sensitive KV channels in the carotid body. Respir. Physiol. Neurobiol. 2007, 157, 65–74. [Google Scholar] [CrossRef]
- Sweeney, M.; Yuan, J.X. Hypoxic pulmonary vasoconstriction: Role of voltage-gated potassium channels. Respir. Res. 2000, 1, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef]
- Lopez-Barneo, J.; Benot, A.R.; Urena, J. Oxygen Sensing and the Electrophysiology of Arterial Chemoreceptor Cells. News Physiol Sci. 1993, 8, 191–195. [Google Scholar] [CrossRef]
- Montoro, R.J.; Urena, J.; Fernandez-Chacon, R.; Alvarez de Toledo, G.; López-Barneo, J. Oxygen sensing by ion channels and chemotransduction in single glomus cells. J. Gen. Physiol. 1996, 107, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kim, D. Activation of voltage-dependent K+ channels strongly limits hypoxia-induced elevation of [Ca2+ ]i in rat carotid body glomus cells. J. Physiol. 2018, 596, 3119–3136. [Google Scholar] [CrossRef]
- Latorre, R.; Oberhauser, A.; Labarca, P.; Alvarez, O. Varieties of calcium-activated potassium channels. Annu. Rev. Physiol. 1989, 51, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Vergara, C.; Latorre, R.; Marrion, N.V.; Adelman, J.P. Calcium-activated potassium channels. Curr. Opin. Neurobiol. 1998, 8, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Knaus, H.G.; Eberhart, A.; Glossmann, H.; Munujos, P.; Kaczorowski, G.J.; Garcia, M.L. Pharmacology and structure of high conductance calcium-activated potassium channels. Cell. Signal. 1994, 6, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, G.J.; Knaus, H.G.; Leonard, R.J.; McManus, O.B.; Garcia, M.L. High-conductance calcium-activated potassium channels; structure, pharmacology, and function. J. Bioenerg. Biomembr. 1996, 28, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Tsunoda, S.; McCobb, D.P.; Wei, A.; Salkoff, L. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channel. Science 1993, 261, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, M.; Salkoff, L. A novel calcium-sensing domain in the BK channel. Biophys. J. 1997, 73, 1355–1363. [Google Scholar] [CrossRef]
- Cox, R.H. Molecular determinants of voltage-gated potassium currents in vascular smooth muscle. Cell Biochem. Biophys. 2005, 42, 167–195. [Google Scholar] [CrossRef]
- McManus, O.B.; Helms, L.M.; Pallanck, L.; Ganetzky, B.; Swanson, R.; Leonard, R.J. Functional role of the β subunit of high conductance calcium-activated potassium channels. Neuron 1995, 14, 645–650. [Google Scholar] [CrossRef]
- Guntur, D.; Olschewski, H.; Enyedi, P.; Csaki, R.; Olschewski, A.; Nagaraj, C. Revisiting the Large-Conductance Calcium-Activated Potassium (BKCa) Channels in the Pulmonary Circulation. Biomolecules 2021, 11, 1629. [Google Scholar] [CrossRef]
- Wyatt, C.N.; Peers, C. Ca2+-activated K+ channels in isolated type I cells of the neonatal rat carotid body. J. Physiol. 1995, 483, 559–565. [Google Scholar] [CrossRef]
- Wyatt, C.N.; Wright, C.; Bee, D.; Peers, C. O2-sensitive K+ currents in carotid body chemoreceptor cells from normoxic and chronically hypoxic rats and their roles in hypoxic chemotransduction. Proc. Natl. Acad. Sci. USA 1995, 92, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Pardal, R.; Ludewig, U.; Garcia-Hirschfeld, J.; Lopez-Barneo, J. Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc. Natl. Acad. Sci. USA 2000, 97, 2361–2366. [Google Scholar] [CrossRef] [PubMed]
- Riesco-Fagundo, A.M.; Pérez-García, M.T.; González, C.; López-López, J.R. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ. Res. 2001, 89, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Latorre, R.; Castillo, K.; Carrasquel-Ursulaez, W.; Sepulveda, R.V.; Gonzalez-Nilo, F.; Gonzalez, C.; Alvarez, O. Molecular Determinants of BK Channel Functional Diversity and Functioning. Physiol. Rev. 2017, 97, 39–87. [Google Scholar] [CrossRef]
- Lahiri, S.; Roy, A.; Rozanov, C.; Mokashi, A. K current modulated by PO2 in type I cells in rat carotid body is not a chemosensor. Brain Res. 1998, 794, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.F. Are oxygen dependent K+ channels essential for carotid body chemo-transduction? Respir. Physiol. 1997, 110, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Niño, A.; Obeso, A.; Baranda, J.A.; Santo-Domingo, J.; Lopez-Lopez, J.R.; Gonzalez, C. MaxiK potassium channels in the function of chemoreceptor cells of the rat carotid body. Am. J. Physiol. Cell Physiol. 2009, 297, C715–C722. [Google Scholar] [CrossRef]
- Hatton, C.J.; Carpenter, E.; Pepper, D.R.; Kumar, P.; Peers, C. Developmental changes in isolated rat type I carotid body cell K+ currents and their modulation by hypoxia. J. Physiol. 1997, 501, 49–58. [Google Scholar] [CrossRef]
- Peers, C.; Wyatt, C.N. The role of maxiK channels in carotid body chemotransduction. Respir. Physiol. Neurobiol. 2007, 157, 75–82. [Google Scholar] [CrossRef]
- López-Barneo, J. All for one—O2-sensitive K+ channels that mediate carotid body activation. J. Physiol. 2018, 596, 2951–2952. [Google Scholar] [CrossRef]
- Eyzaguirre, C. Chemical and electric transmission in the carotid body chemoreceptor complex. Biol. Res. 2005, 38, 341–345. [Google Scholar] [CrossRef]
- Hou, S.; Heinemann, S.H.; Hoshi, T. Modulation of BKCa channel gating by endogenous signaling molecules. Physiology 2009, 24, 26–35. [Google Scholar] [CrossRef]
- Kyle, B.D.; Braun, A.P. The regulation of BK channel activity by pre- and post-translational modifications. Front. Physiol. 2014, 5, 316. [Google Scholar] [CrossRef]
- Bolotina, V.M.; Najibi, S.; Palacino, J.J.; Pagano, P.J.; Cohen, R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Jaggar, J.H.; Li, A.; Parfenova, H.; Liu, J.; Umstot, E.S.; Dopico, A.M.; Leffler, C.W. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ. Res. 2005, 97, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.E.; Wootton, P.; Mason, H.S.; Bould, J.; Iles, D.E.; Riccardi, D.; Peers, C.; Kemp, P.J. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science 2004, 306, 2093–2097. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Dinerman, J.L.; Agani, F.H.; Snyder, S.H. Carbon monoxide: A role in carotid body chemoreception. Proc. Natl. Acad. Sci. USA 1995, 92, 1994–1997. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Sáenz, P.; Pascual, A.; Gómez-Díaz, R.; López-Barneo, J. Acute oxygen sensing in heme oxygenase-2 null mice. J. Gen. Physiol. 2006, 128, 405–411. [Google Scholar] [CrossRef] [PubMed]
- McCartney, C.E.; McClafferty, H.; Huibant, J.M.; Rowan, R.G.; Shipston, M.J.; Rowe, I.C.M. A cysteine-rich motif confers hypoxia sensitivity to mammalian large conductance voltage- and Ca-activated K (BK) channel α-subunits. Proc. Natl. Acad. Sci. USA 2005, 102, 17870–17876. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.A.; Rafferty, J.N.; Dallas, M.L.; Ogunbayo, O.; Ikematsu, N.; McClafferty, H.; Tian, L.; Widmer, H.; Rowe, I.C.; Wyatt, C.N.; et al. Selective expression in carotid body type I cells of a single splice variant of the large conductance calcium- and voltage-activated potassium channel confers regulation by AMP-activated protein kinase. J. Biol. Chem. 2011, 286, 11929–11936. [Google Scholar] [CrossRef]
- Evans, A.M.; Mustard, K.J.; Wyatt, C.N.; Peers, C.; Dipp, M.; Kumar, P.; Kinnear, N.P.; Hardie, D.G. Does AMP-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in O2-sensing cells? J. Biol. Chem. 2005, 280, 41504–41511. [Google Scholar] [CrossRef]
- Evans, A.M.; Hardie, D.G.; Peers, C.; Wyatt, C.N.; Viollet, B.; Kumar, P.; Dallas, M.L.; Ross, F.; Ikematsu, N.; Jordan, H.L.; et al. Ion channel regulation by AMPK: The route of hypoxia-response coupling in thecarotid body and pulmonary artery. Ann. N. Y. Acad. Sci. 2009, 1177, 89–100. [Google Scholar] [CrossRef]
- Wyatt, C.N.; Mustard, K.J.; Pearson, S.A.; Dallas, M.L.; Atkinson, L.; Kumar, P.; Peers, C.; Hardie, D.G.; Evans, A.M. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J. Biol. Chem. 2007, 282, 8092–8098. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.D.; Lewis, S.; Juričić, L.; Udoh, U.A.; Hartmann, S.; Jansen, M.A.; Ogunbayo, O.A.; Puggioni, P.; Holmes, A.P.; Kumar, P.; et al. AMP-activated Protein Kinase Deficiency Blocks the Hypoxic Ventilatory Response and Thus Precipitates Hypoventilation and Apnea. Am. J. Respir. Crit. Care Med. 2016, 193, 1032–1043. [Google Scholar] [CrossRef]
- MacMillan, S.; Holmes, A.P.; Dallas, M.L.; Mahmoud, A.D.; Shipston, M.J.; Peers, C.; Hardie, D.G.; Kumar, P.; Evans, A.M. LKB1 is the gatekeeper of carotid body chemosensing and the hypoxic ventilatory response. Commun. Biol. 2022, 5, 642. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Dombkowski, R.A.; Russell, M.J.; Doellman, M.M.; Head, S.K.; Whitfield, N.L.; Madden, J.A. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J. Exp. Biol. 2006, 209, 4011–4023. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Hydrogen sulfide and oxygen sensing: Implications in cardiorespiratory control. J. Exp. Biol. 2008, 211, 2727–2734. [Google Scholar] [CrossRef]
- Yuan, G.; Vasavda, C.; Peng, Y.J.; Makarenko, V.V.; Raghuraman, G.; Nanduri, J.; Gadalla, M.M.; Semenza, G.L.; Kumar, G.K.; Snyder, S.H.; et al. Protein kinase G-regulated production of H2S governs oxygen sensing. Sci. Signal. 2015, 8, ra37. [Google Scholar] [CrossRef]
- Peng, Y.J.; Nanduri, J.; Raghuraman, G.; Souvannakitti, D.; Gadalla, M.M.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. H2S mediates O2 sensing in the carotid body. Proc. Natl. Acad. Sci. USA 2010, 107, 10719–10724. [Google Scholar] [CrossRef]
- Li, Q.; Sun, B.; Wang, X.; Jin, Z.; Zhou, Y.; Dong, L.; Jiang, L.H.; Rong, W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid. Redox Signal. 2010, 12, 1179–1189. [Google Scholar] [CrossRef]
- Telezhkin, V.; Brazier, S.P.; Cayzac, S.H.; Wilkinson, W.J.; Riccardi, D.; Kemp, P.J. Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir. Physiol. Neurobiol. 2010, 172, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, I.; Wang, J.; White, C.; Carroll, J.L. Hydrogen sulfide and hypoxia-induced changes in TASK (K2P3/9) activity and intracellular Ca2+ concentration in rat carotid body glomus cells. Respir. Physiol. Neurobiol. 2015, 215, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hogan, J.O.; Wang, R.; White, C.; Kim, D. Role of cystathionine-γ-lyase in hypoxia-induced changes in TASK activity, intracellular [Ca2+] and ventilation in mice. Respir. Physiol. Neurobiol. 2017, 246, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Are Reactive Sulfur Species the New Reactive Oxygen Species? Antioxid. Redox Signal. 2020, 33, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. A Case for Hydrogen Sulfide Metabolism as an Oxygen Sensing Mechanism. Antioxidants 2021, 10, 1650. [Google Scholar] [CrossRef]
- Buckler, K.J. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflügers Arch. 2012, 463, 743–754. [Google Scholar] [CrossRef]
- Yang, K.; Coburger, I.; Langner, J.M.; Peter, N.; Hoshi, T.; Schonherr, R.; Heinemann, S.H. Modulation of K+ channel N-type inactivation by sulfhydration through hydrogen sulfide and polysulfides. Pflügers Arch. 2019, 471, 557–571. [Google Scholar] [CrossRef]
- Lesage, F.; Lazdunski, M. Molecular and functional properties of two-pore-domain potassium channels. Am. J. Physiol. Renal Physiol. 2000, 279, F793–F801. [Google Scholar] [CrossRef]
- Kim, Y.; Bang, H.; Kim, D. TBAK-1 and TASK-1, two-pore K+ channel subunits: Kinetic properties and expression in rat heart. Am. J. Physiol. 1999, 277, H1669–H1678. [Google Scholar]
- Kim, Y.; Bang, H.; Kim, D. TASK-3, a new member of the tandem pore K+ channel family. J. Biol. Chem. 2000, 275, 9340–9347. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Wischmeyer, E.; Xin Liu, G.; Preisig-Müller, R.; Daut, J.; Karschin, A.; Derst, C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J. Biol. Chem. 2000, 275, 16650–16657. [Google Scholar] [CrossRef] [PubMed]
- Ashmole, I.; Goodwin, P.A.; Stanfield, P.R. TASK-5, a novel member of the tandem pore K+ channel family. Pflügers Arch. 2001, 442, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Decher, N.; Maier, M.; Dittrich, W.; Gassenhuber, J.; Brüggemann, A.; Busch, A.E.; Steinmeyer, K. Characterization of TASK-4, a novel member of the pH-sensitive, two-pore domain potassium channel family. FEBS Lett. 2001, 492, 84–89. [Google Scholar] [CrossRef]
- Medhurst, A.D.; Rennie, G.; Chapman, C.G.; Meadows, H.; Duckworth, M.D.; Kelsell, R.E.; Gloger, I.I.; Pangalos, M.N. Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res. Mol. Brain Res. 2001, 86, 101–114. [Google Scholar] [CrossRef]
- Sepulveda, F.V.; Cid, L.P.; Teulon, J.; Niemeyer, M.I. Molecular aspects of structure, gating, and physiology of pH-sensitive background K2P and Kir K+-transport channels. Physiol. Rev. 2015, 95, 179–217. [Google Scholar] [CrossRef]
- Maingret, F.; Patel, A.J.; Lazdunski, M.; Honoré, E. The endocannabinoid anandamide is a direct and selective blocker of the background K+ channel TASK-1. EMBO J. 2001, 20, 47–54. [Google Scholar] [CrossRef]
- Kindler, C.H.; Yost, C.S.; Gray, A.T. Local anesthetic inhibition of baseline potassium channels with two pore domains in tandem. Anesthesiology 1999, 90, 1092–1102. [Google Scholar] [CrossRef]
- Lesage, F. Pharmacology of neuronal background potassium channels. Neuropharmacology 2003, 44, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J.; Williams, B.A.; Honore, E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J. Physiol. 2000, 525, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.; Buckler, K.J. Biophysical properties and metabolic regulation of a TASK-like potassium channel in rat carotid body type 1 cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L221–L230. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J.; Williams, B.A.; Orozco, R.V.; Wyatt, C.N. The role of TASK-like K+ channels in oxygen sensing in the carotid body. Signalling Pathways in Acute Oxygen Sensing; Novartis Foundation Symposium; Wiley: Hoboken, NJ, USA, 2006; Volume 272, pp. 73–94. [Google Scholar]
- Czirják, G.; Enyedi, P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem. 2002, 277, 5426–5432. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Han, J.; Talley, E.M.; Bayliss, D.A.; Kim, D. Functional expression of TASK-1/TASK-3 heteromers in cerebellar granule cells. J. Physiol. 2004, 554, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kummer, W.; Atoji, Y.; Suzuki, Y. TASK-1, TASK-2, TASK-3 and TRAAK immunoreactivities in the rat carotid body. Brain Res. 2002, 950, 304–307. [Google Scholar] [CrossRef]
- Trapp, S.; Aller, M.I.; Wisden, W.; Gourine, A.V. A role for TASK-1 (KCNK3) channels in the chemosensory control of breathing. J. Neurosci. 2008, 28, 8844–8850. [Google Scholar] [CrossRef]
- Ortega-Sáenz, P.; Levitsky, K.L.; Marcos-Almaraz, M.T.; Bonilla-Henao, V.; Pascual, A.; López-Barneo, J. Carotid body chemosensory responses in mice deficient of TASK channels. J. Gen. Physiol. 2010, 135, 379–392. [Google Scholar] [CrossRef]
- Varas, R.; Wyatt, C.N.; Buckler, K.J. Modulation of TASK-like background potassium channels in rat arterial chemoreceptor cells by intracellular ATP and other nucleotides. J. Physiol. 2007, 583, 521–536. [Google Scholar] [CrossRef]
- Buckler, K.J. TASK channels in arterial chemoreceptors and their role in oxygen and acid sensing. Pflügers Arch. 2015, 467, 1013–1025. [Google Scholar] [CrossRef]
- Kang, D.; Wang, J.; Hogan, J.O.; Vennekens, R.; Freichel, M.; White, C.; Kim, D. Increase in cytosolic Ca2+ produced by hypoxia and other depolarizing stimuli activates a non-selective cation channel in chemoreceptor cells of rat carotid body. J. Physiol. 2014, 592, 1975–1992. [Google Scholar] [CrossRef]
- Ganfornina, M.D.; López-Barneo, J. Single K+ channels in membrane patches of arterial chemoreceptor cells are modulated by O2 tension. Proc. Natl. Acad. Sci. USA 1991, 88, 2927–2930. [Google Scholar] [CrossRef]
- Lewis, A.; Peers, C.; Ashford, M.L.; Kemp, P.J. Hypoxia inhibits human recombinant large conductance, Ca2+-activated K+ (maxi-K) channels by a mechanism which is membrane delimited and Ca2+ sensitive. J. Physiol. 2002, 540, 771–780. [Google Scholar] [CrossRef]
- Gonzalez, C.; Vaquero, L.M.; López-López, J.R.; Pérez-García, M.T. Oxygen-sensitive potassium channels in chemoreceptor cell physiology: Making a virtue of necessity. Ann. N. Y. Acad. Sci. 2009, 1177, 82–88. [Google Scholar] [CrossRef]
- Acker, H. Cellular oxygen sensors. Ann. N. Y. Acad. Sci. 1994, 718, 3–12. [Google Scholar] [CrossRef]
- Cross, A.R.; Henderson, L.; Jamesm, T.G.; Delpiano, A.; Hentschel, J.; Acker, H. Involvement of an NAD(P)H oxidase as a PO2 sensor protein in the rat carotid body. Biochem. J. 1990, 272, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Acker, H.; Bölling, B.; Delpiano, M.A.; Dufau, E.; Görlach, A.; Holtermann, G. The meaning of H2O2 generation in carotid body cells for PO2 chemoreception. J. Auton. Nerv. Syst. 1992, 41, 41–51. [Google Scholar] [CrossRef]
- Obeso, A.; Gómez-Niño, A.; Gonzalez, C. NADPH oxidase inhibition does not interfere with low PO2 transduction in rat and rabbit CB chemoreceptor cells. Am. J. Physiol. 1999, 276, C593–601. [Google Scholar] [CrossRef]
- Hutchinson, D.S.; Csikasz, R.I.; Yamamoto, D.L.; Shabalina, I.G.; Wikstrom, P.; Wilcke, M.; Bengtsson, T. Diphenylene iodonium stimulates glucose uptake in skeletal muscle cells through mitochondrial complex I inhibition and activation of AMP-activated protein kinase. Cell. Signal. 2007, 19, 1610–1620. [Google Scholar] [CrossRef]
- He, L.; Dinger, B.; Sanders, K.; Hoidal, J.; Obeso, A.; Stensaas, L.; Fidone, S.; Gonzalez, C. Effect of p47phox gene deletion on ROS production and oxygen sensing in mouse carotid body chemoreceptor cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L916–L924. [Google Scholar] [CrossRef]
- Dinger, B.; He, L.; Chen, J.; Liu, X.; Gonzalez, C.; Obeso, A.; Sanders, K.; Hoidal, J.; Stensaas, L.; Fidone, S. The role of NADPH oxidase in carotid body arterial chemoreceptors. Respir. Physiol. Neurobiol. 2007, 157, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Sanz-Alyayate, G.; Agapito, M.T.; Obeso, A. Effects of reducing agents on glutathione metabolism and the function of carotid body chemoreceptor cells. Biol. Chem. 2004, 385, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Agapito, M.T.; Sanz-Alfayate, G.; Gomez-Niño, A.; Gonzalez, C.; Obeso, A. General redox environment and carotid body chemoreceptor function. Am. J. Physiol. Cell Physiol. 2009, 296, C620–C631. [Google Scholar]
- Mkrtchian, S.; Kahlin, J.; Ebberyd, A.; Gonzalez, C.; Sanchez, D.; Balbir, A.; Kostuk, E.W.; Shirahata, M.; Fagerlund, M.J.; Eriksson, L.I. The human carotid body transcriptome with focus on oxygen sensing and inflammation—A comparative analysis. J. Physiol. 2012, 590, 3807–3819. [Google Scholar] [CrossRef]
- Yuan, G.; Khan, S.A.; Luo, W.; Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell. Physiol. 2011, 226, 2925–2933. [Google Scholar] [CrossRef]
- Obeso, A.; Almaraz, L.; Gonzalez, C. Effects of cyanide and uncouplers on chemoreceptor activity and ATP content of the cat carotid body. Brain Res. 1989, 481, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F.; Mokashi, A.; Chugh, D.; Vinogradov, S.; Osanai, S.; Lahiri, S. The primary oxygen sensor of the cat carotid body is cytochrome a3 of the mitochondrial respiratory chain. FEBS Lett. 1994, 351, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Mosqueira, M.; Iturriaga, R. Carotid body chemosensory excitation induced by nitric oxide: Involvement of oxidative metabolism. Respir. Physiol. Neurobiol. 2002, 131, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Sáenz, P.; Pardal, R.; García-Fernandez, M.; López-Barneo, J. Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. J. Physiol. 2003, 548, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J.; Vaughan-Jones, R.D. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J. Physiol. 1998, 513, 819–833. [Google Scholar] [PubMed]
- Wyatt, C.N.; Buckler, K.J. The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type I cells. J. Physiol. 2004, 556, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Agüera, M.C.; Gao, L.; González-Rodríguez, P.; Pintado, C.O.; Arias-Mayenco, I.; García-Flores, P.; García-Pergañeda, A.; Pascual, A.; Ortega-Sáenz, P.; López-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Arias-Mayenco, I.; González-Rodríguez, P.; Torres-Torrelo, H.; Gao, L.; Fernández-Agüera, M.C.; Bonilla-Henao, V.; Ortega-Sáenz, P.; López-Barneo, J. Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab. 2018, 28, 145–158. [Google Scholar] [CrossRef]
- Chang, A.J.; Ortega, F.E.; Riegler, J.; Madison, D.V.; Krasnow, M.A. Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 2015, 527, 240–244. [Google Scholar] [CrossRef]
- Holmes, A.P.; Swiderska, A.; Nathanael, D.; Aldossary, H.S.; Ray, C.J.; Coney, A.M.; Kumar, P. Are Multiple Mitochondrial Related Signalling Pathways Involved in Carotid Body Oxygen Sensing? Front. Physiol. 2022, 13, 908617. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Duranteau, J.; Chandel, N.S.; Kulisz, A.; Shao, Z.; Schumacker, P.T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 1998, 273, 11619–11624. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Waypa, G.B.; Chandel, N.S.; Schumacker, P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ. Res. 2001, 88, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Swiderska, A.; Coney, A.M.; Alzahrani, A.A.; Aldossary, H.S.; Batis, N.; Ray, C.J.; Kumar, P.; Holmes, A.P. Mitochondrial Succinate Metabolism and Reactive Oxygen Species Are Important but Not Essential for Eliciting Carotid Body and Ventilatory Responses to Hypoxia in the Rat. Antioxidants 2021, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Sáenz, P.; Lopez-Barneo, J. Physiology of the Carotid Body: From Molecules to Disease. Annu. Rev. Physiol. 2020, 82, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.J.; Makarenko, V.V.; Gridina, A.; Chupikova, I.; Zhang, X.; Kumar, G.K.; Fox, A.P.; Prabhakar, N.R. H2S mediates carotid body response to hypoxia but not anoxia. Respir. Physiol. Neurobiol. 2019, 259, 75–85. [Google Scholar] [CrossRef]
- Torres-Torrelo, H.; Ortega-Sáenz, P.; Macías, D.; Omura, M.; Zhou, T.; Matsunami, H.; Johnson, R.S.; Mombaerts, P.; López-Barneo, J. The role of Olfr78 in the breathing circuit of mice. Nature 2018, 561, E33–E40. [Google Scholar] [CrossRef] [PubMed]
- Delpiano, M.A.; Acker, H. Extracellular pH changes in the superfused cat carotid body during hypoxia and hypercapnia. Brain Res. 1985, 342, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, A.; Wolf, A.; Brockmeier, U.; Riffkin, H.; Metzen, E.; Acker-Palmer, A.; Fandrey, J.; Acker, H. Carotid body type I cells engage flavoprotein and Pin1 for oxygen sensing. Am. J. Physiol. Cell Physiol. 2020, 318, C719–C731. [Google Scholar] [CrossRef] [PubMed]
- Monti-Bloch, L.; Abudara, V.; Eyzaguirre, C. Electrical communication between glomus cells of the rat carotid body. Brain Res. 1993, 622, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.J.; Nanduri, J.; Wang, N.; Kumar, G.K.; Bindokas, V.; Paul, B.D.; Chen, X.; Fox, A.P.; Vignane, T.; Filipovic, M.R.; et al. Hypoxia sensing requires H2S-dependent persulfidation of olfactory receptor 78. Sci. Adv. 2023, 9, eadf3026. [Google Scholar] [CrossRef]
- Holmes, A.P.; Ray, C.J.; Coney, A.M.; Kumar, P. Is Carotid Body Physiological O2 Sensitivity Determined by a Unique Mitochondrial Phenotype? Front. Physiol. 2018, 9, 562. [Google Scholar] [CrossRef] [PubMed]
- Rakoczy, R.J.; Wyatt, C.N. Acute oxygen sensing by the carotid body: A rattlebag of molecular mechanisms. J. Physiol. 2018, 596, 2969–2976. [Google Scholar] [CrossRef]
- Gonzalez, C.; Sanz-Alfayate, G.; Agapito, M.T.; Gomez-Niño, A.; Rocher, A.; Obeso, A. Significance of ROS in oxygen sensing in cell systems with sensitivity to physiological hypoxia. Respir. Physiol. Neurobiol. 2002, 132, 17–41. [Google Scholar] [CrossRef]
- Kilfoil, P.J.; Tipparaju, S.M.; Barski, O.A.; Bhatnagar, A. Regulation of ion channels by pyridine nucleotides. Circ. Res. 2013, 112, 721–741. [Google Scholar] [CrossRef]
- Otsubo, T.; Kostuk, E.W.; Balbir, A.; Fujii, K.; Shirahata, M. Differential Expression of Large-Conductance Ca2+-Activated K Channels in the Carotid Body between DBA/2J and A/J Strains of Mice. Front. Cell. Neurosci. 2011, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Hartness, M.E.; Brazier, S.P.; Peers, C.; Bateson, A.N.; Ashford, M.L.; Kemp, P.J. Post-transcriptional control of human maxiK potassium channel activity and acute oxygen sensitivity by chronic hypoxia. J. Biol. Chem. 2003, 278, 51422–51432. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Schumacker, P.T. Cellular oxygen sensing by mitochondria: Old questions, new insight. J. Appl. Physiol. 2000, 88, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, D.; Chance, B.; Cadenas, E.; Boveris, A. The relation of free radical production to hyperoxia. Annu. Rev. Physiol. 1986, 48, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P. Sensing hypoxia in the carotid body: From stimulus to response. Essays Biochem. 2007, 43, 43–60. [Google Scholar]
- Sanz-Alfayate, G.; Obeso, A.; Agapito, M.T.; González, C. Reduced to oxidized glutathione ratios and oxygen sensing in calf and rabbit carotid body chemoreceptor cells. J. Physiol. 2001, 537, 209–220. [Google Scholar] [CrossRef]
- Yamamoto, Y.; König, P.; Henrich, M.; Dedio, J.; Kummer, W. Hypoxia induces production of nitric oxide and reactive oxygen species in glomus cells of rat carotid body. Cell Tissue Res. 2006, 325, 3–11. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Biological Production, Detection, and Fate of Hydrogen Peroxide. Antioxid. Redox Signal. 2018, 29, 541–551. [Google Scholar] [CrossRef]
- Netto, L.E.; Antunes, F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol. Cells. 2016, 39, 65–71. [Google Scholar] [CrossRef]
- Grayson, C.; Mailloux, R.J. Coenzyme Q10 and nicotinamide nucleotide transhydrogenase: Sentinels for mitochondrial hydrogen peroxide signaling. Free Radic. Biol. Med. 2023, 208, 260–271. [Google Scholar] [CrossRef]
- Gonzalez, C.; Agapito, M.T.; Rocher, A.; Gomez-Niño, A.; Rigual, R.; Castañeda, J.; Conde, S.V.; Obeso, A. A revisit to O2 sensing and transduction in the carotid body chemoreceptors in the context of reactive oxygen species biology. Respir. Physiol. Neurobiol. 2010, 174, 317–330. [Google Scholar] [CrossRef]
- Papreck, J.R.; Martin, E.A.; Lazzarini, P.; Kang, D.; Kim, D. Modulation of K2P3.1 (TASK-1), K2P9.1 (TASK-3), and TASK-1/3 heteromer by reactive oxygen species. Pflügers Arch. 2012, 464, 471–480. [Google Scholar] [CrossRef]
- Rakoczy, R.J.; Schiebrel, C.M.; Wyatt, C.N. Acute Oxygen-Sensing via Mitochondria-Generated Temperature Transients in Rat Carotid Body Type I Cells. Front. Physiol. 2022, 13, 874039. [Google Scholar] [CrossRef] [PubMed]
- Conrad, P.W.; Conforti, L.; Kobayashi, S.; Beitner-Johnson, D.; Rust, R.T.; Yuan, Y.; Kim, H.W.; Kim, R.H.; Seta, K.; Millhorn, D.E. The molecular basis of O2-sensing and hypoxia tolerance in pheochromocytoma cells. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2001, 128, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Robertson, T.P.; Hague, D.; Aaronson, P.I.; Ward, J.P. Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J. Physiol. 2000, 525, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Dietrich, A.; Fuchs, B.; Kalwa, H.; Ay, M.; Dumitrascu, R.; Olschewski, A.; Storch, U.; Mederos-Schnitzler, M.; Ghofrani, H.A.; et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. USA 2006, 103, 19093–19098. [Google Scholar] [CrossRef]
- Archer, S.L.; Wu, X.C.; Thebaud, B.; Nsair, A.; Bonnet, S.; Tyrrell, B.; McMurtry, M.S.; Hashimoto, K.; Harry, G.; Michelakis, E.D. Preferential expression and function of voltage-gated, O2-sensitive K+ channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: Ionic diversity in smooth muscle cells. Circ. Res. 2004, 95, 308–318. [Google Scholar] [CrossRef]
- Neo, B.H.; Patel, D.; Kandhi, S.; Wolin, M.S. Roles for cytosolic NADPH redox in regulating pulmonary artery relaxation by thiol oxidation-elicited subunit dimerization of protein kinase G1α. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H330–H343. [Google Scholar] [CrossRef]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef]
- Gupte, R.S.; Rawat, D.K.; Chettimada, S.; Cioffi, D.L.; Wolin, M.S.; Gerthoffer, W.T.; McMurtry, I.F.; Gupte, S.A. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J. Biol. Chem. 2010, 285, 19561–19571. [Google Scholar] [CrossRef]
- Cogolludo, A.; Moreno, L.; Frazziano, G.; Moral-Sanz, J.; Menendez, C.; Castaneda, J.; Gonzalez, C.; Villamor, E.; Perez-Vizcaino, F. Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstriction. Cardiovasc. Res. 2009, 82, 296–302. [Google Scholar] [CrossRef]
- Sommer, N.; Huttemann, M.; Pak, O.; Scheibe, S.; Knoepp, F.; Sinkler, C.; Malczyk, M.; Gierhardt, M.; Esfandiary, A.; Kraut, S.; et al. Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing. Circ. Res. 2017, 121, 424–438. [Google Scholar] [CrossRef]
- Olschewski, A.; Li, Y.; Tang, B.; Hanze, J.; Eul, B.; Bohle, R.M.; Wilhelm, J.; Morty, R.E.; Brau, M.E.; Weir, E.K.; et al. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ. Res. 2006, 98, 1072–1080. [Google Scholar] [CrossRef]
- Nagaraj, C.; Tang, B.; Balint, Z.; Wygrecka, M.; Hrzenjak, A.; Kwapiszewska, G.; Stacher, E.; Lindenmann, J.; Weir, E.K.; Olschewski, H.; et al. Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur. Respir. J. 2013, 41, 85–95. [Google Scholar] [CrossRef]
- Sedivy, V.; Joshi, S.; Ghaly, Y.; Mizera, R.; Zaloudikova, M.; Brennan, S.; Novotna, J.; Herget, J.; Gurney, A.M. Role of KV7 channels in responses of the pulmonary circulation to hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L48–L57. [Google Scholar] [CrossRef]
- Peng, W.; Karwande, S.V.; Hoidal, J.R.; Farrukh, I.S. Potassium currents in cultured human pulmonary arterial smooth muscle cells. J. Appl. Physiol. 1996, 80, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ. Res. 1995, 77, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Wiener, C.M.; Banta, M.R.; Dowless, M.S.; Flavahan, N.A.; Sylvester, J.T. Mechanisms of hypoxic vasodilation in ferret pulmonary arteries. Am. J. Physiol. 1995, 269, L351–L357. [Google Scholar] [CrossRef] [PubMed]
- Osipenko, O.N.; Evans, A.M.; Gurney, A.M. Regulation of the resting potential of rabbit pulmonary artery myocytes by a low threshold, O2-sensing potassium current. Br. J. Pharmacol 1997, 120, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Sylvester, J.T.; Sham, J.S. Chronic hypoxia alters effects of endothelin and angiotensin on K+ currents in pulmonary arterial myocytes. Am. J. Physiol. 1999, 277, L431–L439. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, C.; Li, Y.; Tang, B.; Bordag, N.; Guntur, D.; Enyedi, P.; Olschewski, H.; Olschewski, A. Potassium Channels in the Transition from Fetal to the Neonatal Pulmonary Circulation. Int. J. Mol. Sci. 2022, 23, 4681. [Google Scholar] [CrossRef]
- Post, J.M.; Hume, J.R.; Archer, S.L.; Weir, E.K. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am. J. Physiol. 1992, 262, C882–C890. [Google Scholar] [CrossRef]
- McMurtry, I.F.; Davidson, A.B.; Reeves, J.T.; Grover, R.F. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ. Res. 1976, 38, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Harder, D.R.; Madden, J.A.; Dawson, C. A membrane electrical mechanism for hypoxic vasoconstriction of small pulmonary arteries from cat. Chest 1985, 88, 233S–235S. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J.; Goldman, W.F.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am. J. Physiol. 1993, 264, L116–L123. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J.; Goldman, W.F.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. Ionic currents in rat pulmonary and mesenteric arterial myocytes in primary culture and subculture. Am. J. Physiol. 1993, 264, L107–L115. [Google Scholar] [CrossRef] [PubMed]
- Post, J.; Gelband, C.H.; Hume, J.R. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ. Res. 1995, 77, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; Osipenko, O.N.; Gurney, A.M. Properties of a novel K+ current that is active at resting potential in rabbit pulmonary artery smooth muscle cells. J. Physiol. 1996, 496, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Lazdunski, M.; Honore, E. KV2.1/KV9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J. 1997, 16, 6615–6625. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Souil, E.; Dinh-Xuan, A.T.; Schremmer, B.; Mercier, J.C.; El Yaagoubi, A.; Nguyen-Huu, L.; Reeve, H.L.; Hampl, V. Molecular identification of the role of voltage-gated K+ channels, KV1.5 and KV2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J. Clin. Investig. 1998, 101, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; London, B.; Hampl, V.; Wu, X.; Nsair, A.; Puttagunta, L.; Hashimoto, K.; Waite, R.E.; Michelakis, E.D. Impairment of hypoxic pulmonary vasoconstriction in mice lacking the voltage-gated potassium channel KV1.5. FASEB J. 2001, 15, 1801–1803. [Google Scholar] [CrossRef]
- London, B.; Guo, W.; Pan, X.; Lee, J.S.; Shusterman, V.; Rocco, C.J.; Logothetis, D.A.; Nerbonne, J.M.; Hill, J.A. Targeted replacement of KV1.5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of IK,slow and resistance to drug-induced qt prolongation. Circ. Res. 2001, 88, 940–946. [Google Scholar] [CrossRef]
- Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Yu, Y.; Remillard, C.V.; Yuan, J.X. Acute hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2006, 290, C907–C916. [Google Scholar] [CrossRef]
- Hulme, J.T.; Coppock, E.A.; Felipe, A.; Martens, J.R.; Tamkun, M.M. Oxygen sensitivity of cloned voltage-gated K+ channels expressed in the pulmonary vasculature. Circ. Res. 1999, 85, 489–497. [Google Scholar] [CrossRef]
- Platoshyn, O.; Yu, Y.; Ko, E.A.; Remillard, C.V.; Yuan, J.X. Heterogeneity of hypoxia-mediated decrease in IK(V) and increase in [Ca2+]cyt in pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L402–L416. [Google Scholar] [CrossRef]
- Gurney, A.M.; Osipenko, O.N.; MacMillan, D.; McFarlane, K.M.; Tate, R.J.; Kempsill, F.E. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ. Res. 2003, 93, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Sedivy, V.; Hodyc, D.; Herget, J.; Gurney, A.M. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J. Pharmacol. Exp. Ther. 2009, 329, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Huang, J.; Henry, T.; Peterson, D.; Weir, E.K. A redox-based O2 sensor in rat pulmonary vasculature. Circ. Res. 1993, 73, 1100–1112. [Google Scholar] [CrossRef]
- Firth, A.L.; Gordienko, D.V.; Yuill, K.H.; Smirnov, S.V. Cellular localization of mitochondria contributes to KV channel-mediated regulation of cellular excitability in pulmonary but not mesenteric circulation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L347–L360. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Wu, D.; Potus, F.; Sykes, E.A.; Mewburn, J.D.; Charles, R.L.; Eaton, P.; Sultanian, R.A.; Archer, S.L. Ndufs2, a Core Subunit of Mitochondrial Complex I, Is Essential for Acute Oxygen-Sensing and Hypoxic Pulmonary Vasoconstriction. Circ. Res. 2019, 124, 1727–1746. [Google Scholar] [CrossRef]
- Olschewski, A.; Hong, Z.; Peterson, D.A.; Nelson, D.P.; Porter, V.A.; Weir, E.K. Opposite effects of redox status on membrane potential, cytosolic calcium, and tone in pulmonary arteries and ductus arteriosus. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L15–L22. [Google Scholar] [CrossRef]
- Park, M.K.; Lee, S.H.; Ho, W.K.; Earm, Y.E. Redox agents as a link between hypoxia and the responses of ionic channels in rabbit pulmonary vascular smooth muscle. Exp. Physiol. 1995, 80, 835–842. [Google Scholar] [CrossRef]
- Park, M.K.; Bae, Y.M.; Lee, S.H.; Ho, W.K.; Earm, Y.E. Modulation of voltage-dependent K+ channel by redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflügers Arch. 1997, 434, 764–771. [Google Scholar] [CrossRef]
- Reeve, H.L.; Weir, E.K.; Nelson, D.P.; Peterson, D.A.; Archer, S.L. Opposing effects of oxidants and antioxidants on K+ channel activity and tone in rat vascular tissue. Exp. Physiol. 1995, 80, 825–834. [Google Scholar] [CrossRef]
- Schach, C.; Xu, M.; Platoshyn, O.; Keller, S.H.; Yuan, J.X. Thiol oxidation causes pulmonary vasodilation by activating K+ channels and inhibiting store operated Ca2+ channels. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L685–L698. [Google Scholar] [CrossRef]
- Weir, E.K.; Hong, Z.; Porter, V.A.; Reeve, H.L. Redox signaling in oxygen sensing by vessels. Respir. Physiol. NeuroBiol. 2002, 132, 121–130. [Google Scholar] [CrossRef]
- Olschewski, A.; Weir, E.K. Redox regulation of ion channels in the pulmonary circulation. Antioxid. Redox Signal. 2015, 22, 465–485. [Google Scholar] [CrossRef] [PubMed]
- Frazziano, G.; Moreno, L.; Moral-Sanz, J.; Menendez, C.; Escolano, L.; Gonzalez, C.; Villamor, E.; Alvarez-Sala, J.L.; Cogolludo, A.L.; Perez-Vizcaino, F. Neutral sphingomyelinase, NADPH oxidase and reactive oxygen species. Role in acute hypoxic pulmonary vasoconstriction. J. Cell. Physiol. 2011, 226, 2633–2640. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Moreno, L.; Bosca, L.; Tamargo, J.; Perez-Vizcaino, F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: Role of protein kinase Czeta. Circ. Res. 2003, 93, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Frazziano, G.; Cobeno, L.; Moreno, L.; Lodi, F.; Villamor, E.; Tamargo, J.; Perez-Vizcaino, F. Role of reactive oxygen species in KV channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann. N. Y. Acad. Sci. 2006, 1091, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Frazziano, G.; Cogolludo, A.; Cobeno, L.; Tamargo, J.; Perez-Vizcaino, F. Role of protein kinase Czeta and its adaptor protein p62 in voltage-gated potassium channel modulation in pulmonary arteries. Mol. Pharmacol. 2007, 72, 1301–1309. [Google Scholar] [CrossRef]
- Moral-Sanz, J.; Gonzalez, T.; Menendez, C.; David, M.; Moreno, L.; Macias, A.; Cortijo, J.; Valenzuela, C.; Perez-Vizcaino, F.; Cogolludo, A. Ceramide inhibits KV currents and contributes to TP-receptor-induced vasoconstriction in rat and human pulmonary arteries. Am. J. Physiol. Cell Physiol. 2011, 301, C186–C194. [Google Scholar] [CrossRef]
- Desireddi, J.R.; Farrow, K.N.; Marks, J.D.; Waypa, G.B.; Schumacker, P.T. Hypoxia increases ROS signaling and cytosolic Ca2+ in pulmonary artery smooth muscle cells of mouse lungs slices. Antioxid. Redox Signal. 2010, 12, 595–602. [Google Scholar] [CrossRef]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. Hypoxic pulmonary vasoconstriction. J. Appl. Physiol. 2005, 98, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Caouette, D.; Dongmo, C.; Berube, J.; Fournier, D.; Daleau, P. Hydrogen peroxide modulates the KV1.5 channel expressed in a mammalian cell line. Naunyn Schmiedeberg’s Arch. Pharmacol. 2003, 368, 479–486. [Google Scholar] [CrossRef]
- Yang, L.; Ma, J.H.; Zhang, P.H.; Zou, A.R.; Tu, D.N. Quercetin activates human KV1.5 channels by a residue I502 in the S6 segment. Clin. Exp. Pharmacol. Physiol. 2009, 36, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Duprat, F.; Guillemare, E.; Romey, G.; Fink, M.; Lesage, F.; Lazdunski, M.; Honore, E. Susceptibility of cloned K+ channels to reactive oxygen species. Proc. Natl. Acad. Sci. USA 1995, 92, 11796–11800. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Noh, H.J.; Sung, D.J.; Kim, J.G.; Kim, J.M.; Ryu, S.Y.; Kang, K.; Kim, B.; Bae, Y.M.; Cho, H. Hydrogen peroxide induces vasorelaxation by enhancing 4-aminopyridine-sensitive KV currents through S-glutathionylation. Pflügers Arch. 2015, 467, 285–297. [Google Scholar] [CrossRef]
- Svoboda, L.K.; Reddie, K.G.; Zhang, L.; Vesely, E.D.; Williams, E.S.; Schumacher, S.M.; O’Connell, R.P.; Shaw, R.; Day, S.M.; Anumonwo, J.M.; et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel KV1.5. Circ. Res. 2012, 111, 842–853. [Google Scholar] [CrossRef]
- Gamper, N.; Zaika, O.; Li, Y.; Martin, P.; Hernandez, C.C.; Perez, M.R.; Wang, A.Y.; Jaffe, D.B.; Shapiro, M.S. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006, 25, 4996–5004. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.J.; Wang, J.; Juhaszova, M.; Golovina, V.A.; Rubin, L.J. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. Am. J. Physiol. 1998, 274, L621–L635. [Google Scholar] [CrossRef]
- Bahring, R.; Milligan, C.J.; Vardanyan, V.; Engeland, B.; Young, B.A.; Dannenberg, J.; Waldschutz, R.; Edwards, J.P.; Wray, D.; Pongs, O. Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of KVβ subunits. J. Biol. Chem 2001, 276, 22923–22929. [Google Scholar] [CrossRef]
- Dwenger, M.M.; Raph, S.M.; Reyzer, M.L.; Lisa Manier, M.; Riggs, D.W.; Wohl, Z.B.; Ohanyan, V.; Mack, G.; Pucci, T.; Moore, J.B.; et al. Pyridine nucleotide redox potential in coronary smooth muscle couples myocardial blood flow to cardiac metabolism. Nat. Commun. 2022, 13, 2051. [Google Scholar] [CrossRef]
- Raph, S.M.; Dwenger, M.M.; Hu, X.; Nystoriak, M.A. Basal NAD(H) redox state permits hydrogen peroxide-induced mesenteric artery dilatation. J. Physiol. 2023, 601, 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Remillard, C.V.; Fantozzi, I.; Mandegar, M.; Sison, T.T.; Zhang, S.; Burg, E.; Yuan, J.X. Diversity of voltage-dependent K+ channels in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L226–L238. [Google Scholar] [CrossRef] [PubMed]
- Dwenger, M.M.; Raph, S.M.; Baba, S.P.; Moore, J.B.; Nystoriak, M.A. Diversification of Potassium Currents in Excitable Cells via KVβ Proteins. Cells 2022, 11, 2230. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Hampl, V.; Nsair, A.; Wu, X.; Harry, G.; Haromy, A.; Gurtu, R.; Archer, S.L. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ. Res. 2002, 90, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Rebeyka, I.; Wu, X.; Nsair, A.; Thebaud, B.; Hashimoto, K.; Dyck, J.R.; Haromy, A.; Harry, G.; Barr, A.; et al. O2 sensing in the human ductus arteriosus: Regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ. Res. 2002, 91, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S.H.; Ho, W.K. Hydrogen peroxide selectively increases TREK-2 currents via myosin light chain kinases. Front Biosci. 2007, 12, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.B.; Langford, T.F.; Huang, B.K.; Deen, W.M.; Sikes, H.D. A reaction-diffusion model of cytosolic hydrogen peroxide. Free Radic. Biol. Med. 2016, 90, 85–90. [Google Scholar] [CrossRef]
- Lee, Y.M.; Kim, B.J.; Chun, Y.S.; So, I.; Choi, H.; Kim, M.S.; Park, J.W. NOX4 as an oxygen sensor to regulate TASK-1 activity. Cell. Signal. 2006, 18, 499–507. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef]
- Stuart, J.A.; Fonseca, J.; Moradi, F.; Cunningham, C.; Seliman, B.; Worsfold, C.R.; Dolan, S.; Abando, J.; Maddalena, L.A. How Supraphysiological Oxygen Levels in Standard Cell Culture Affect Oxygen-Consuming Reactions. Oxidative Med. Cell. Longev. 2018, 2018, 8238459. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Kraut, S.; Wilhelm, J.; Sommer, N.; Quanz, K.; Seeger, W.; Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm. Circ. 2016, 6, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, C.; Tabeling, C.; Nagy, B.M.; Jain, P.P.; Marsh, L.M.; Papp, R.; Pienn, M.; Witzenrath, M.; Ghanim, B.; Klepetko, W.; et al. Hypoxic vascular response and ventilation/perfusion matching in end-stage COPD may depend on p22phox. Eur. Respir. J. 2017, 50, 1601651. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wang, J.; Shimoda, L.A.; Sylvester, J.T. Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+ responses to hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L104–L113. [Google Scholar] [CrossRef]
- Wang, J.; Shimoda, L.A.; Sylvester, J.T. Ca2+ responses of pulmonary arterial myocytes to acute hypoxia require release from ryanodine and inositol trisphosphate receptors in sarcoplasmic reticulum. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L161–L168. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.X.; Xu, X.Y.; Zhao, Z.; Zhao, F.Y.; Gao, Y.M.; Yan, X.H.; Wan, Y. Hydrogen peroxide is a critical regulator of the hypoxia-induced alterations of store-operated Ca2+ entry into rat pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L477–L487. [Google Scholar] [CrossRef]
- Lin, M.J.; Yang, X.R.; Cao, Y.N.; Sham, J.S. Hydrogen peroxide induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1598–L1608. [Google Scholar] [CrossRef]
- Yadav, V.R.; Song, T.; Joseph, L.; Mei, L.; Zheng, Y.M.; Wang, Y.X. Important role of PLC-gamma1 in hypoxic increase in intracellular calcium in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L143–L151. [Google Scholar] [CrossRef]
- Moral-Sanz, J.; Mahmoud, A.D.; Ross, F.A.; Eldstrom, J.; Fedida, D.; Hardie, D.G.; Evans, A.M. AMP-activated protein kinase inhibits Kv1.5 channel currents of pulmonary arterial myocytes in response to hypoxia and inhibition of mitochondrial oxidative phosphorylation. J. Physiol. 2016, 594, 4901–4915. [Google Scholar] [CrossRef] [PubMed]
- Moral-Sanz, J.; Lewis, S.A.; MacMillan, S.; Ross, F.A.; Thomson, A.; Viollet, B.; Foretz, M.; Moran, C.; Hardie, D.G.; Evans, A.M. The LKB1-AMPK-α1 signaling pathway triggers hypoxic pulmonary vasoconstriction downstream of mitochondria. Sci. Signal. 2018, 11, eaau0296. [Google Scholar] [CrossRef]
- Del Gaudio, F.; Liu, D.; Lendahl, U. Notch signalling in healthy and diseased vasculature. Open Biol. 2022, 12, 220004. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Voiriot, G.; Tang, H.; Fraidenburg, D.R.; Song, S.; Yamamura, H.; Yamamura, A.; Guo, Q.; Wan, J.; Pohl, N.M.; et al. Notch Activation of Ca2+ Signaling in the Development of Hypoxic Pulmonary Vasoconstriction and Pulmonary Hypertension. Am. J. Respir. Cell Mol Biol. 2015, 53, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.P.; Hosokawa, S.; Xiong, M.; Babicheva, A.; Zhao, T.; Rodriguez, M.; Rahimi, S.; Pourhashemi, K.; Balistrieri, F.; Lai, N.; et al. Revisiting the mechanism of hypoxic pulmonary vasoconstriction using isolated perfused/ventilated mouse lung. Pulm. Circ. 2020, 10, 2045894020956592. [Google Scholar] [CrossRef]
- Song, S.; Babicheva, A.; Zhao, T.; Ayon, R.J.; Rodriguez, M.; Rahimi, S.; Balistrieri, F.; Harrington, A.; Shyy, J.Y.; Thistlethwaite, P.A.; et al. Notch enhances Ca2+ entry by activating calcium-sensing receptors and inhibiting voltage-gated K+ channels. Am. J. Physiol. Cell Physiol. 2020, 318, C954–C968. [Google Scholar] [CrossRef]
- Knock, G.A.; Snetkov, V.A.; Shaifta, Y.; Drndarski, S.; Ward, J.P.; Aaronson, P.I. Role of src-family kinases in hypoxic vasoconstriction of rat pulmonary artery. Cardiovasc. Res. 2008, 80, 453–462. [Google Scholar] [CrossRef]
- MacKay, C.E.; Shaifta, Y.; Snetkov, V.V.; Francois, A.A.; Ward, J.P.T.; Knock, G.A. ROS-dependent activation of RhoA/Rho-kinase in pulmonary artery: Role of Src-family kinases and ARHGEF1. Free Radic. Biol. Med. 2017, 110, 316–331. [Google Scholar] [CrossRef]
- Pak, O.; Nolte, A.; Knoepp, F.; Giordano, L.; Pecina, P.; Huttemann, M.; Grossman, L.I.; Weissmann, N.; Sommer, N. Mitochondrial oxygen sensing of acute hypoxia in specialized cells—Is there a unifying mechanism? Biochim. Biophys. Acta Bioenerg. 2022, 1863, 148911. [Google Scholar] [CrossRef]
- Alruwaili, N.; Kandhi, S.; Sun, D.; Wolin, M.S. Metabolism and Redox in Pulmonary Vascular Physiology and Pathophysiology. Antioxid. Redox Signal. 2019, 31, 752–769. [Google Scholar] [CrossRef] [PubMed]
- Kemp, P.J.; Telezhkin, V. Oxygen sensing by the carotid body: Is it all just rotten eggs? Antioxid. Redox Signal. 2014, 20, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Bilan, D.S.; Belousov, V.V. New tools for redox biology: From imaging to manipulation. Free Radic. Biol. Med. 2017, 109, 167–188. [Google Scholar] [CrossRef]
- Lukyanov, K.A.; Belousov, V.V. Genetically encoded fluorescent redox sensors. Biochim. Biophys. Acta 2014, 1840, 745–756. [Google Scholar] [CrossRef]
- Pouvreau, S. Genetically encoded reactive oxygen species (ROS) and redox indicators. Biotechnol. J. 2014, 9, 282–293. [Google Scholar] [CrossRef]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Type | Sensor | Mediator | Effector K+ Channel | References |
|---|---|---|---|---|
| CBCC | Mitochondria/oxidative phosphorylation | ↑ ROS and/or NADH | KV | [50,161] |
| ↓ ATP | TASK 1/3 | [129] | ||
| Thermal transients | TASK 1/3 | [185] | ||
| ↑ Lactate causes acidification | TASK 1/3(?) | [166] | ||
| ↑ Lactate activates olfr 78 | ?? | [154] | ||
| Heme-oxygenase 2 | ↓ CO binds to heme | BKCa | [86,87] | |
| ↓ CO causes ↑H2S | BKCa | [100,101,102] | ||
| NADPH oxidase | ↓ ROS | ?? | [136] | |
| Stress-regulated exon (STREX) causes channel closure via unknown mechanism | BKCa | [90] | ||
| O2 interacts directly with channel-associated site to inhibit it via unknown mechanism | KV, BKCa | [47,51,74,133] | ||
| PASMC | Mitochondria/oxidative phosphorylation | ↓ ROS | KV1.5 | [33,204,207] |
| ↑ ROS | KV1.5 | [193,194] | ||
| ↑ AMP activates AMPK | KV1.5 | [263,264] | ||
| ?? | ↑ [Ca2+]cyt | KV | [209] | |
| ?? | Inhibition of src family kinase | TASK 1 | [195,196] | |
| ?? | ?? | KV7 | [197,219] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocher, A.; Aaronson, P.I. The Thirty-Fifth Anniversary of K+ Channels in O2 Sensing: What We Know and What We Don’t Know. Oxygen 2024, 4, 53-89. https://doi.org/10.3390/oxygen4010004
Rocher A, Aaronson PI. The Thirty-Fifth Anniversary of K+ Channels in O2 Sensing: What We Know and What We Don’t Know. Oxygen. 2024; 4(1):53-89. https://doi.org/10.3390/oxygen4010004
Chicago/Turabian StyleRocher, Asuncion, and Philip I. Aaronson. 2024. "The Thirty-Fifth Anniversary of K+ Channels in O2 Sensing: What We Know and What We Don’t Know" Oxygen 4, no. 1: 53-89. https://doi.org/10.3390/oxygen4010004