
The Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases

 , , ,
, , ,  and
and
Abstract
:1. Introduction
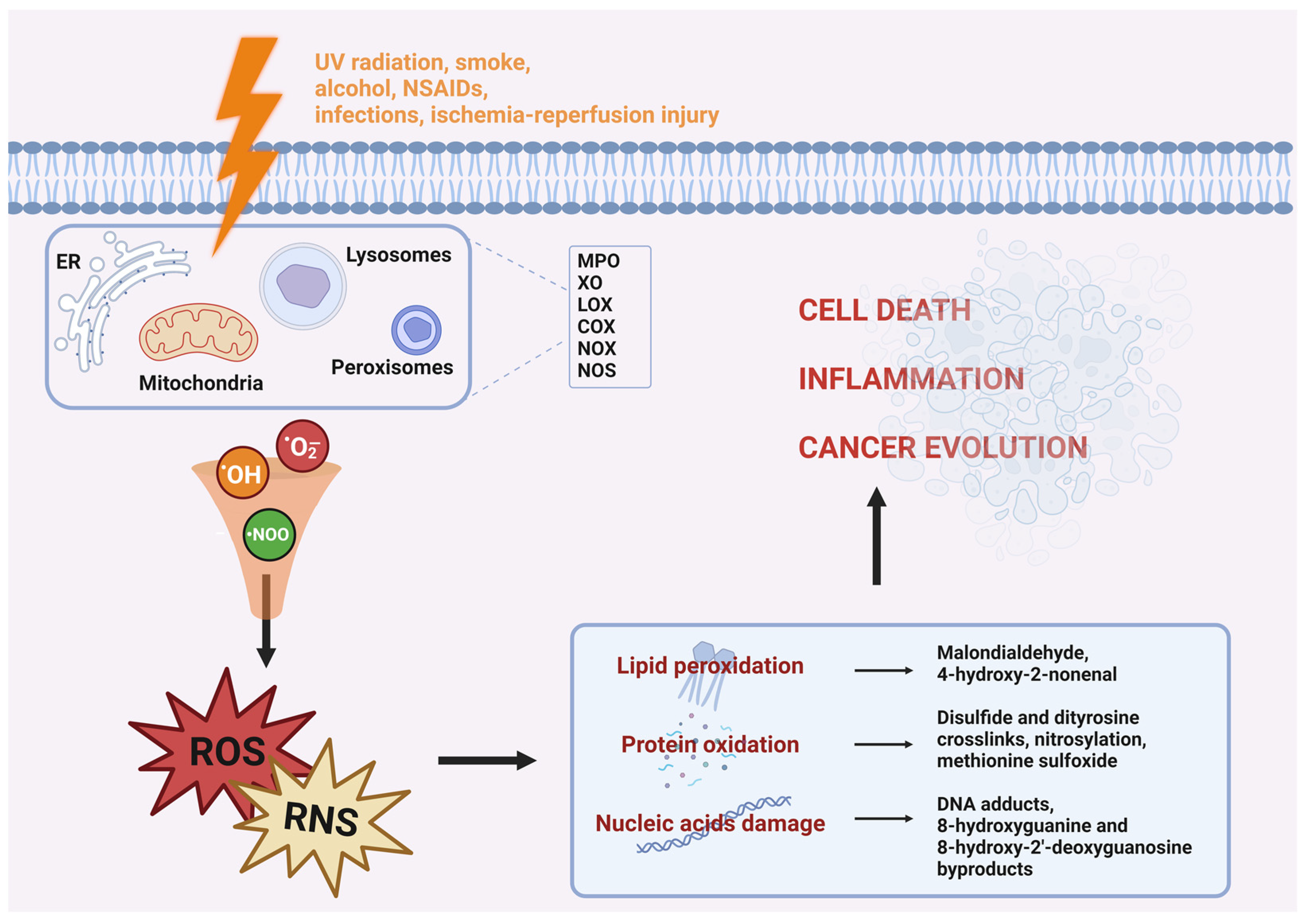
1.1. Oxidative Stress
1.2. Reactive Oxygen and Nitrogen Species
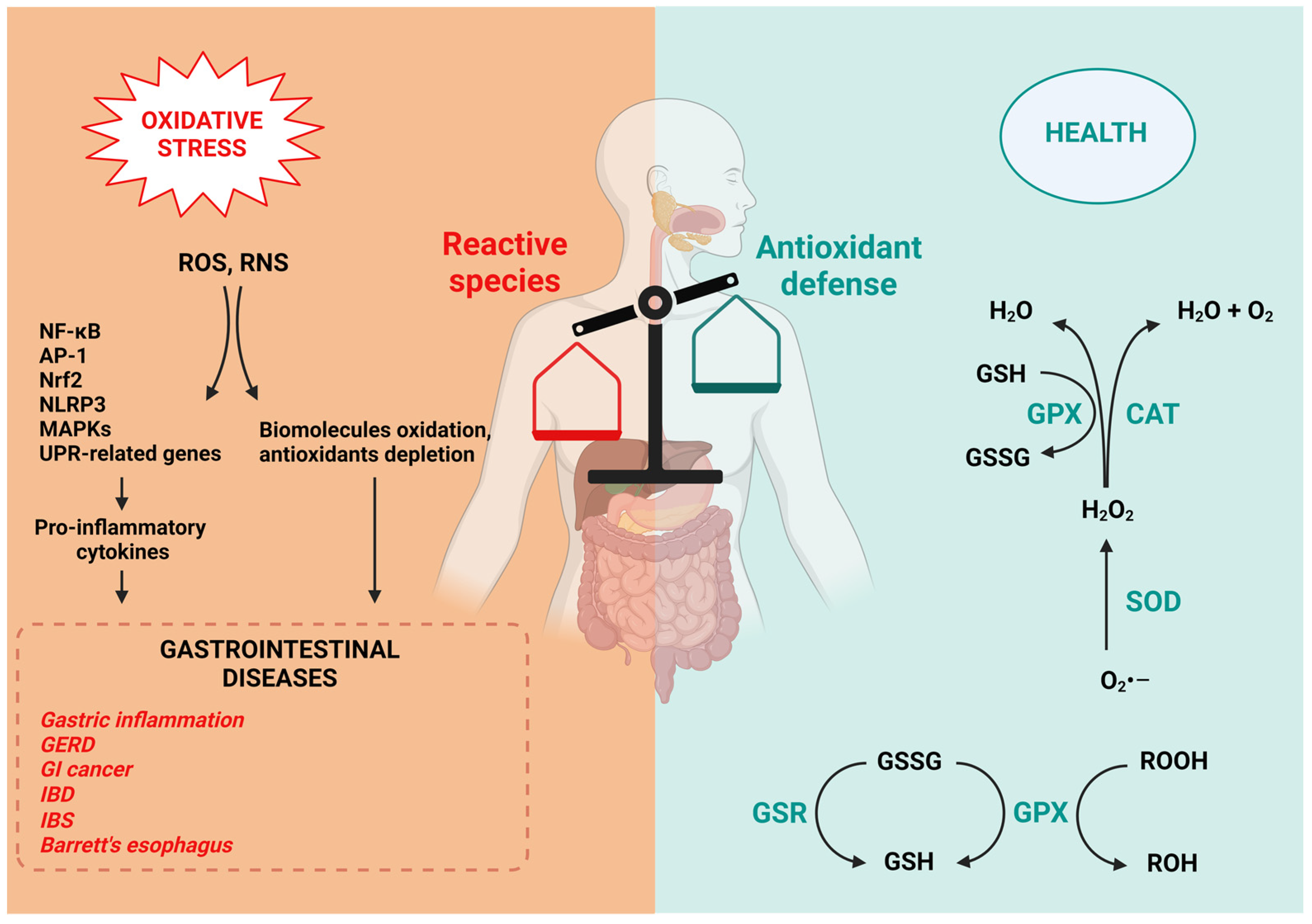
1.3. Antioxidant Defense in the GI Tract
1.4. Oxidative Stress in GI Diseases
2. Ion Channels and Oxidative Stress
2.1. Ion Channels Involved in Oxidative Stress-Related GI Diseases
2.2. Calcium Channels
2.2.1. Voltage-Gated Ca2+ Channels
2.2.2. Calcium-Permeable Non-Selective Ion Channels
TRPM2
TRPV1
TRPA1
2.3. Chloride Channels
2.3.1. Cystic Fibrosis Transmembrane Conductance Regulator
2.3.2. Calcium-Activated Chloride Channels
2.3.3. Chloride Intracellular Channels
2.4. Potassium Channels
2.4.1. The ATP-Sensitive Potassium Channels
2.4.2. Kv Channels
3. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Ion Channels | Localization | Function | Gastrointestinal Disease | References | |
|---|---|---|---|---|---|
| Calcium channels | Voltage-gated Ca2+ channels | ||||
| L-type Ca2+ channels | Smooth muscle cells, interstitial cells of Cajal | Smooth muscle contractility | Colon inflammation | [99,101,102,104,105,106,107,108,109] | |
| Calcium-permeable non-selective ion channels | TRP channels | ||||
| TRPM2 | Enterocytes, enteroendocrine cells, pancreatic β-cells, immune cells present in the intestinal mucosa | Maturation and chemotaxis of dendritic cells, regulating endothelial permeability and insulin secretion | Diabetes, IBD | [120,126,127,128,130,131] | |
| TRPV1 | Enteric neurons, enterocytes, smooth muscle cells, immune cells | Pain perception, regulation of motility, secretion of digestive enzymes | IBS, GERD | [134,135,136,137,138,139,140,141,142] | |
| TRPA1 | Sensory neurons of GI mucous, enteroendocrine cells | Cold perception, inflammatory pain | IBD, GERD | [145,147,148,158] | |
| Chloride channels | CFTR | Epithelial cells of pancreas, intestine, salivary glands, bile ducts and vas deferens | Fluid transport in epithelial tissues | Cystic fibrosis, colorectal cancer, IBD | [159,160,163,164,165,166,167,168,169,172,173,174,175] |
| CaCCs | |||||
| CLCA | Intestinal epithelial cells, interstitial cells of Cajal | Sensory transduction, epithelial secretion, smooth muscle contraction | IBD, cystic fibrosis | [180,181,182,184,185] | |
| BEST | |||||
| TMEM16 | |||||
| CLIC1 | Intestinal epithelial cells, enteroendocrine cells, pancreatic β-cells | Ion homeostasis, cell cycle regulation | Gastric carcinoma, colorectal cancer, gallbladder carcinoma | [186,187,188,192,193,194,196] | |
| Potassium channels | KATP channels | Smooth muscle cells, pancreatic β-cells | Regulation of insulin secretion, protection against cellular stress, adaptation to metabolic changes | Colon inflammation, type 2 diabetes | [203,204,205,206,207,208,209,210,211,212,213,214,215,216] |
| Kv channels | Smooth muscle cells, enteric neurons, Langerhans cells | Secretion of hormones, generation and propagation of electrical impulses, motility of the gastrointestinal tract | Abdominal pain, intestinal motility disorders | [210,217,218,219,224,227] | |
References
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 26, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar] [CrossRef]
- Sies, H. Strategies of antioxidant defense. Eur. J. Biochem. 1993, 215, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef]
- Tian, T.; Wang, Z.; Zhang, J. Pathomechanisms of Oxidative Stress in Inflammatory Bowel Disease and Potential Antioxidant Therapies. Oxidative Med. Cell. Longev. 2017, 2017, 4535194. [Google Scholar] [CrossRef]
- Ponnampalam, E.N.; Kiani, A.; Santhiravel, S.; Holman, B.W.B.; Lauridsen, C.; Dunshea, F.R. The Importance of Dietary Antioxidants on Oxidative Stress, Meat and Milk Production, and Their Preservative Aspects in Farm Animals: Antioxidant Action, Animal Health, and Product Quality-Invited Review. Animals 2022, 12, 3279. [Google Scholar] [CrossRef] [PubMed]
- Udenigwe, C.C.; Aluko, R.E. Food protein-derived bioactive peptides: Production, processing, and potential health benefits. J. Food Sci. 2012, 1, R11–R24. [Google Scholar] [CrossRef] [PubMed]
- Pepe, G.; Basilicata, M.G.; Carrizzo, A.; Adesso, S.; Ostacolo, C.; Sala, M.; Sommella, E.; Ruocco, M.; Cascioferro, S.; Ambrosio, M.; et al. β-Lactoglobulin Heptapeptide Reduces Oxidative Stress in Intestinal Epithelial Cells and Angiotensin II-Induced Vasoconstriction on Mouse Mesenteric Arteries by Induction of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Translocation. Oxidative Med. Cell. Longev. 2019, 2019, 1616239. [Google Scholar] [CrossRef] [PubMed]
- Basilicata, M.G.; Pepe, G.; Rapa, S.F.; Merciai, F.; Ostacolo, C.; Manfra, M.; Di Sarno, V.; Autore, G.; De Vita, D.; Marzocco, S.; et al. Anti-Inflammatory and Antioxidant Properties of Dehydrated Potato-Derived Bioactive Compounds in Intestinal Cells. Int. J. Mol. Sci. 2019, 20, 6087. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Samson, S.E.; Grover, A.K. Antioxidant Supplements and Gastrointestinal Diseases: A Critical Appraisal. Med. Princ. Pract. 2017, 26, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Herrero de la Parte, B.; Rodeño-Casado, M.; Iturrizaga Correcher, S.; Mar Medina, C.; García-Alonso, I. Curcumin Reduces Colorectal Cancer Cell Proliferation and Migration and Slows In Vivo Growth of Liver Metastases in Rats. Biomedicines 2021, 9, 1183. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Singh, A.; Kumar, A.; Kumar, R.; Pal, R.; Sachan, A.K.; Dixit, R.K.; Nath, R. Effect of Curcumin and Coenzyme Q10 Alone and in Combination on Learning and Memory in an Animal Model of Alzheimer’s Disease. Biomedicines 2023, 11, 1422. [Google Scholar] [CrossRef]
- Vestuto, V.; Amodio, G.; Pepe, G.; Basilicata, M.G.; Belvedere, R.; Napolitano, E.; Guarnieri, D.; Pagliara, V.; Paladino, S.; Rodriquez, M.; et al. Cocoa Extract Provides Protection against 6-OHDA Toxicity in SH-SY5Y Dopaminergic Neurons by Targeting PERK. Biomedicines 2022, 10, 2009. [Google Scholar] [CrossRef]
- Marino, P.; Pepe, G.; Basilicata, M.G.; Vestuto, V.; Marzocco, S.; Autore, G.; Procino, A.; Gomez-Monterrey, I.M.; Manfra, M.; Campiglia, P. Potential Role of Natural Antioxidant Products in Oncological Diseases. Antioxidants 2023, 12, 704. [Google Scholar] [CrossRef]
- Quagliariello, V.; Basilicata, M.G.; Pepe, G.; De Anseris, R.; Di Mauro, A.; Scognamiglio, G.; Palma, G.; Vestuto, V.; Buccolo, S.; Luciano, A.; et al. Combination of Spirulina platensis, Ganoderma lucidum and Moringa oleiferaImproves Cardiac Functions and Reduces Pro-Inflammatory Biomarkers in Preclinical Models of Short-Term Doxorubicin-Mediated Cardiotoxicity: New Frontiers in Cardioncology? J. Cardiovasc. Dev. Dis. 2022, 9, 423. [Google Scholar] [CrossRef]
- Dziąbowska-Grabias, K.; Sztanke, M.; Zając, P.; Celejewski, M.; Kurek, K.; Szkutnicki, S.; Korga, P.; Bulikowski, W.; Sztanke, K. Antioxidant Therapy in Inflammatory Bowel Diseases. Antioxidants 2021, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The Impact of Oxidative Stress in Human Pathology: Focus on Gastrointestinal Disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef]
- Yoshida, N. Inflammation and oxidative stress in gastroesophageal reflux disease. J. Clin. Biochem. Nutr. 2007, 40, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Kourie, J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998, 275, C1–C24. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Halliwell, B.; Chirico, S. Lipid peroxidation: Its mechanism, measurement, and significance. Am. J. Clin. Nutr. 1993, 57, 715S–724S. [Google Scholar] [CrossRef]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium stress: An oxidative challenge. Biometals 2010, 23, 927–940. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S. Role of oxidative stress and protein oxidation in the aging process. Free Radic. Biol. Med. 2002, 33, 37–44. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein oxidation and Aging. Free Radic. Res. 2006, 40, 1250–1258. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. Oxidative DNA damage, antioxidants, and cancer. Bioessays 1999, 21, 238–246. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Chiorcea-Paquim, A.M. 8-oxoguanine and 8-oxodeoxyguanosine Biomarkers of Oxidative DNA Damage: A Review on HPLC-ECD Determination. Molecules 2022, 27, 1620. [Google Scholar] [CrossRef]
- Pereira, C.; Grácio, D.; Teixeira, J.P.; Magro, F. Oxidative Stress and DNA Damage: Implications in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 2403–2417. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Swindle, E.J.; Metcalfe, D.D. The role of reactive oxygen species and nitric oxide in mast cell-dependent inflammatory processes. Immunol. Rev. 2007, 217, 186–205. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Kulkarni, A.C.; Kuppusamy, P.; Parinandi, N. Oxygen, the lead actor in the pathophysio- logic drama: Enactment of the trinity of normoxia, hypoxia, and hyperoxia in disease and therapy. Antioxid. Redox Signal. 2007, 9, 1717–1730. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Geiszt, M.; Leto, T.L. The Nox family of NAD(P)H oxidases: Host defense and beyond. J. Biol. Chem. 2004, 279, 51715–51718. [Google Scholar] [CrossRef]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef]
- O’Neill, S.; Brault, J.; Stasia, M.J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015, 6, 135–156. [Google Scholar] [CrossRef]
- Cerqua, I.; Musella, S.; Peltner, L.K.; D’Avino, D.; Di Sarno, V.; Granato, E.; Vestuto, V.; Di Matteo, R.; Pace, S.; Ciaglia, T.; et al. Discovery and Optimization of Indoline-Based Compounds as Dual 5-LOX/sEH Inhibitors: In Vitro and In Vivo Anti-Inflammatory Characterization. J. Med. Chem. 2022, 65, 14456–14480. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yang, L.; Zhou, B.; Yu, R.; Tang, N.; Wang, B. Myeloperoxidase G-463A polymorphism and the risk of gastric cancer: A case-control study. Carcinogenesis 2006, 27, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar] [CrossRef]
- Peek, R.M., Jr.; Fiske, C.; Wilson, K.T. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 2010, 90, 831–858. [Google Scholar] [CrossRef]
- Kaneko, K.; Akuta, T.; Sawa, T.; Kim, H.W.; Fujii, S.; Okamoto, T.; Nakayama, H.; Ohigashi, H.; Murakami, A.; Akaike, T. Mutagenicity of 8-nitroguanosine, a product of nitrative nucleoside modification by reactive nitrogen oxides, in mammalian cells. Cancer Lett. 2008, 262, 239–247. [Google Scholar] [CrossRef]
- Muñoz, M.; Sánchez, A.; Pilar Martínez, M.; Benedito, S.; López-Oliva, M.E.; García-Sacristán, A.; Hernández, M.; Prieto, D. COX-2 is involved in vascular oxidative stress and endothelial dysfunction of renal interlobar arteries from obese Zucker rats. Free Radic. Biol. Med. 2015, 84, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. The COXIB experience: A look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 265–290. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef]
- Kruidenier, L.; Kuiper, I.; Van Duijn, W.; Marklund, S.L.; Van Hogezand, R.A.; Lamers, C.B.; Verspaget, H.W. Differential mucosal expression of three superoxide dismutase isoforms in inflammatory bowel disease. J. Pathol. 2003, 201, 7–16. [Google Scholar] [CrossRef]
- Janssen, A.M.; Bosman, C.B.; Van Duijn, W.; Oostendorp-van de Ruit, M.M.; Kubben, F.J.; Griffioen, G.; Lamers, C.B.; van Krieken, J.H.; van de Velde, C.J.; Verspaget, H.W. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin. Cancer Res. 2000, 6, 3183–3192. [Google Scholar]
- Komatsu, H.; Okayasu, I.; Mitomi, H.; Imai, H.; Nakagawa, Y.; Obata, F. Immunohistochemical detection of human gastrointestinal glutathione peroxidase in normal tissues and cultured cells with novel mouse monoclonal antibodies. J. Histochem. Cytochem. 2001, 49, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Lan, S. Implications of Antioxidant Systems in Inflammatory Bowel Disease. BioMed Res. Int. 2018, 2018, 1290179. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar] [PubMed]
- Pérez, S.; Taléns-Visconti, R.; Rius-Pérez, S.; Finamor, I.; Sastre, J. Redox signaling in the gastrointestinal tract. Free Radic. Biol. Med. 2017, 104, 75–103. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Hong, J.; Li, D.; Cao, W. Rho Kinase ROCK2 Mediates Acid-Induced NADPH Oxidase NOX5-S Expression in Human Esophageal Adenocarcinoma Cells. PLoS ONE 2016, 11, e0149735. [Google Scholar] [CrossRef]
- Li, D.; Cao, W. Role of intracellular calcium and NADPH oxidase NOX5-S in acid-induced DNA damage in Barrett’s cells and Barrett’s esophageal adenocarcinoma cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G863–G872. [Google Scholar] [CrossRef]
- Zhang, Q.B.; Nakashabendi, I.M.; Mokhashi, M.S.; Dawodu, J.B.; Gemmell, C.G.; Russell, R.I. Association of cytotoxin production and neutrophil activation by strains of Helicobacter pylori isolated from patients with peptic ulceration and chronic gastritis. Gut 1996, 38, 841–845. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Barry, D.P.; Frye, J.W.; Casero, R.A., Jr.; Wilson, K.T. Spermine oxidase is a regulator of macrophage host response to Helicobacter pylori: Enhancement of antimicrobial nitric oxide generation by depletion of spermine. Amino Acids 2014, 46, 531–542. [Google Scholar] [CrossRef]
- Colgan, S.P.; Taylor, C.T. Hypoxia: An alarm signal during intestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 281–287. [Google Scholar] [CrossRef]
- Iborra, M.; Moret, I.; Rausell, F.; Bastida, G.; Aguas, M.; Cerrillo, E.; Nos, P.; Beltran, B. Role of oxidative stress and antioxidant enzymes in Crohn’s disease. Biochem. Soc. Trans. 2011, 39, 1102–1106. [Google Scholar] [CrossRef]
- Singer, I.I.; Kawka, D.W.; Scott, S.; Weidner, J.R.; Mumford, R.A.; Riehl, T.E.; Stenson, W.F. Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology 1996, 111, 871–885. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Hypochlorous Acid Chemistry in Mammalian Cells—Influence on Infection and Role in Various Pathologies. Int. J. Mol. Sci. 2022, 23, 10735. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, B.A.; Gokhale, R.; Cho, J.H. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin. Microbiol. Rev. 2002, 15, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, L.F.; Santos, A.R.O.D.; Carvalho, A.C.A.; Bechara, M.D.; Guiguer, E.L.; Goulart, R.A.; Vargas Sinatora, R.; Araújo, A.C.; Barbalho, S.M. Phytochemicals and Regulation of NF-kB in Inflammatory Bowel Diseases: An Overview of In Vitro and In Vivo Effects. Metabolites 2023, 13, 96. [Google Scholar] [CrossRef]
- Coskun, M.; Olsen, J.; Seidelin, J.B.; Nielsen, O.H. MAP kinases in inflammatory bowel disease. Clin. Chim. Acta 2011, 412, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Zhai, Y.; Liang, S.; Mori, Y.; Han, R.; Sutterwala, F.S.; Qiao, L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 2013, 4, 1611. [Google Scholar] [CrossRef]
- Hirota, S.A.; Ng, J.; Lueng, A.; Khajah, M.; Parhar, K.; Li, Y.; Lam, V.; Potentier, M.S.; Ng, K.; Bawa, M.; et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 2011, 17, 1359–1372. [Google Scholar] [CrossRef]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Eugene, S.P.; Reddy, V.S.; Trinath, J. Endoplasmic Reticulum Stress and Intestinal Inflammation: A Perilous Union. Front. Immunol. 2020, 11, 543022. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Qayyum, N.; Haseeb, M.; Kim, M.S.; Choi, S. Role of Thioredoxin-Interacting Protein in Diseases and Its Therapeutic Outlook. Int. J. Mol. Sci. 2021, 22, 2754. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Leng, W.; Zhang, J.; Zhang, G.; Liu, D.; Zhao, Z.; Chen, F.; Shi, Y.; Hao, Y.; Lv, J.; et al. miR-146a-5p/TXNIP axis attenuates intestinal ischemia-reperfusion injury by inhibiting autophagy via the PRKAA/mTOR signaling pathway. Biochem. Pharmacol. 2022, 197, 114839. [Google Scholar] [CrossRef]
- Kiselyov, K.; Muallem, S. ROS and intracellular ion channels. Cell Calcium. 2016, 60, 108–114. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Ramírez, A.; Vázquez-Sánchez, A.Y.; Carrión-Robalino, N.; Camacho, J. Ion Channels and Oxidative Stress as a Potential Link for the Diagnosis or Treatment of Liver Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 3928714. [Google Scholar] [CrossRef]
- Annunziato, L.; Pannaccione, A.; Cataldi, M.; Secondo, A.; Castaldo, P.; Di Renzo, G.; Taglialatela, M. Modulation of ion channels by reactive oxygen and nitrogen species: A pathophysiological role in brain aging? Neurobiol. Aging 2002, 23, 819–834. [Google Scholar] [CrossRef]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, S.M. Oxidative stress, autophagy and airway ion transport. Am. J. Physiol. Cell Physiol. 2019, 316, C16–C32. [Google Scholar] [CrossRef]
- Beyder, A.; Farrugia, G. Ion channelopathies in functional GI disorders. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G581–G586. [Google Scholar] [CrossRef]
- Akbarali, H.I.; Hawkins, E.G.; Ross, G.R.; Kang, M. Ion channel remodeling in gastrointestinal inflammation. Neurogastroenterol. Motil. 2010, 22, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Zhao, Y.; Ma, Z.; Zhang, M.; Wang, H.; Yi, Z.; Tuo, B.; Li, T.; Liu, X. Pathophysiological role of ion channels and transporters in gastrointestinal mucosal diseases. Cell. Mol. Life Sci. 2021, 78, 8109–8125. [Google Scholar] [CrossRef]
- Hill-Eubanks, D.C.; Werner, M.E.; Heppner, T.J.; Nelson, M.T. Calcium signaling in smooth muscle. Cold Spring Harb. Perspect. Biol. 2011, 3, a004549. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Drumm, B.T.; Hwang, S.J.; Baker, S.A.; Ward, S.M.; Sanders, K.M. Ca2+ signalling behaviours of intramuscular interstitial cells of Cajal in the murine colon. J. Physiol. 2019, 597, 3587–3617. [Google Scholar] [CrossRef]
- Chen, H.; Ordög, T.; Chen, J.; Young, D.L.; Bardsley, M.R.; Redelman, D.; Ward, S.M.; Sanders, K.M. Differential gene expression in functional classes of interstitial cells of Cajal in murine small intestine. Physiol. Genom. 2007, 31, 492–509. [Google Scholar] [CrossRef]
- Terlau, H.; Stühmer, W. Structure and function of voltage-gated ion channels. Naturwissenschaft 1998, 85, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Beyder, A.; Farrugia, G. Targeting ion channels for the treatment of gastrointestinal motility disorders. Therap. Adv. Gastroenterol. 2012, 5, 5–21. [Google Scholar] [CrossRef]
- Todorovic, S.M.; Jevtovic-Todorovic, V. Redox regulation of neuronal voltage-gated calcium channels. Antioxid. Redox Signal. 2014, 21, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef]
- Shi, X.Z.; Sarna, S.K. Impairment of Ca2+ mobilization in circular muscle cells of the inflamed colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G234–G242. [Google Scholar] [CrossRef]
- Liu, X.; Rusch, N.J.; Striessnig, J.; Sarna, S.K. Down-regulation of L-type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology 2001, 120, 480–489. [Google Scholar] [CrossRef]
- Ross, G.R.; Kang, M.; Shirwany, N.; Malykhina, A.P.; Drozd, M.; Akbarali, H.I. Nitrotyrosylation of Ca2+ channels prevents c-Src kinase regulation of colonic smooth muscle contractility in experimental colitis. J. Pharmacol. Exp. Ther. 2007, 322, 948–956. [Google Scholar] [CrossRef]
- Ross, G.R.; Kang, M.; Akbarali, H.I. Colonic inflammation alters Src kinase-dependent gating properties of single Ca2+ channels via tyrosine nitration. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G976–G984. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.G.; Barrett, C.F.; Groth, R.D.; Safa, P.; Tsien, R.W. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol. 2008, 183, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Ross, G.R.; Akbarali, H.I. The effect of tyrosine nitration of L-type Ca2+ channels on excitation-transcription coupling in colonic inflammation. Br. J. Pharmacol. 2010, 159, 1226–1235. [Google Scholar] [CrossRef]
- Akbarali, H.I. Oxidative Stress and Ion Channels. In Systems Biology of Free Radicals and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar] [CrossRef]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacology 2014, 66, 676–814. [Google Scholar] [CrossRef]
- Ciaglia, T.; Vestuto, V.; Bertamino, A.; González-Muñiz, R.; Gómez-Monterrey, I. On the modulation of TRPM channels: Current perspectives and anticancer therapeutic implications. Front. Oncol. 2023, 12, 1065935. [Google Scholar] [CrossRef]
- Daisuke, K.; Nozomi, O.; Yasuo, M. Redox Regulation of Transient Receptor Potential Channels. Antioxid. Redox Signal. 2014, 21, 971–986. [Google Scholar] [CrossRef]
- Simon, F.; Varela, D.; Cabello-Verrugio, C. Oxidative stress-modulated TRPM ion channels in cell dysfunction and pathological conditions in humans. Cell. Signal. 2013, 25, 1614–1624. [Google Scholar] [CrossRef]
- Holzer, P. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacology 2011, 131, 142–170. [Google Scholar] [CrossRef]
- Chen, Y.; Mu, J.; Zhu, M.; Mukherjee, A.; Zhang, H. Transient Receptor Potential Channels and Inflammatory Bowel Disease. Front. Immunol. 2020, 11, 180. [Google Scholar] [CrossRef]
- Du, Y.; Chen, J.; Shen, L.; Wang, B. TRP channels in inflammatory bowel disease: Potential therapeutic targets. Biochem. Pharmacol. 2022, 203, 115195. [Google Scholar] [CrossRef]
- Wehrhahn, J.; Kraft, R.; Harteneck, C.; Hauschildt, S. Transient Receptor Potential Melastatin 2 Is Required for Lipopolysaccharide-Induced Cytokine Production in Human Monocytes. J. Immunol. 2010, 184, 2386–2393. [Google Scholar] [CrossRef]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef]
- Perraud, A.L.; Schmitz, C.; Scharenberg, A.M. TRPM2 Ca2+ permeable cation channels: From gene to biological function. Cell Calcium. 2003, 33, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.; Li, Y.; Perraud, A.L. The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol. Res. 2013, 55, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, Ü.S.; Nazıroğlu, M.; Şenol, N.; Ghazizadeh, V. Hypericum perforatum Attenuates Spinal Cord Injury-Induced Oxidative Stress and Apoptosis in the Dorsal Root Ganglion of Rats: Involvement of TRPM2 and TRPV1 Channels. Mol. Neurobiol. 2016, 53, 3540–3551. [Google Scholar] [CrossRef]
- Buelow, B.; Song, Y.; Scharenberg, A.M. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 2008, 283, 24571–24583. [Google Scholar] [CrossRef] [PubMed]
- Vestuto, V.; Di Sarno, V.; Musella, S.; Di Dona, G.; Moltedo, O.; Gomez-Monterrey, I.M.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Ciaglia, T. New Frontiers on ER Stress Modulation: Are TRP Channels the Leading Actors? Int. J. Mol. Sci. 2023, 24, 185. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Takahashi, N.; Mori, Y. Chemical physiology of oxidative stress-activated TRPM2 and TRPC5 channels. Prog. Biophys. Mol. Biol. 2010, 103, 18–27. [Google Scholar] [CrossRef]
- Oda, S.; Uchida, K.; Wang, X.; Lee, J.; Shimada, Y.; Tominaga, M.; Kadowaki, M. TRPM2 contributes to antigen-stimulated Ca2+ influx in mucosal mast cells. Mol. Cell. Mech. Dis. 2013, 465, 1023–1030. [Google Scholar] [CrossRef]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Takagi, K.; Kato, A.; Ishibashi, T.; Mori, Y.; Tashima, K.; Mitsumoto, A.; Kato, S.; Horie, S. Role of transient receptor potential melastatin 2 (TRPM2) channels in visceral nociception and hypersensitivity. Exp. Neurol. 2016, 285, 41–50. [Google Scholar] [CrossRef]
- Olah, M.E.; Jackson, M.F.; Li, H.; Perez, Y.; Sun, H.S.; Kiyonaka, S.; Mori, Y.; Tymianski, M.; MacDonald, J.F. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J. Physiol. 2009, 587, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Kawanaka, H.; Hori, M.; Kusamori, K.; Utsumi, D.; Tsukahara, T.; Amagase, K.; Horie, S.; Yamamoto, A.; Ozaki, H.; et al. Role of transient receptor potential melastatin 2 in surgical inflammation and dysmotility in a mouse model of postoperative ileus. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G104–G116. [Google Scholar] [CrossRef]
- Uchida, K.; Tominaga, M. The role of TRPM2 in pancreatic β-cells and the development of diabetes. Cell Calcium 2014, 56, 332–339. [Google Scholar] [CrossRef]
- Holzer, P. TRP channels in the digestive system. Curr. Pharm. Biotechnol. 2011, 12, 24–34. [Google Scholar] [CrossRef]
- Frias, B.; Merighi, A. Capsaicin, Nociception and Pain. Molecules 2016, 21, 797. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Liao, Q.; Chen, C.; Yang, X.; Xie, R.; Xu, J. The Role of Transient Receptor Potential Vanilloid 1 in Common Diseases of the Digestive Tract and the Cardiovascular and Respiratory System. Front. Physiol. 2019, 10, 1064. [Google Scholar] [CrossRef]
- Akbar, A.; Yiangou, Y.; Facer, P.; Walters, J.R.; Anand, P.; Ghosh, S. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut 2008, 57, 923–929. [Google Scholar] [CrossRef]
- Chuang, H.H.; Lin, S. Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 20097–20102. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Eder, C. Importance of the non-selective cation channel TRPV1 for microglial reactive oxygen species generation. J. Neuroimmunol. 2009, 216, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, E.; Naito, Y.; Handa, O.; Okada, H.; Mizushima, K.; Hirai, Y.; Nakabe, N.; Uchiyama, K.; Ishikawa, T.; Takagi, T.; et al. Oxidative stress-induced posttranslational modification of TRPV1 expressed in esophageal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G230–G238. [Google Scholar] [CrossRef]
- Bhat, Y.M.; Bielefeldt, K. Capsaicin receptor (TRPV1) and non-erosive reflux disease. Eur. J. Gastroenterol. Hepatol. 2006, 18, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Matthews, P.J.; Aziz, Q.; Facer, P.; Davis, J.B.; Thompson, D.G.; Anand, P. Increased capsaicin receptor TRPV1 nerve fibres in the inflamed human oesophagus. Eur. J. Gastroenterol. Hepatol. 2004, 16, 897–902. [Google Scholar] [CrossRef]
- Clarrett, D.M.; Hachem, C. Gastroesophageal Reflux Disease (GERD). MO Med. 2018, 115, 214–218. [Google Scholar]
- Cheng, L.; de la Monte, S.; Ma, J.; Hong, J.; Tong, M.; Cao, W.; Behar, J.; Biancani, P.; Harnett, K.M. HCl− Activated Neural and Epithelial Vanilloid Receptors (TRPV1) in Cat Esophageal Mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G135–G143. [Google Scholar] [CrossRef]
- Banerjee, B.; Medda, B.K.; Lazarova, Z.; Bansal, N.; Shaker, R.; Sengupta, J.N. Effect of reflux-induced inflammation on transient receptor potential vanilloid one (TRPV1) expression in primary sensory neurons innervating the oesophagus of rats. Neurogastroenterol. Motil. 2007, 19, 681–691. [Google Scholar] [CrossRef]
- Bautista, D.M. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; Kawabata-Shoda, E.; Doihara, H.; Kojima, R.; Okada, H.; Mochizuki, S.; Sano, Y.; Inamura, K.; Matsushime, H.; Koizumi, T.; et al. TRPA1 regulates gastrointestinal motility through serotonin release from enterochromaffin cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3408–3413. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Duggan, A.; Kumar, G.; Garcia-Anoveros, J. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J. Neurosci. 2005, 25, 4052–4061. [Google Scholar] [CrossRef]
- Hinman, A.; Chuang, H.H.; Bautista, D.M.; Julius, D. TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA 2006, 103, 19564–19568. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, L.J.; Dubin, A.E.; Evans, M.J.; Marr, F.; Schultz, P.G.; Cravatt, B.F.; Patapoutian, A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 2007, 445, 541–545. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 20, 363–383. [Google Scholar] [CrossRef]
- Liu, M.; Gao, L.; Zhang, N. Berberine reduces neuroglia activation and inflammation in streptozotocin-induced diabetic mice. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419866379. [Google Scholar] [CrossRef]
- Zou, B.; Cao, C.; Fu, Y.; Pan, D.; Wang, W.; Kong, L. Berberine Alleviates Gastroesophageal Reflux-Induced Airway Hyperresponsiveness in a Transient Receptor Potential A1-Dependent Manner. Evid. Based Complement. Altern. Med. 2022, 2022, 7464147. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer cells co-opt the neuronal redox-sensing channel TRPA1 to promote oxidative-stress tolerance. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Herrmann, A.K.; Wüllner, V.; Moos, S.; Graf, J.; Chen, J.; Kieseier, B.; Kurschus, F.C.; Albrecht, P.; Vangheluwe, P.; Methner, A. Dimethyl fumarate alters intracellular Ca2+ handling in immune cells by redox-mediated pleiotropic effects. Free Radic. Biol. Med. 2019, 141, 338–347. [Google Scholar] [CrossRef]
- Sawada, Y.; Hosokawa, H.; Matsumura, K.; Kobayashi, S. Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur. J. Neurosci. 2008, 27, 1131–1142. [Google Scholar] [CrossRef]
- Liu, X.L.; Zhen, C.; Chen, Y.X.; Fan, X.R.; Wu, Z.Y.; Ma, F.C.; Rong, J.; Di, G.F.; Jiang, X.C. Inhibition of TRPA1 Attenuates Oxidative Stress-induced Damage After Traumatic Brain Injury via the ERK/AKT Signaling Pathway. Neuroscience 2022, 494, 51–68. [Google Scholar] [CrossRef]
- Eid, S.R.; Crown, E.D.; Moore, E.L.; Liang, H.A.; Choong, K.C.; Dima, S.; Henze, D.A.; Kane, S.A.; Urban, M.O. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol. Pain 2008, 4, 48. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca2+ as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef]
- Csekő, K.; Beckers, B.; Keszthelyi, D.; Helyes, Z. Role of TRPV1 and TRPA1 Ion Channels in Inflammatory Bowel Diseases: Potential Therapeutic Targets? Pharmaceuticals 2019, 12, 48. [Google Scholar] [CrossRef]
- Gibson-Corley, K.N.; Meyerholz, D.K.; Engelhardt, J.F. Pancreatic pathophysiology in cystic fibrosis. J. Pathol. 2016, 238, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Olivier, A.K.; Gibson-Corley, K.N.; Meyerholz, D.K. Animal models of gastrointestinal and liver diseases. Animal models of cystic fibrosis: Gastrointestinal, pancreatic, and hepatobiliary disease and pathophysiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G459–G471. [Google Scholar] [CrossRef]
- Trouvé, P.; Férec, C.; Génin, E. The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells 2021, 10, 2980. [Google Scholar] [CrossRef] [PubMed]
- Kerbiriou, M.; Le Drévo, M.A.; Férec, C.; Trouvé, P. Coupling cystic fibrosis to endoplasmic reticulum stress: Differential role of Grp78 and ATF6. Biochim. Biophys. Acta 2007, 1772, 1236–1249. [Google Scholar] [CrossRef]
- Bresso, F.; Askling, J.; Astegiano, M.; Demarchi, B.; Sapone, N.; Rizzetto, M.; Gionchetti, P.; Lammers, K.M.; de Leone, A.; Riegler, G.; et al. Potential role for the common cystic fibrosis F508 mutation in Crohn’s disease. Inflamm. Bowel Dis. 2007, 13, 531–536. [Google Scholar] [CrossRef]
- Collobert, M.; Bocher, O.; Le Nabec, A.; Génin, E.; Férec, C.; Moisan, S. CFTR Cooperative Cis-Regulatory Elements in Intestinal Cells. Int. J. Mol. Sci. 2021, 22, 2599. [Google Scholar] [CrossRef] [PubMed]
- Chappe, V.; Hinkson, D.A.; Zhu, T.; Chang, X.B.; Riordan, J.R.; Hanrahan, J.W. Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J. Physiol. 2003, 548, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Mathews, C.J.; Hanrahan, J.W. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J. Biol. Chem. 1997, 272, 4978–4984. [Google Scholar] [CrossRef] [PubMed]
- Moliteo, E.; Sciacca, M.; Palmeri, A.; Papale, M.; Manti, S.; Parisi, G.F.; Leonardi, S. Cystic Fibrosis and Oxidative Stress: The Role of CFTR. Molecules 2022, 27, 5324. [Google Scholar] [CrossRef]
- Favia, M.; de Bari, L.; Bobba, A.; Atlante, A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. J. Clin. Med. 2019, 8, 1890. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Ekbom, A.; Askling, J.; Johannesson, M.; Montgomery, S.M. Cystic fibrosis gene mutations and gastrointestinal diseases. J. Cyst. Fibros. 2010, 9, 288–291. [Google Scholar] [CrossRef]
- Zhang, Z.; Leir, S.-H.; Harris, A. Oxidative stress regulates CFTR gene expression in human airway epithelial cells through a distal antioxidant response element. Am. J. Respir. Cell Mol. Biol. 2015, 52, 387–396. [Google Scholar] [CrossRef]
- Borcherding, D.C.; Siefert, M.E.; Lin, S.; Brewington, J.; Sadek, H.; Clancy, J.P.; Plafker, S.M.; Ziady, A.G. Clinically approved CFTR modulators rescue Nrf2 dysfunction in cystic fibrosis airway epithelia. J. Clin. 2019, 129, 3448–3463. [Google Scholar] [CrossRef]
- Duranton, C.; Rubera, I.; Cougnon, M.; Melis, N.; Chargui, A.; Mograbi, B.; Tauc, M. CFTR is involved in the fine tuning of intracellular redox status: Physiological implications in cystic fibrosis. Am. J. Pathol. 2012, 181, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Lubelska, K.; Wiktorska, K.; Mielczarek, L.; Milczarek, M.; Zbroińska-Bregisz, I.; Chilmonczyk, Z. Sulforaphane regulates NFE2L2/Nrf2-dependent xenobiotic metabolism phase II and phase III enzymes differently in human colorectal cancer and untransformed epithelial colon cells. Nutr. Cancer 2016, 68, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Blankenheim, Z.; Scott, P.M.; Cormier, R.T. CFTR and Gastrointestinal Cancers: An Update. J. Pers. Med. 2022, 12, 868. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef]
- Zheng, W.; Kuhlicke, J.; Jäckel, K.; Eltzschig, H.K.; Singh, A.; Sjoblöm, M.; Riederer, B.; Weinhold, C.; Seidler, U.; Colgan, S.P. Hypoxia inducible factor-1 (HIF-l)-mediated repression of cystic fibrosis transmembrane conductance regulator (CFTR) in the intestinal epithelium. FASEB J. 2009, 23, 204–213. [Google Scholar] [CrossRef]
- Swahn, H.; Harris, A. Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics. Genes 2019, 10, 235. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Y.; You, B.; Peng, Y.; Chen, Y.; Yang, Z.; Zhang, Y.; Chen, J. Molecular mechanism mediating enteric bacterial translocation after severe burn: The role of cystic fibrosis transmembrane conductance regulator. Burn 2021, 9, tkaa042. [Google Scholar] [CrossRef]
- Hartzell, C.; Putzier, I.; Arreola, J. Calcium-activated chloride channels. Annu. Rev. Physiol. 2005, 167, 719–758. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Kongsuphol, P.; Chootip, K.; Toledo, C.; Martins, J.R.; Almaca, J.; Tian, Y.; Witzgall, R.; Ousingsawat, J.; Schreiber, R. Role of the Ca2+ -activated Cl- channels bestrophin and anoctamin in epithelial cells. Biol. Chem. 2011, 392, 125–134. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Kongsuphol, P.; Aldehni, F.; Tian, Y.; Ousingsawat, J.; Warth, R.; Schreiber, R. Bestrophin and TMEM16-Ca(2+) activated Cl− channels with different functions. Cell Calcium. 2009, 46, 233–241. [Google Scholar] [CrossRef]
- Barrett, K.E.; Keely, S.J. Chloride secretion by the intestinal epithelium: Molecular basis and regulatory aspects. Annu. Rev. Physiol. 2000, 62, 535–572. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.M.; Zhu, M.H.; Britton, F.; Koh, S.D.; Ward, S.M. Anoctamins and gastrointestinal smooth muscle excitability. Exp. Physiol. 2012, 97, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Jeulin, C.; Guadagnini, R.; Marano, F. Oxidant stress stimulates Ca2+-activated chloride channels in the apical activated membrane of cultured nonciliated human nasal epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L636–L646. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.R.; Thoreson, W.B.; Beck, C.L. Molecular and functional analyses of two new calcium-activated chloride channel family members from mouse eye and intestine. J. Biol. Chem. 2004, 279, 41792–41800. [Google Scholar] [CrossRef]
- Ulmasov, B.; Bruno, J.; Woost, P.G.; Edwards, J.C. Tissue and subcellular distribution of CLIC1. BMC Cell Biol. 2007, 8, 8. [Google Scholar] [CrossRef]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef]
- Uretmen Kagiali, Z.C.; Saner, N.; Akdag, M.; Sanal, E.; Degirmenci, B.S.; Mollaoglu, G.; Ozlu, N. CLIC4 and CLIC1 bridge plasma membrane and cortical actin network for a successful cytokinesis. Life Sci. Alliance 2020, 3, e201900558. [Google Scholar] [CrossRef]
- Argenzio, E.; Moolenaar, W.H. Emerging biological roles of Cl- intracellular channel proteins. J. Cell Sci. 2016, 129, 4165–4174. [Google Scholar] [CrossRef]
- Tulk, B.M.; Kapadia, S.; Edwards, J.C. CLIC1 inserts from the aqueous phase into phospholipid membranes, where it functions as an anion channel. Am. J. Physiol. Cell Physiol. 2002, 282, C1103–C1112. [Google Scholar] [CrossRef]
- Novarino, G.; Fabrizi, C.; Tonini, R.; Denti, M.A.; Malchiodi-Albedi, F.; Lauro, G.M.; Sacchetti, B.; Paradisi, S.; Ferroni, A.; Curmi, P.M.; et al. Involvement of the intracellular ion channel CLIC1 in microglia-mediated beta-amyloid-induced neurotoxicity. J. Neurosci. 2004, 24, 5322–5330. [Google Scholar] [CrossRef]
- Chen, C.D.; Wang, C.S.; Huang, Y.H.; Chien, K.Y.; Liang, Y.; Chen, W.J.; Lin, K.H. Overexpression of CLIC1 in human gastric carcinoma and its clinicopathological significance. Proteomics 2007, 7, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Petrova, D.T.; Asif, A.R.; Armstrong, V.W.; Dimova, I.; Toshev, S.; Yaramov, N.; Oellerich, M.; Toncheva, D. Expression of chloride intracellular channel protein 1 (CLIC1) and tumor protein D52 (TPD52) as potential biomarkers for colorectal cancer. Clin. Biochem. 2008, 41, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Le, Y.; Ding, J.; Dou, X.; Mao, W.; Zhu, J. CLIC1 Inhibition Protects Against Cellular Senescence and Endothelial Dysfunction Via the Nrf2/HO-1 Pathway. Cell Biochem. Biophys. 2021, 79, 239–252. [Google Scholar] [CrossRef]
- Meng, M. CLIC1 facilitates cancer associated fibroblast activation and gastric cancer progression via integrins/NF-κB pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G836. [Google Scholar] [CrossRef]
- Li, B.P.; Mao, Y.T.; Wang, Z.; Chen, Y.Y.; Wang, Y.; Zhai, C.Y.; Shi, B.; Liu, S.Y.; Liu, J.L.; Chen, J.Q. CLIC1 Promotes the Progression of Gastric Cancer by Regulating the MAPK/AKT Pathways. Cell. Physiol. Biochem. 2018, 46, 907–924. [Google Scholar] [CrossRef]
- Wang, J.W.; Peng, S.Y.; Li, J.T.; Wang, Y.; Zhang, Z.P.; Cheng, Y.; Cheng, D.Q.; Weng, W.H.; Wu, X.S.; Fei, X.Z.; et al. Identification of metastasis-associated proteins involved in gallbladder carcinoma metastasis by proteomic analysis and functional exploration of chloride intracellular channel 1. Cancer Lett. 2009, 281, 71–81. [Google Scholar] [CrossRef]
- Pardo, L.A.; Stühmer, W. The roles of K+ channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef]
- González, C.; Baez-Nieto, D.; Valencia, I.; Oyarzún, I.; Rojas, P.; Naranjo, D.; Latorre, R. K+ channels: Function-structural overview. Compr. Physiol. 2012, 2, 2087–2149. [Google Scholar] [CrossRef]
- Coetzee, W.A.; Amarillo, Y.; Chiu, J.; Chow, A.; Lau, D.; McCormack, T.; Moreno, H.; Nadal, M.S.; Ozaita, A.; Pountney, D.; et al. Molecular diversity of K+ channels. Ann. N. Y. Acad. Sci. 1999, 868, 233–285. [Google Scholar] [CrossRef]
- Pardo, L.A.; Contreras-Jurado, C.; Zientkowska, M.; Alves, F.; Stühmer, W. Role of voltage-gated potassium channels in cancer. J. Membr. Biol. 2005, 205, 115–124. [Google Scholar] [CrossRef]
- Taglialatela, M.; Castaldo, P.; Iossa, S.; Pannaccione, A.; Fresi, A.; Ficker, E.; Annunziato, L. Regulation of the human ether-a-gogo related gene (HERG) K+ channels by reactive oxygen species. Proc. Natl. Acad. Sci. USA 1997, 94, 11698–11703. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Limberis, J.; Martin, R.L.; Xu, R.; Kolbe, K.; Heinemann, S.H.; Hoshi, T.; Cox, B.F.; Gintant, G.A. Functional consequences of methionine oxidation of hERG potassium channels. Biochem. Pharmacol. 2007, 74, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; He, H.J.; Tanaka, O.; Suzuki, R.; Sekiguchi, M.; Yasuoka, Y.; Kawahara, K.; Itoh, H.; Abe, H. Localization of the sulphonylurea receptor subunits, SUR2A and SUR2B, in rat renal tubular epithelium. Tohoku J. Exp. Med. 2008, 214, 247–256. [Google Scholar] [CrossRef]
- Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Seino, S. Roles of KATP channels as metabolic sensors in acute metabolic changes. J. Mol. Cell. Cardiol. 2005, 38, 917–925. [Google Scholar] [CrossRef]
- Cui, Y.; Fan, Z. Mechanism of Kir6.2 channel inhibition by sulfhydryl modification: Pore block or allosteric gating? J. Physiol. 2002, 540, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Zoga, V.; Kimura, M.; Liang, M.Y.; Wu, H.E.; Gemes, G. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: Action by direct S-nitrosylation. Mol. Pain 2009, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef]
- Tonooka, N.; Oseid, E.; Zhou, H.; Harmon, J.S.; Robertson, R.P. Glutathione peroxidase protein expression and activity in human islets isolated for transplantation. Clin. Transplant. 2007, 21, 767–772. [Google Scholar] [CrossRef]
- Welsh, N.; Margulis, B.; Borg, L.A.; Wiklund, H.J.; Saldeen, J.; Flodström, M.; Mello, M.A.; Andersson, A.; Pipeleers, D.G.; Hellerström, C. Differences in the expression of heat-shock proteins and antioxidant enzymes between human and rodent pancreatic islets: Implications for the pathogenesis of insulin-dependent diabetes mellitus. Mol. Med. 1995, 1, 806–820. [Google Scholar] [CrossRef]
- Gier, B.; Krippeit-Drews, P.; Sheiko, T.; Aguilar-Bryan, L.; Bryan, J.; Düfer, M.; Drews, G. Suppression of KATP channel activity protects murine pancreatic beta cells against oxidative stress. J. Clin. Investig. 2009, 119, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Fridlyand, L.E.; Philipson, L.H. Does the glucose-dependent insulin secretion mechanism itself cause oxidative stress in pancreatic beta-cells? Diabetes 2004, 53, 1942–1948. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef]
- Kang, M.; Hashimoto, A.; Gade, A.; Akbarali, H.I. Interaction between hydrogen sulfide-induced sulfhydration and tyrosine nitration in the KATP channel complex. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G532–G539. [Google Scholar] [CrossRef] [PubMed]
- Gade, A.R.; Kang, M.; Akbarali, H.I. Hydrogen sulfide as an allosteric modulator of ATP-sensitive potassium channels in colonic inflammation. Mol. Pharmacol. 2013, 83, 294–306. [Google Scholar] [CrossRef]
- Jin, X.; Malykhina, A.P.; Lupu, F.; Akbarali, H.I. Altered gene expression and increased bursting activity of colonic smooth muscle ATP-sensitive K+ channels in experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G274–G285. [Google Scholar] [CrossRef] [PubMed]
- Heitzmann, D.; Warth, R. Physiology and pathophysiology of potassium channels in gastrointestinal epithelia. Physiol. Rev. 2008, 88, 1119–1182. [Google Scholar] [CrossRef]
- Ottschytsch, N.; Raes, A.; Van Hoorick, D.; Snyders, D.J. Obligatory heterotetramerization of three previously uncharacterized Kv channel alpha-subunits identified in the human genome. Proc. Natl. Acad. Sci. USA 2002, 99, 7986–7991. [Google Scholar] [CrossRef]
- Peri, R.; Wible, B.A.; Brown, A.M. Mutations in the Kv beta 2 binding site for NADPH and their effects on Kv1. 4. J. Biol. Chem. 2001, 276, 738–741. [Google Scholar] [CrossRef]
- Pan, Y.; Weng, J.; Cao, Y.; Bhosle, R.C.; Zhou, M. Functional coupling between the Kv1.1 channel and aldoketoreductase Kvbeta1. J. Biol. Chem. 2008, 283, 8634–8642. [Google Scholar] [CrossRef] [PubMed]
- Anderson, U.A.; Carson, C.; McCloskey, K.D. KCNQ currents and their contribution to resting membrane potential and the excitability of interstitial cells of Cajal from the guinea pig bladder. J. Urol. 2009, 182, 330–336. [Google Scholar] [CrossRef]
- Musella, S.; Carotenuto, L.; Iraci, N.; Baroli, G.; Ciaglia, T.; Nappi, P.; Basilicata, M.G.; Salviati, E.; Barrese, V.; Vestuto, V.; et al. Beyond Retigabine: Design, Synthesis, and Pharmacological Characterization of a Potent and Chemically Stable Neuronal Kv7 Channel Activator with Anticonvulsant Activity. J. Med. Chem. 2022, 65, 11340–11364. [Google Scholar] [CrossRef]
- Roche, J.P.; Westenbroek, R.; Sorom, A.J.; Hille, B.; Mackie, K.; Shapiro, M.S. Antibodies and a cysteine-modifying reagent show correspondence of M current in neurons to KCNQ2 and KCNQ3 K+ channels. Br. J. Pharmacol. 2002, 137, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.; Hockley, J.R.; Reed, D.E.; Smith, E.S.J.; Bulmer, D.C.; Blackshaw, L.A. Peripheral K(v)7 channels regulate visceral sensory function in mouse and human colon. Mol. Pain 2017, 13, 1744806917709371. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, A.J.; Rottgen, T.S.; Rajendran, V.M. Activation of KCNQ (KV7) K+ channels in enteric neurons inhibits epithelial Cl- secretion in mouse distal colon. Am. J. Physiol. Cell Physiol. 2021, 320, C1074–C1087. [Google Scholar] [CrossRef]
- Gamper, N.; Zaika, O.; Li, Y.; Martin, P.; Hernandez, C.C.; Perez, M.R.; Wang, A.Y.; Jaffe, D.B.; Shapiro, M.S. Oxidative modification of M-type K(+) channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006, 25, 4996–5004. [Google Scholar] [CrossRef]
- Suh, B.C.; Hille, B. PIP2 is a necessary cofactor for ion channel function: How and why? Annu. Rev. Biophys. 2008, 37, 175–195. [Google Scholar] [CrossRef]
- Kim, H.J.; Jeong, M.H.; Kim, K.R.; Jung, C.Y.; Lee, S.Y.; Kim, H.; Koh, J.; Vuong, T.A.; Jung, S.; Yang, H.; et al. Protein arginine methylation facilitates KCNQ channel-PIP2 interaction leading to seizure suppression. Elife 2016, 5, e17159. [Google Scholar] [CrossRef]
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic. Biol. Med. 2004, 37, 1951–1962. [Google Scholar] [CrossRef]
- Tétard, C.; Mittaine, M.; Bui, S.; Beaufils, F.; Maumus, P.; Fayon, M.; Burgel, P.R.; Lamireau, T.; Delhaes, L.; Mas, E.; et al. Reduced Intestinal Inflammation with Lumacaftor/Ivacaftor in Adolescents with Cystic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.Y.; Syed, S.A.; Rossi, L.; Garg, M.; Needham, B.; Avolio, J.; Young, K.; Surette, M.G.; Gonska, T. Impact of CFTR modulation with Ivacaftor on Gut Microbiota and Intestinal Inflammation. Sci. Rep. 2018, 8, 17834. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Miwa, H. Potent Potassium-competitive Acid Blockers: A New Era for the Treatment of Acid-related Diseases. J. Neurogastroenterol. Motil. 2018, 24, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Park, I.S.; Kim, S.; Ma, H.W.; Kim, J.H.; Kim, T.I.; Kim, W.H.; Han, J.; Kim, S.W.; Cheon, J.H. Novel Potassium-Competitive Acid Blocker, Tegoprazan, Protects Against Colitis by Improving Gut Barrier Function. Front. Immunol. 2022, 13, 870817. [Google Scholar] [CrossRef]
- Frolov, R.V.; Singh, S. Celecoxib and ion channels: A story of unexpected discoveries. Eur. J. Pharmacol. 2014, 730, 61–71. [Google Scholar] [CrossRef]
- Han, J.; Lee, S.H.; Giebisch, G.; Wang, T. Potassium Channelopathies and Gastrointestinal Ulceration. Gut Liver 2016, 10, 881–889. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda, M.R.; Vestuto, V.; Moltedo, O.; Manfra, M.; Campiglia, P.; Pepe, G. The Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases. Oxygen 2023, 3, 336-365. https://doi.org/10.3390/oxygen3030022
Miranda MR, Vestuto V, Moltedo O, Manfra M, Campiglia P, Pepe G. The Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases. Oxygen. 2023; 3(3):336-365. https://doi.org/10.3390/oxygen3030022
Chicago/Turabian StyleMiranda, Maria Rosaria, Vincenzo Vestuto, Ornella Moltedo, Michele Manfra, Pietro Campiglia, and Giacomo Pepe. 2023. "The Ion Channels Involved in Oxidative Stress-Related Gastrointestinal Diseases" Oxygen 3, no. 3: 336-365. https://doi.org/10.3390/oxygen3030022