1. Introduction
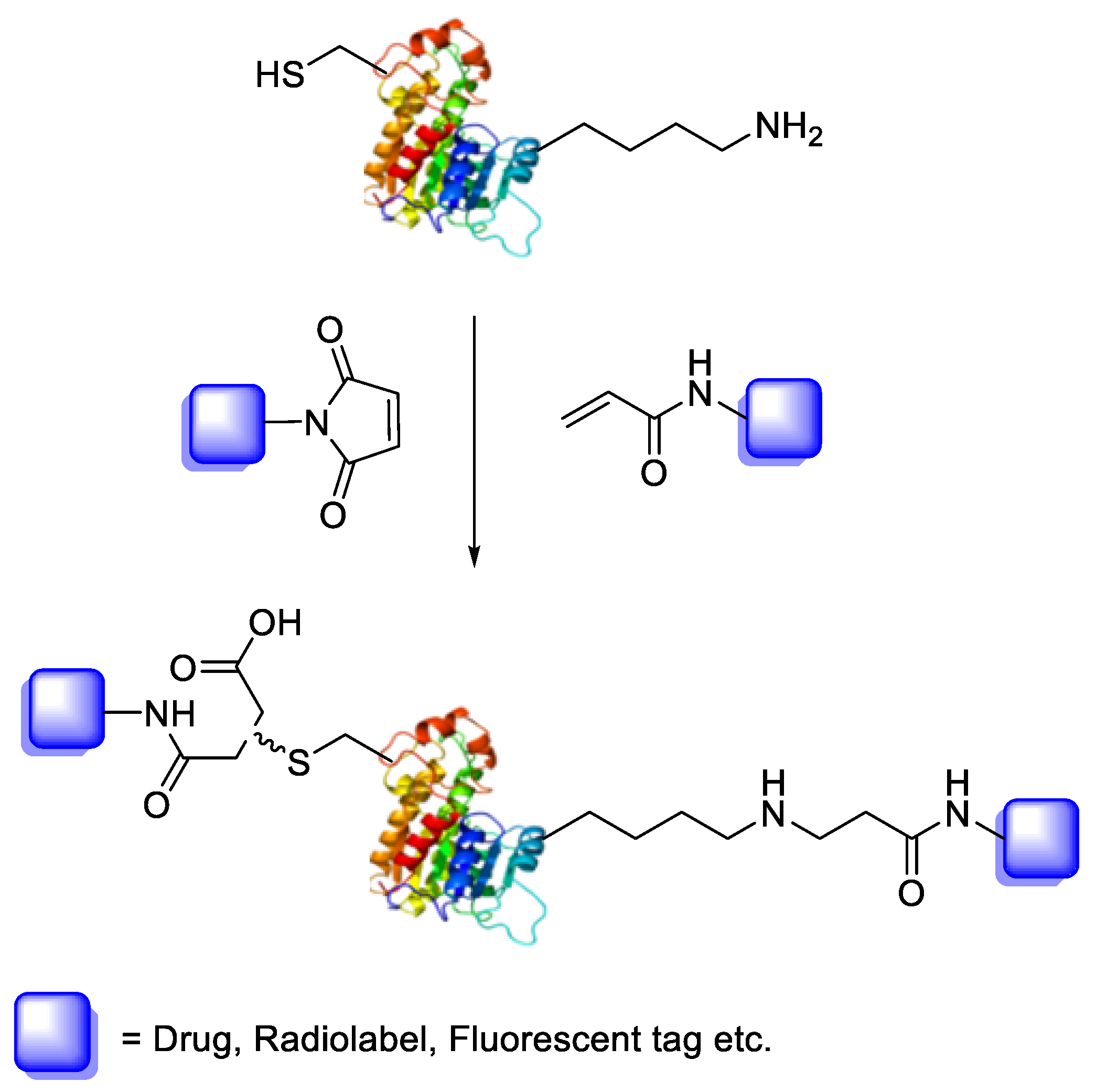
Bioconjugation is the coupling of a biologically active molecule, such as a peptide, protein or antibody, with a fragment bearing a particular function, such as a drug, nanoparticle, fluorescent tag or radiolabel. Conjugation between the two components is achieved via an intermediary covalent linkage established by a reactive and chemo-selective prosthetic group (
Scheme 1); it is, however, appropriate to speak of “bioconjugation” when the reaction conditions are compatible with the use of biologics (e.g., aqueous buffer conditions).
Our group has previously demonstrated that the 3- and 4-nitrophenyl sulphur pentafluoride can undergo
18F/
19F radioisotopic exchange [
1]. In addition to its potential as an [
18F]F radio-synthon, the -SF
5 group also demonstrates unique pharmacological properties owing to its high lipophilicity (π = 1.51, relative to the –CF
3 group; π = 1.09) and electron withdrawing effect (σ
p = 0.68, relative to the –CF
3 group; σ
p = 0.54) [
2,
3]. Furthermore, the groups high fluorine content shows promise for its potential application as an
19F-MRI tag [
4].
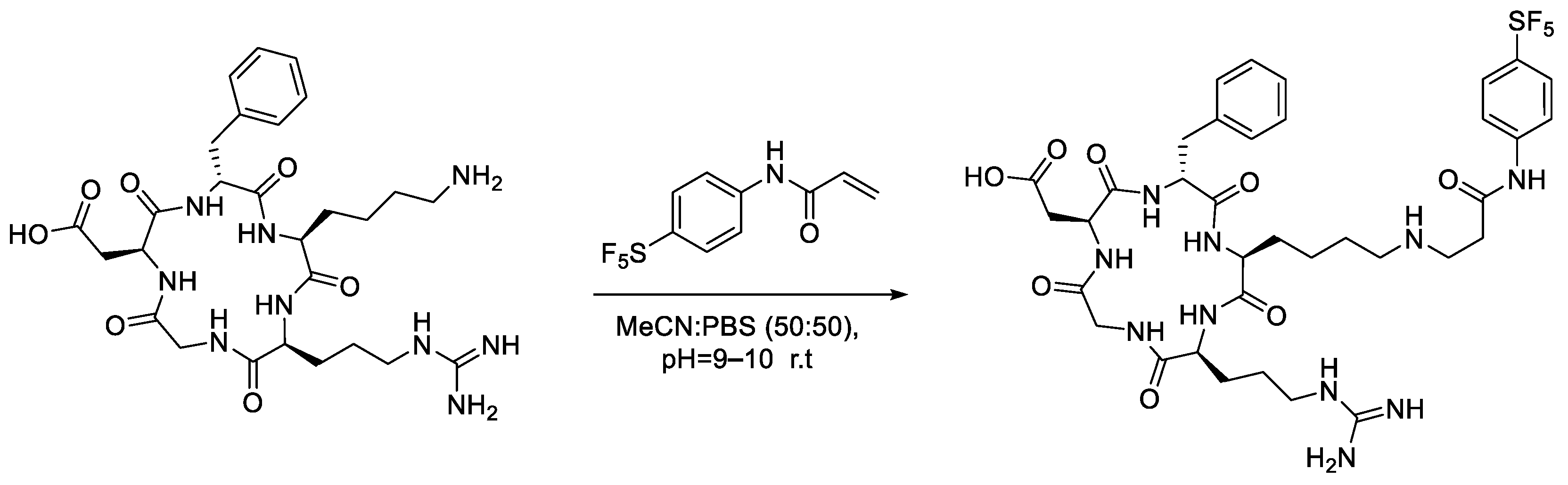
In light of these attributes, we investigated the PTAD–tyrosine conjugation as a potential means of selectively introducing an -[
18F]SF
5 tag onto the side chains of tyrosine residues [
5]. However, it was found that the aqueous stability of the SF
5-bearing PTAD derivative was a significant limitation and could only be used for bio-conjugation successfully under highly specialised conditions. For this reason, our investigation changed focus to the more robust maleimide and acrylamide moieties for selective bioconjugation with cysteine and lysine residues, respectively.
The thiol side chain of cysteine residues is a common target for bioconjugate reactions due to the unique nucleophilicity of the thiolate anion and its relatively low abundance in peptides and proteins [
6]. This abundance enables selective bioconjugation to be performed with a high degree of chemo- and regio-selectivity with the appropriate prosthetic group. Maleimides have been used extensively as prosthetic groups for cysteine selective bioconjugation, owing to their fast rate kinetics, selectivity and lack of by-products. The impressive selectivity of the maleimide towards thiols is imparted in two ways: firstly, the pKa of the thiol group (cysteine intrinsic pKa = ~8.6) allows for reactions to be performed in a pH range of 6.5–7.5, whereby potentially competing side chains, such as the ε-amine of lysine residues, remain predominantly protonated and thus unreactive [
7].
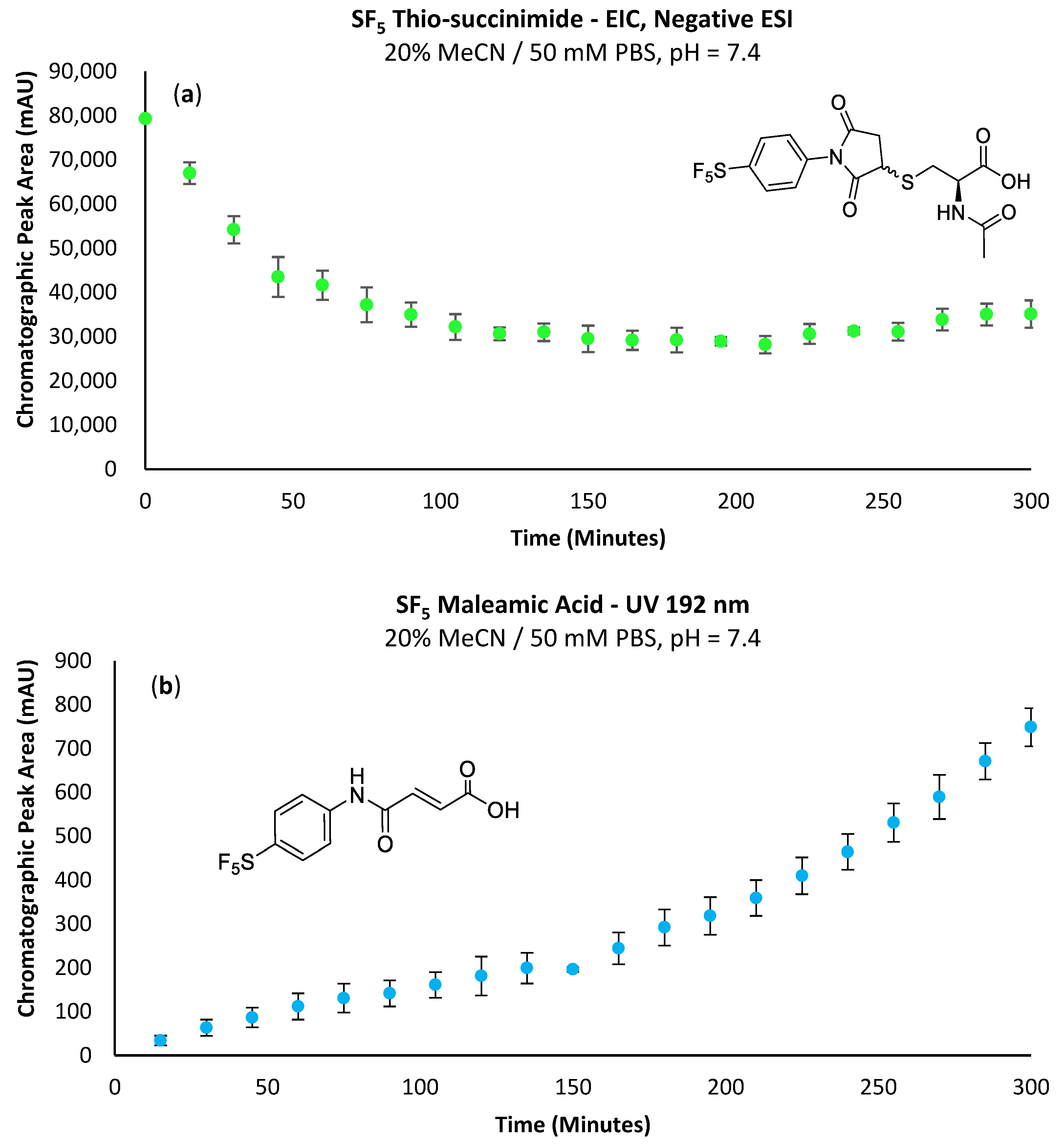
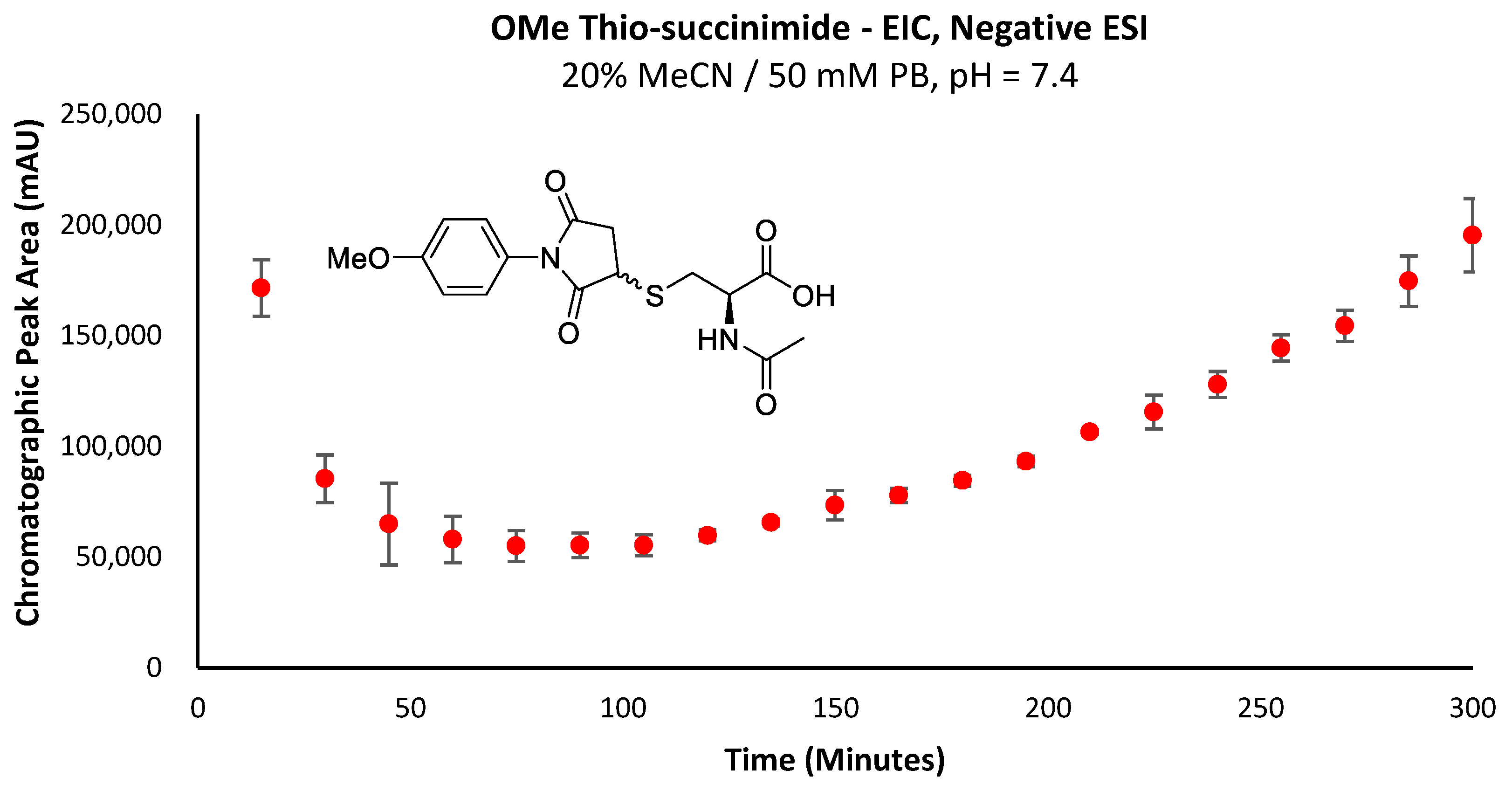
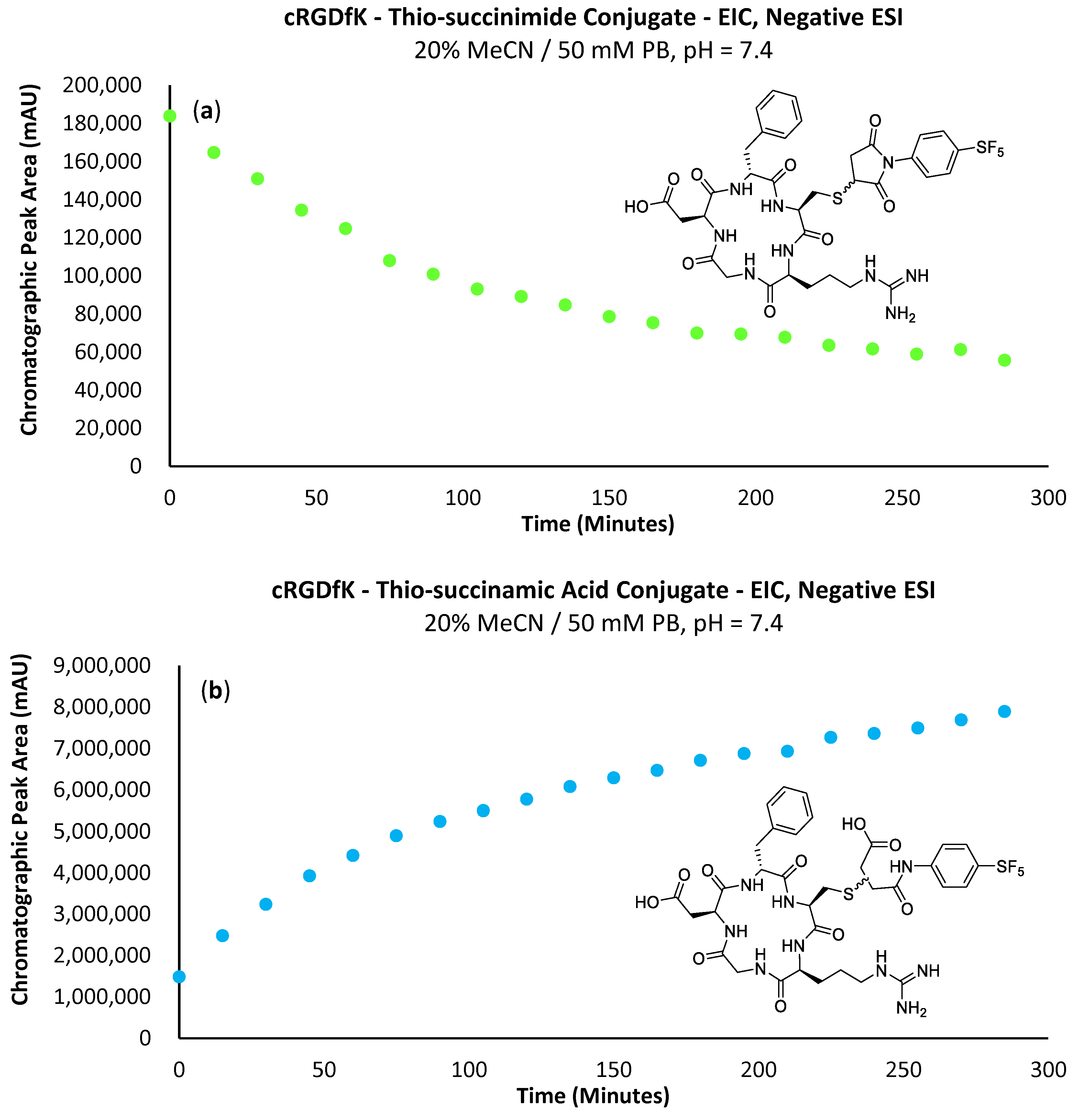
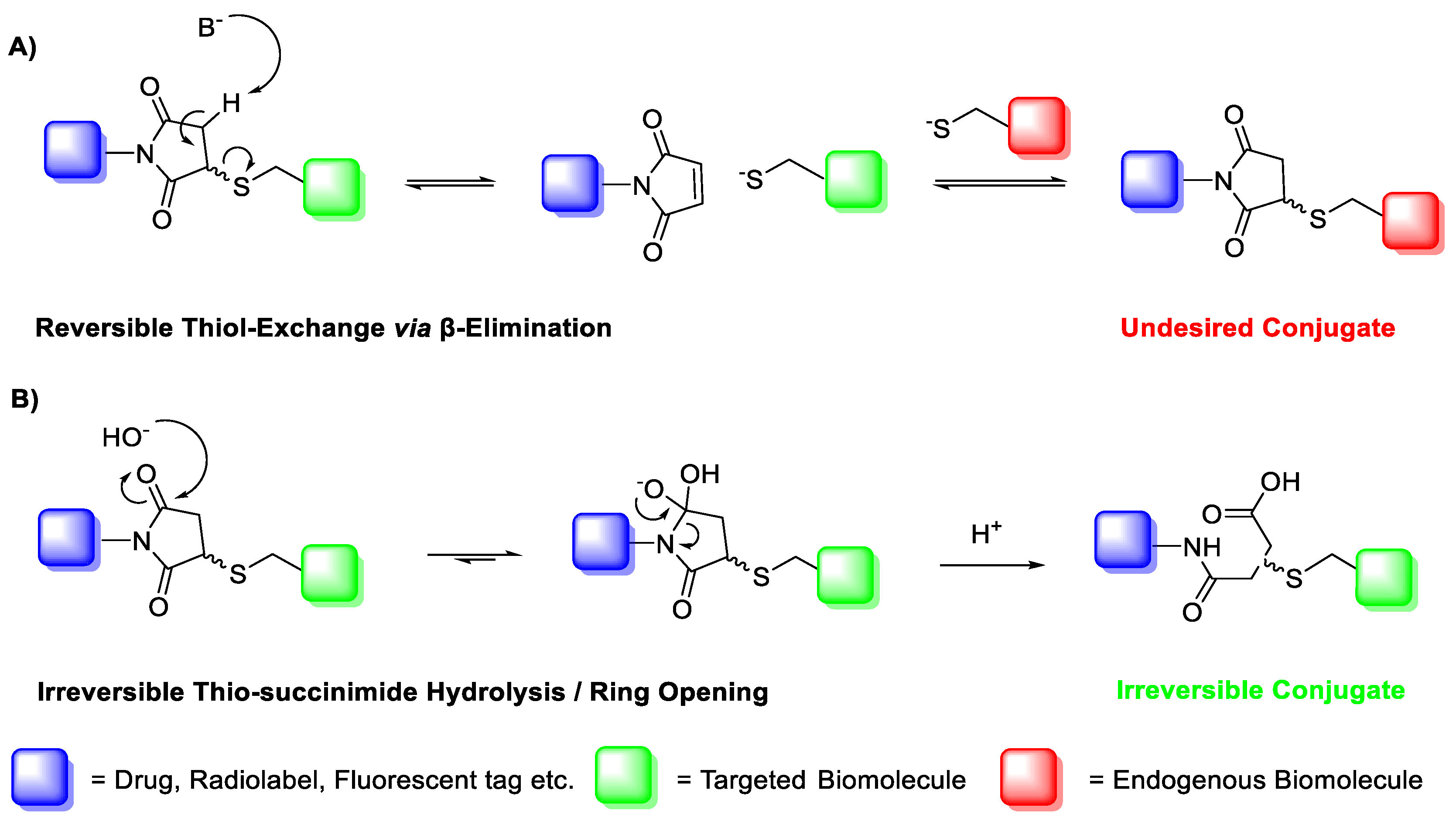
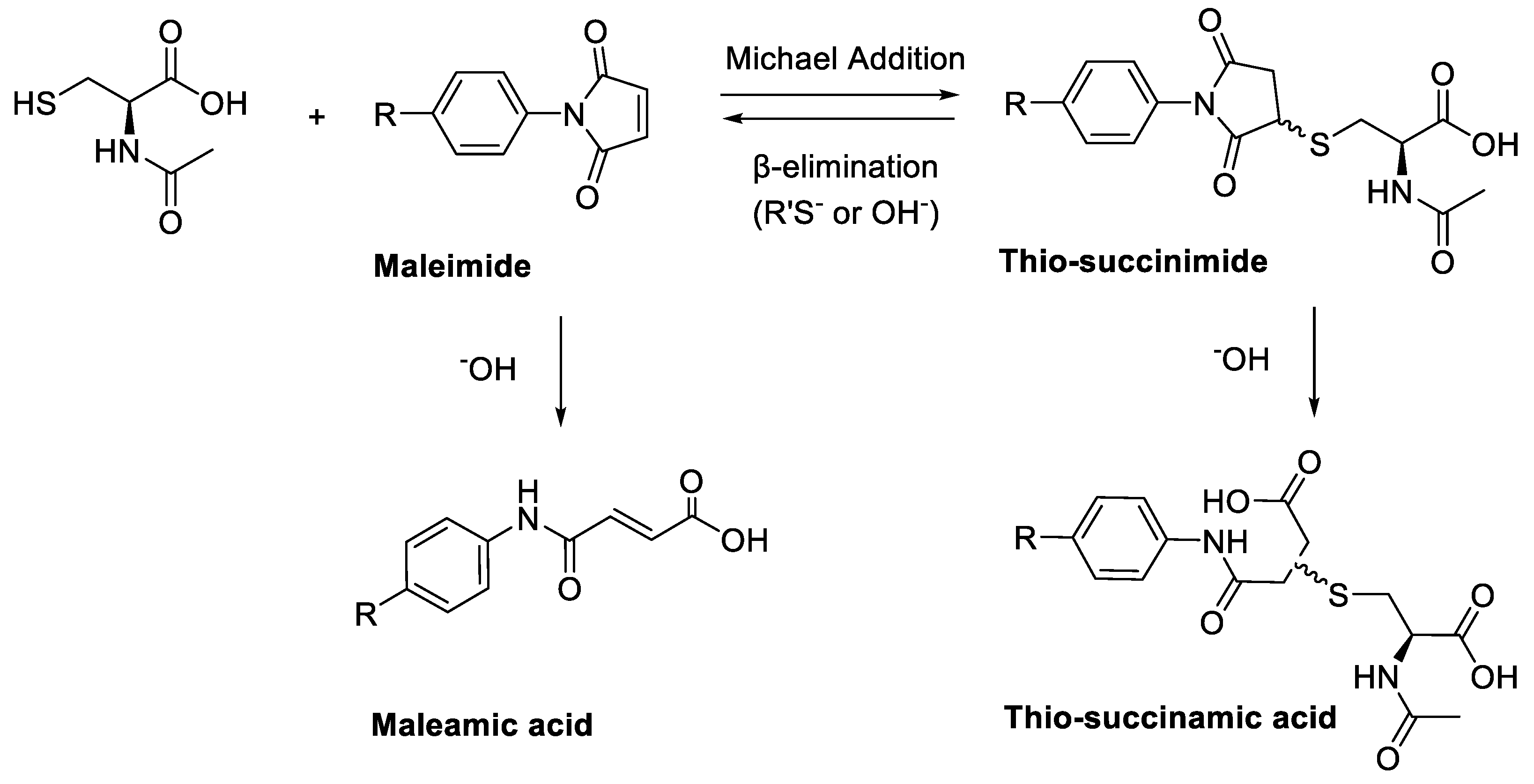
A major limitation of maleimide–cysteine conjugation is the propensity for the intermediate thio-succinimide ether to undergo β-elimination through the action of either base or nucleophile [
8]. This regenerates the maleimide, which can consequently undergo conjugation to non-targeted endogenous biomolecules, presenting a significant issue for in vitro applications such as PET imaging of biomolecule–radiolabel conjugates (
Scheme 2). Thiol exchange has been sparingly reported in the literature. For example, Alley et al. observed thiol exchange by mass spectrometry between thio-succinimide-linked antibody–drug conjugates and rat serum albumin in vivo, consequently forming albumin–drug conjugates [
9]. Lewis et al. utilised a
bis-maleimide linker to conjugate a monoclonal antibody with a DOTA chelating agent for radiolabelling with
111In(III) and
90Y(III); much like Alley et al., the authors observed that the conjugate underwent significant thiol exchange with human serum albumin in vivo [
10].
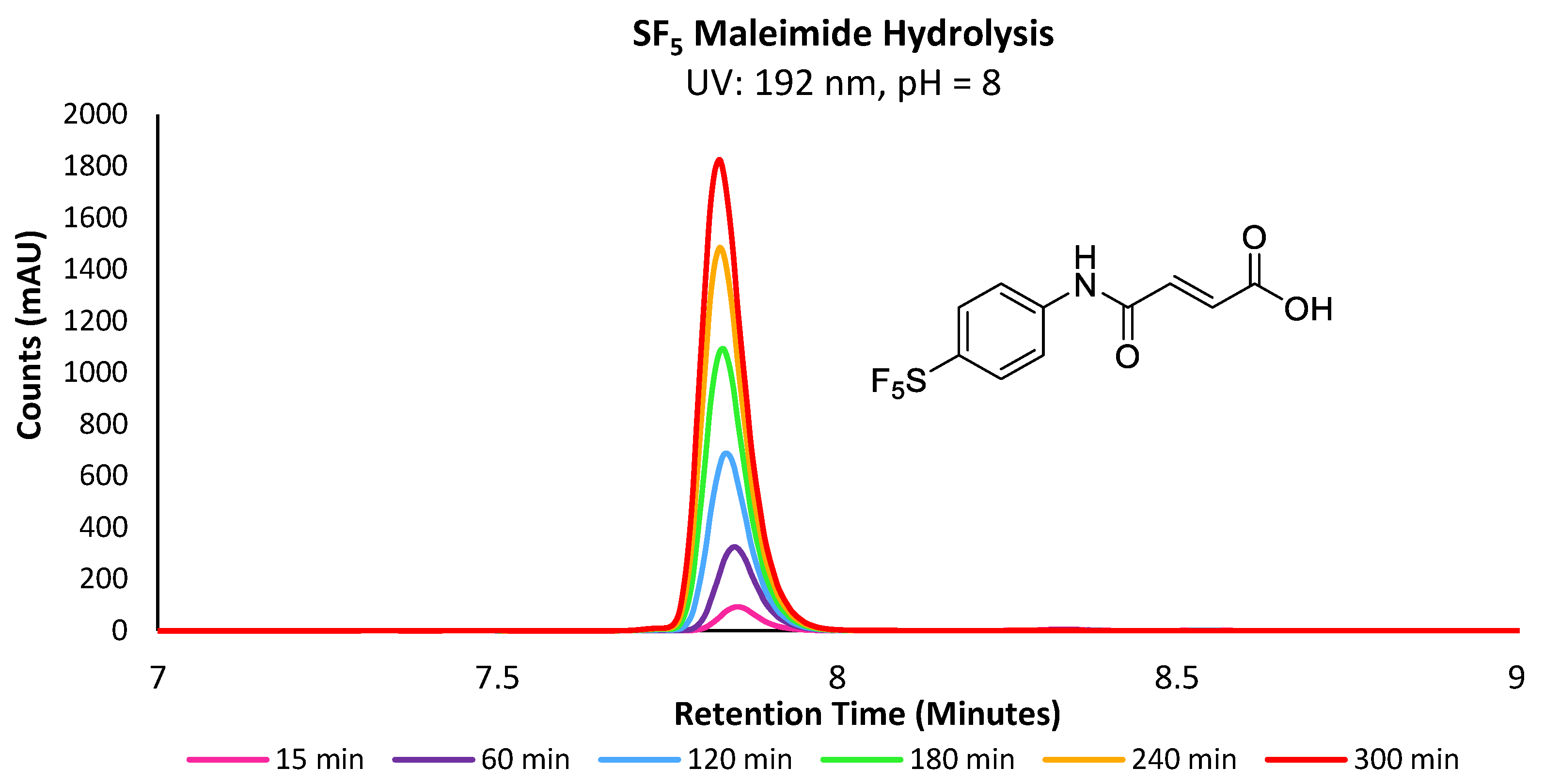
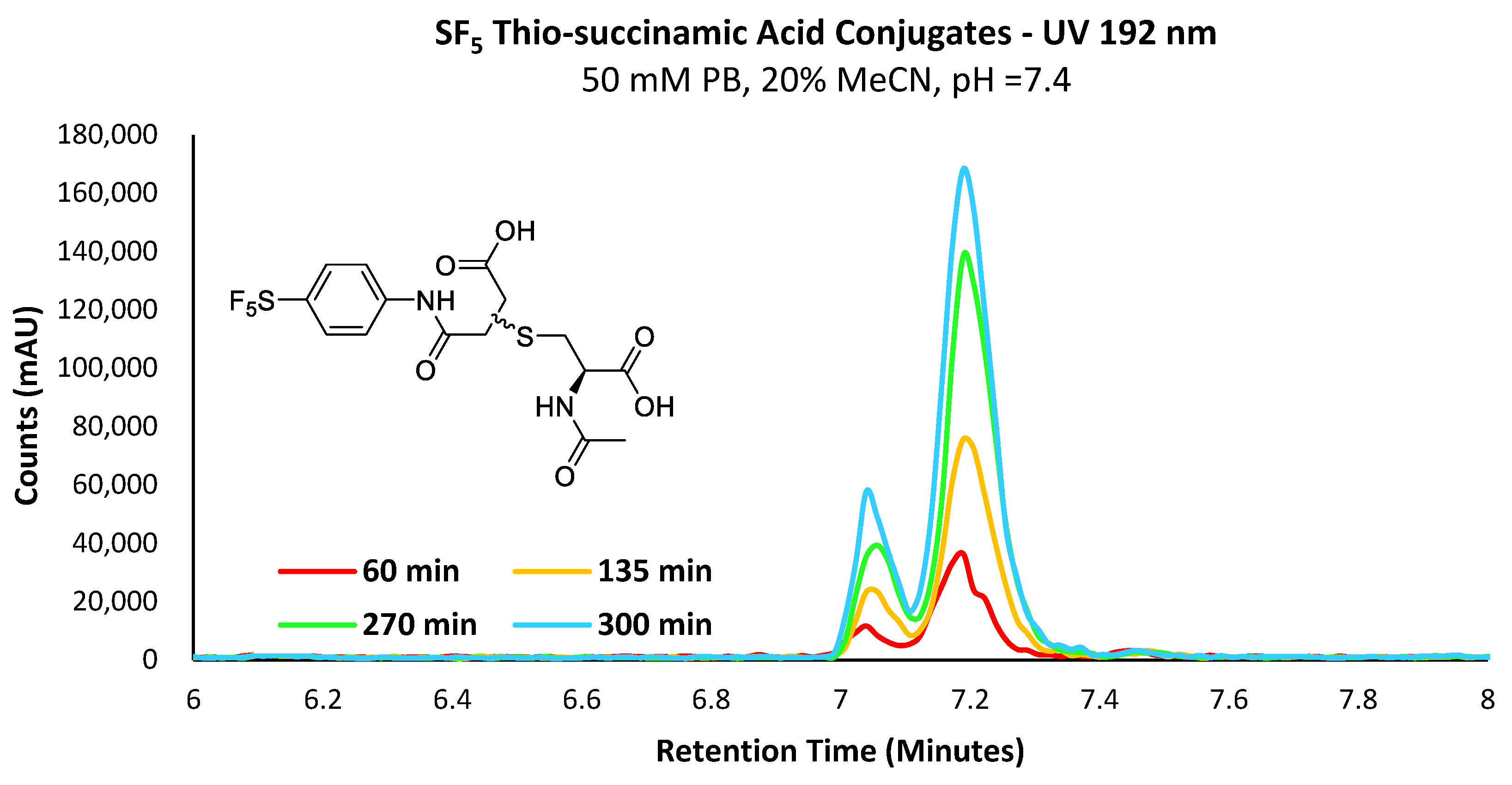
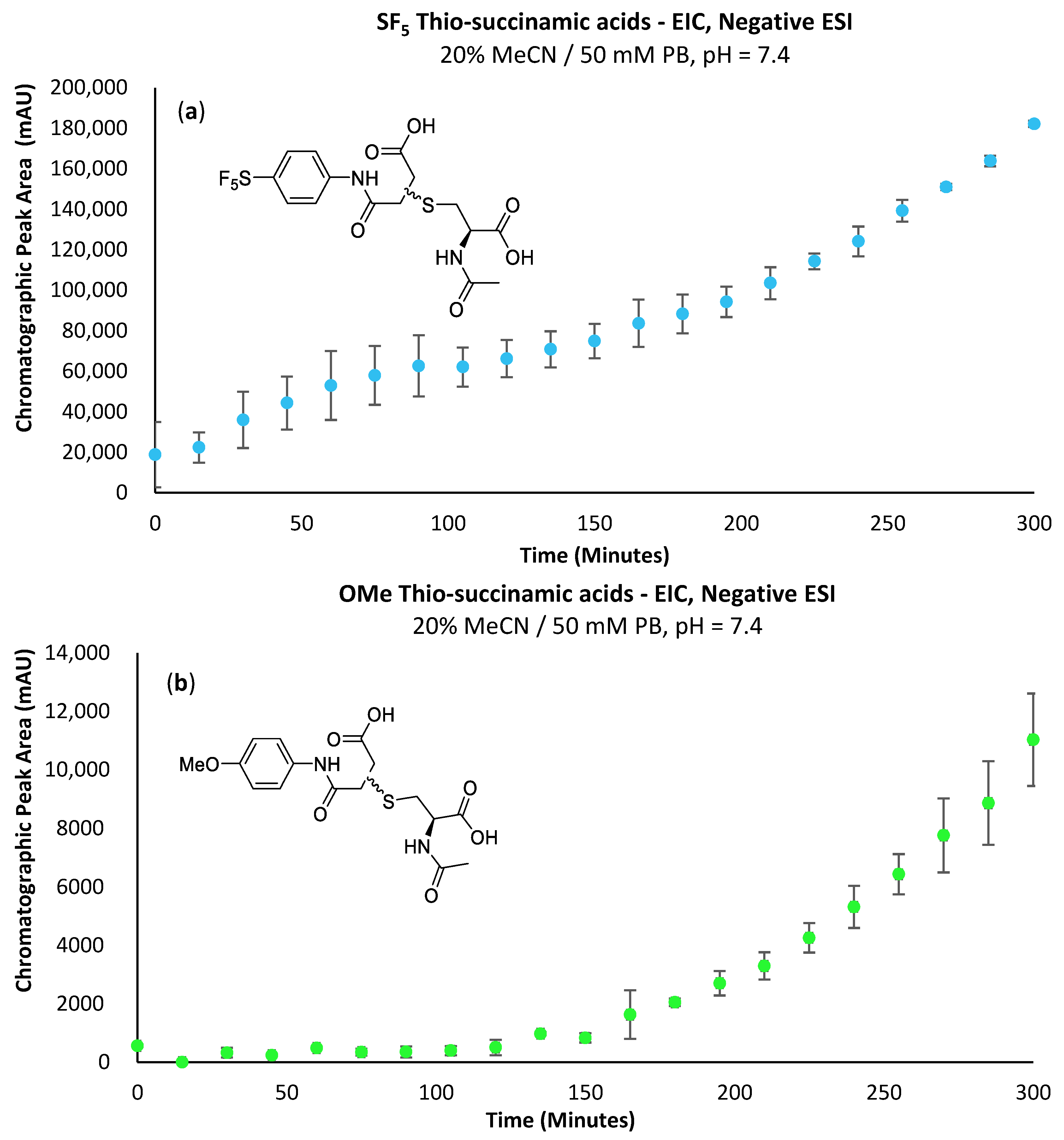
The propensity for the thio-succinimide ether to undergo β-elimination and subsequent thiol exchange is driven by the acidity of the α proton, which is increased by electron-withdrawing groups. On the other hand, this same electron-withdrawing effect also increases the electrophilicity of the carbonyl carbons, thus encouraging ring hydrolysis to a stable and irreversible thio-succinamic acid conjugate, a well-documented occurrence [
11,
12,
13]. The influence of electronic effects on thio-succinimide conjugate stability was first reported by Baldwin et al. in 2011, who found that electron-withdrawing substituents increase the rate constant for thio-succinimide ring opening, thus minimising the conjugates susceptibility towards thiol exchange [
14]. This affect was investigated further by Fontaine et al., who deduced the rate constants for these competing reactions across a wide variety of substituted
N-aliphatic maleimides [
15]. It was found that favourability for thio-succinimide ring hydrolysis over thiol exchange at pH = 7.4 was significantly improved with increasingly electron-withdrawing
N-alkyl substituents, indicating that electron-withdrawing groups are ultimately beneficial in preventing thiol exchange.
In 2014, an alternative approach was utilised by Lyon and co-workers for inducing thio-succinimide ring hydrolysis, whereby a diaminopropionic acid linker was utilised between the maleimide head group and drug payload, the primary amine of which was found to act as an intermolecular catalyst to rapidly induce ring hydrolysis in the subsequent thio-succinimide antibody–drug conjugates [
16]. Later, in 2015, Christie et al. evaluated the stability of a variety of antibody–drug conjugates formed via maleimides bearing either
N-aryl or
N-alkyl linkers. From their results, researchers observed substantially faster rates of thio-succinimide ring hydrolysis among the
N-aryl maleimide conjugates [
13]. Furthermore, the researchers found that the
N-aryl maleimide derivatives reacted approximately 2.5 times faster with the thiolate substrates compared with the
N-alkyl derivatives investigated, a desirable quality for time-sensitive applications such as
18F-radiolabelling.
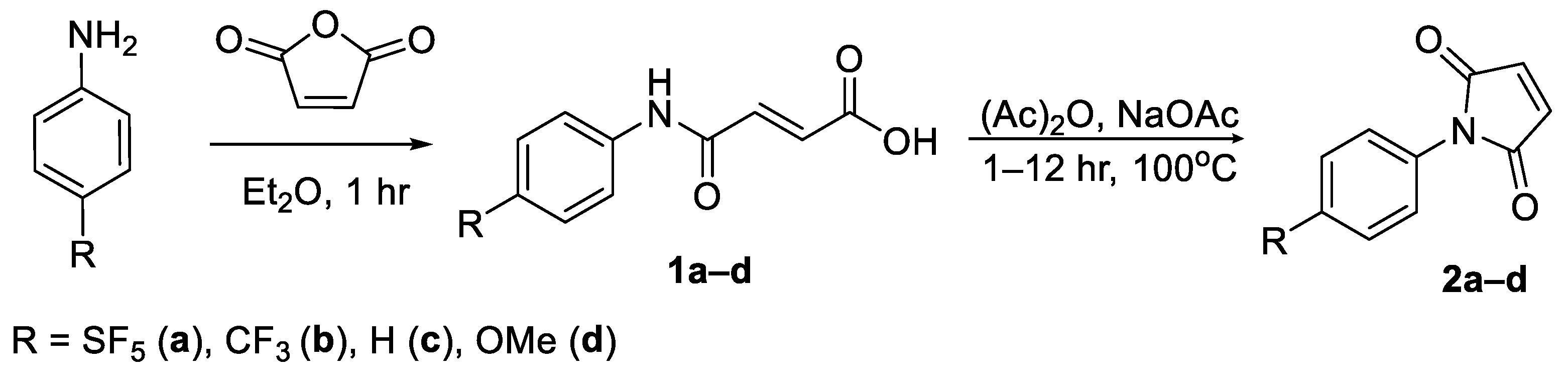
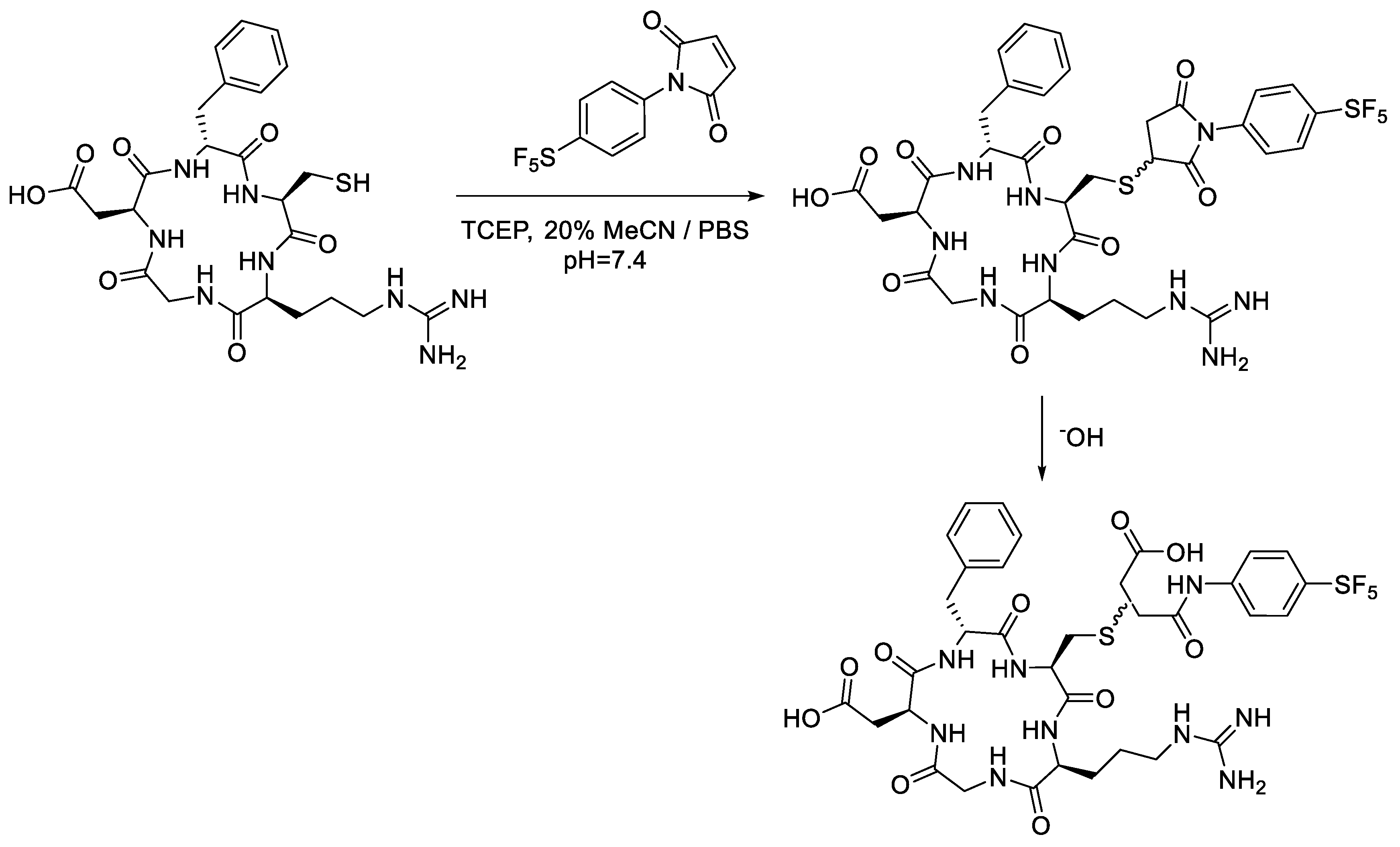
In light of these findings, we set out to determine if an aryl -SF
5 moiety would promote thio-succinimide ring hydrolysis in addition to serving as an efficient means of tagging cysteine residues with the -SF
5 group for
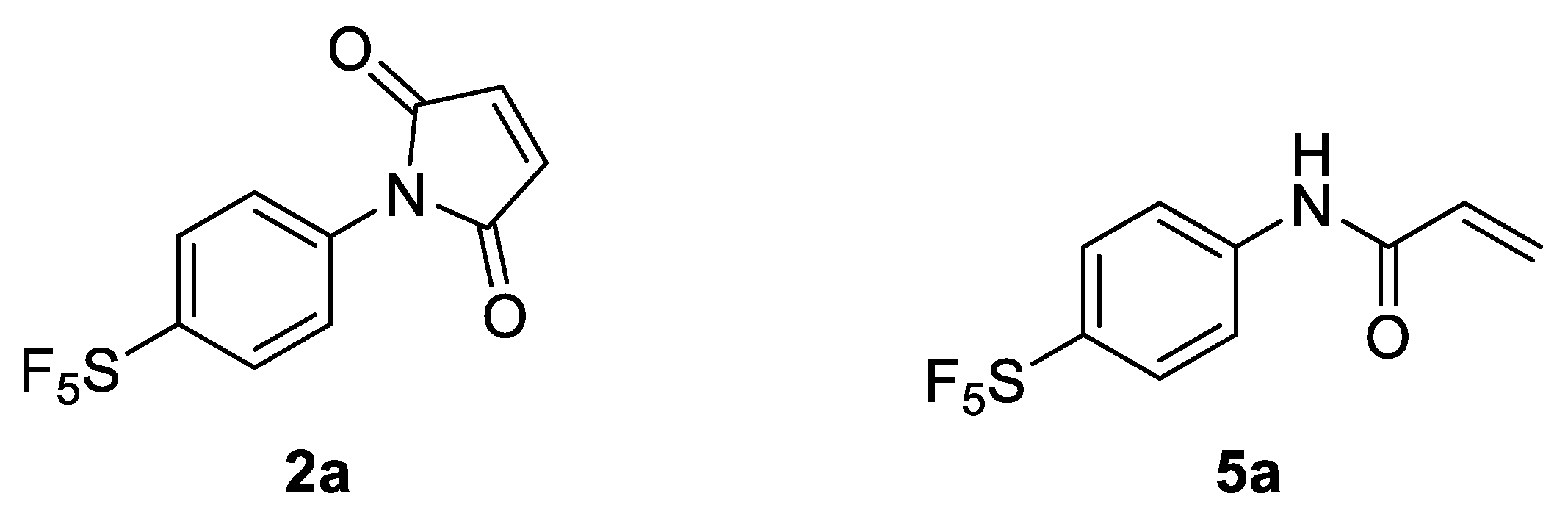
19F-MRI or PET applications. For this reason, 4-[4-(pentafluorosulfanyl)phenyl]pyrrole-2,5-dione (
2a) was envisaged as a potential prosthetic group for incorporating the -SF
5 moiety onto biomolecules of interest (
Figure 1).
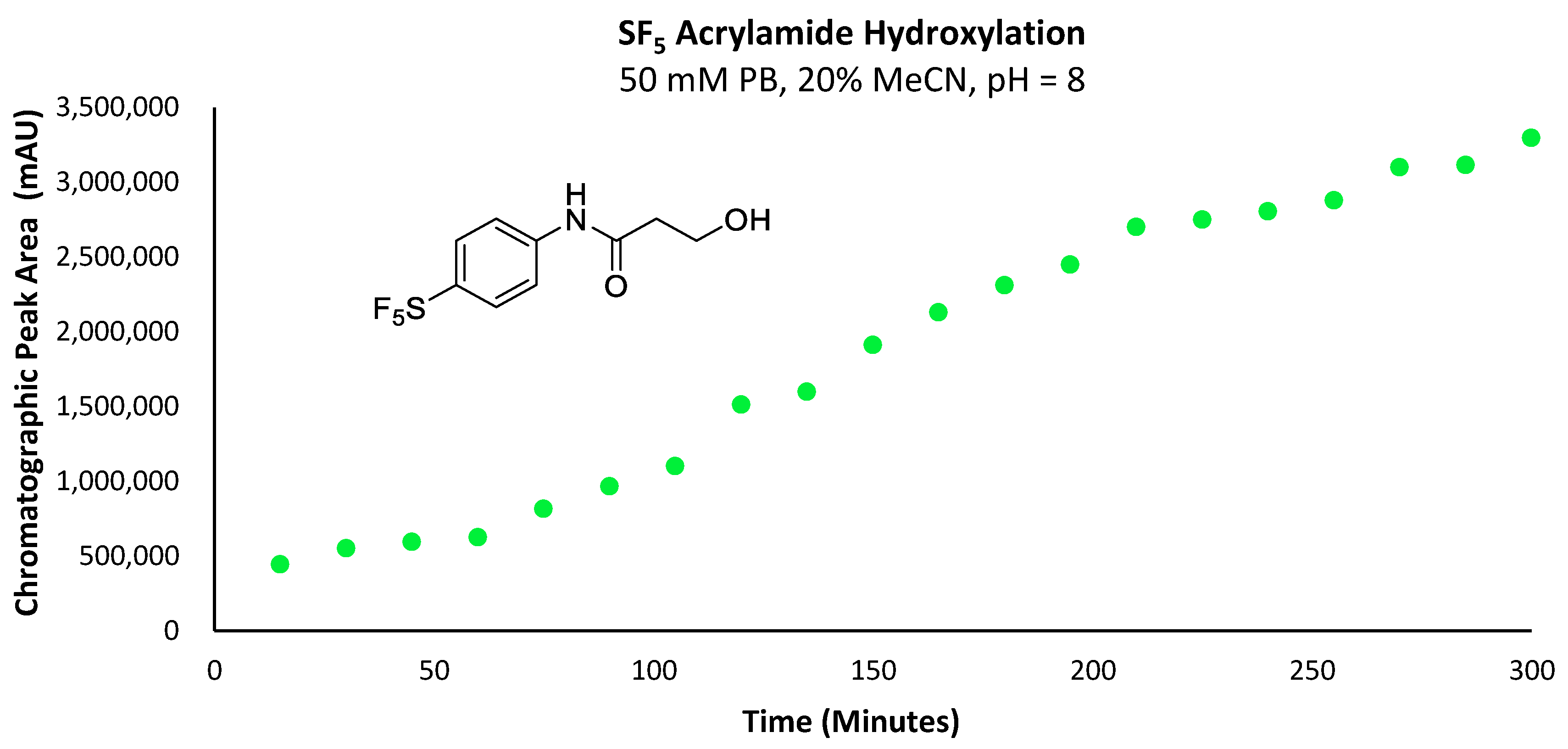
Due to the susceptibility of maleimides towards alkaline hydrolysis, they are substantially less applicable at the pH level required for lysine-selective bioconjugation (intrinsic pKa of lysine’s ε-amine = 10.4) [
17]. However, as the constrained pyrrole-2,5-dione ring significantly promotes hydrolysis; the corresponding acrylamides demonstrate far greater stability under basic aqueous conditions. This difference in stability becomes particularly pronounced in
N-aryl systems. Surprisingly, despite the impressive reactivity of
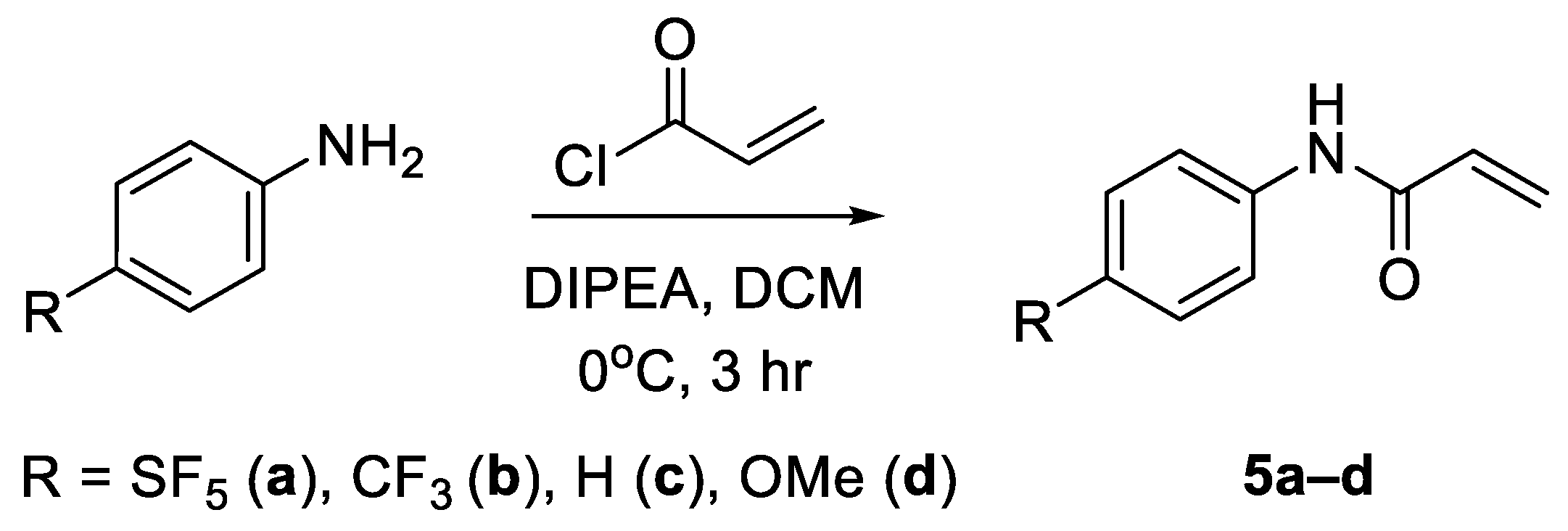
N-phenyl acrylamides towards lysine and cysteine residues, their application has been almost exclusively limited to use as covalent inhibitors for drug development, rather than as a means of introducing tags via bioconjugation [
18]. For this reason, 4-[4-(pentafluorosulfanyl)phenyl]-2-propenamide (
5a) was also investigated as a potential prosthetic group for tagging lysine residues with the -SF
5 moiety (
Figure 1).
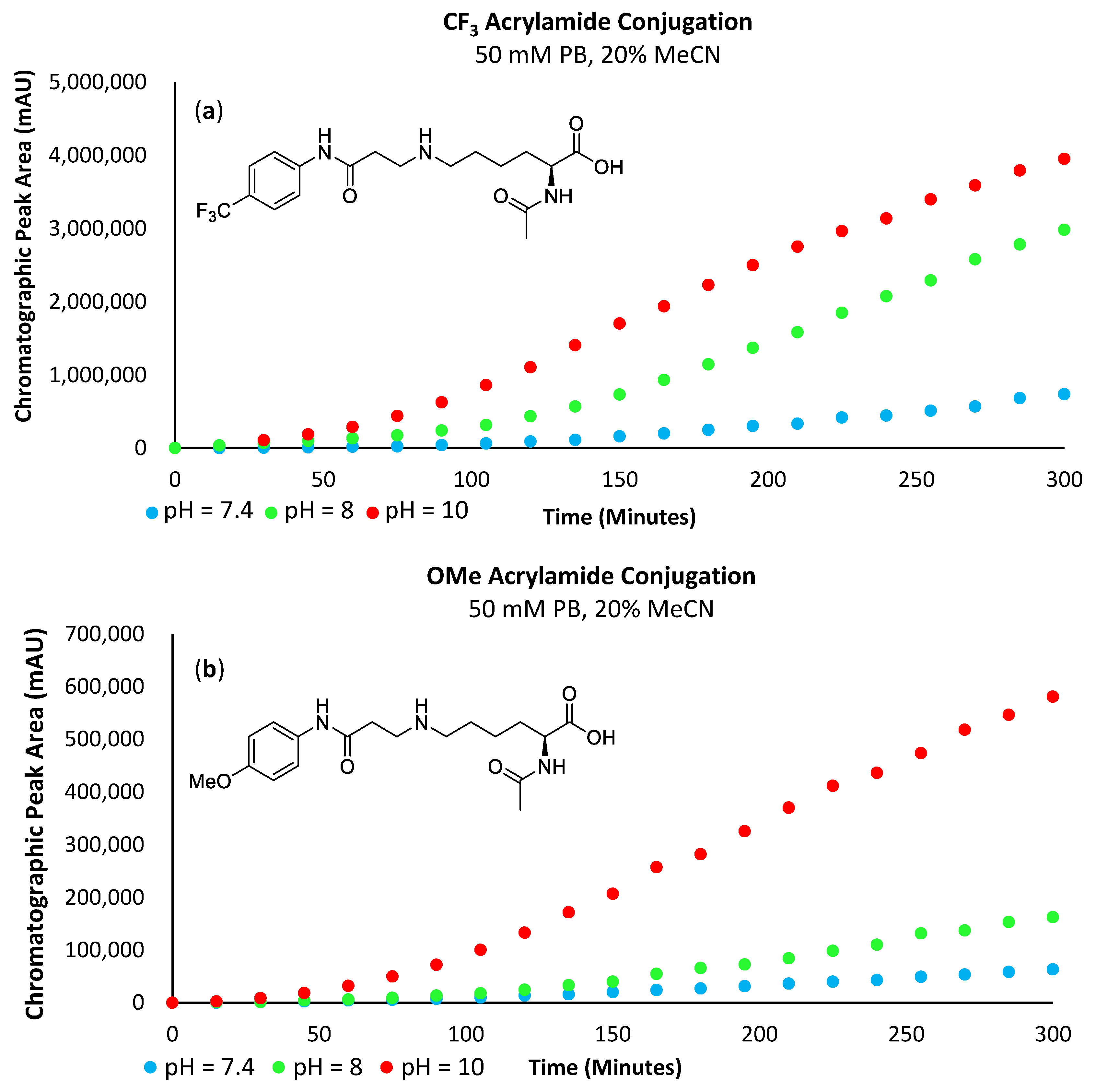
To properly ascertain the influence of the -SF
5 groups considerable electron-withdrawing effect on these bioconjugate approaches (σ
p = 0.68), a number of analogous,
para-substituted
N-aryl maleimide and acrylamide derivatives were also synthesised and subjected to the same bioconjugate reactions for comparison, namely; OMe (σ
p = −0.27), H (σ
p = 0.00) and CF
3 (σ
p = 0.54) [
3].
3. Materials and Methods
General Experimental: All chemical reagents and AR grade or analytical grade solvents were acquired from commercial sources; 4-(pentafluorosulfanyl) aniline was purchased from Fluorochem and all other compounds were purchased from Sigma-Aldrich Merck unless otherwise stated. All reactions were monitored using TLC Silica gel 60 F254 with UV detection at 254 nm. Solvents were removed under reduced pressure using a Buchi rotary evaporator. High-resolution mass spectra were obtained using an Agilent 6510 Q-TOF Mass Spectrometer (ESI) and Agilent 1290 Infinity HPLC system. IR spectra were recorded on a Nicolet 6700 FT-IR spectrometer (KBr). Data for IR were recorded as follows: wavelength (cm−1), absorbance (s = strong, m = medium, w = weak, br = broad). 1H-NMR, 13C-NMR and 19F-NMR spectra were recorded on a Bruker Ascend premium shielded spectrometer 400 MHz (400 MHz 1H, 125 MHz 13C, 376 MHz 19F). Data for 1H, 19F and 13C NMR were recorded as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad).
General LC-MS method: Reactive prosthetic groups were prepared in a 100 mM stock solution of anhydrous acetonitrile. The stock solution (10 µL) was automatically transferred into a secondary vial containing a degassed solution of the relevant buffer (800 µL), acetonitrile (190 µL) of the model amino acid (10 mM) in the respective buffer solution. The reaction mixture was then mixed 20 times using the auto-injector, and the reaction was analysed by LC-MS periodically every 15 min. In experiments investigating cysteine conjugations, amino acid reaction solutions were incubated with TCEP (4 equiv.) for two hours before injecting the prosthetic group stock solution.
Mass spectrometry parameters: negative ESI, gas temperature 350 °C, drying gas 13 L/min, nebuliser 55 psig, capillary voltage 3500 V, fragmentation voltage 100 V. Mobile phase: solvent A: acetonitrile (0.1% TFA), solvent B: Milli-Q water (0.1% TFA); Mobile phase gradient: 2 min 95% A; 10 min 5% A, 12 min 5% A, 15 min 95% A. Flow rate 0.5 mL/min. Injection volume 10 µL. An injector program achieved the automatic transfer of prosthetic group stock solution to reaction vials. Separation was achieved using a Kinetex 2.6 µm XB-C18 column 50 × 4.6 mm.
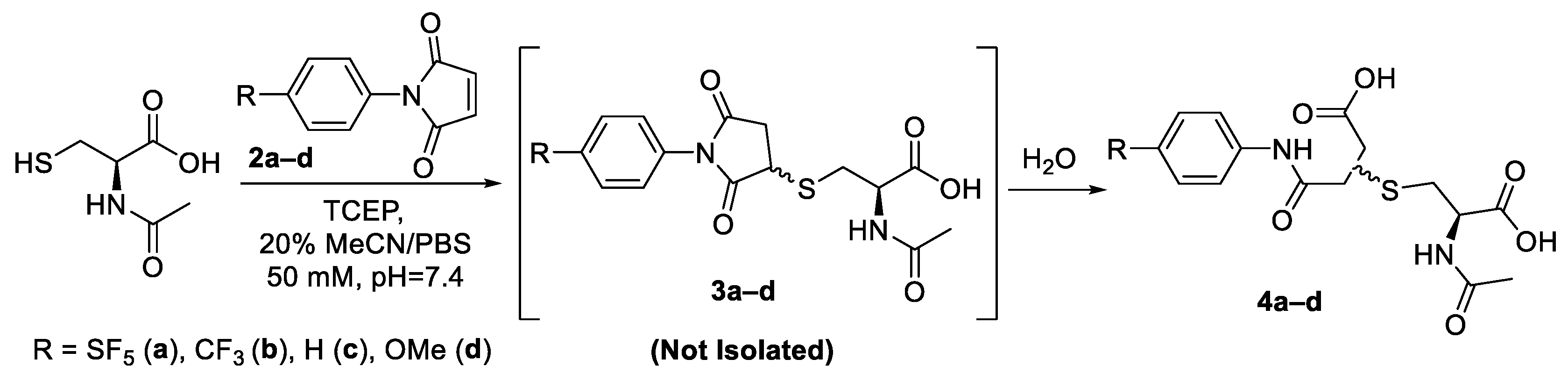
General procedure for the synthesis of (4a–d): N-acetyl-L-cysteine (32 mg, 0.2 mmol) and TCEP.HCl (115 mg, 0.4 mmol, 2 equiv.) were dissolved in a degassed solution of methanol (2 mL) and sodium carbonate (128 mg, 1.2 mmol, 6 equiv.) and the solution was allowed to stir for 30 min. N-aryl maleimide (0.3 mmol, 1.5 equiv) was added portion-wise to the solution, and the reaction was allowed to continue for 5 h. The reaction mixture was then concentrated in vacuo, and the resulting residue was suspended in 0.1 M HCl (5 mL) and filtered. The precipitate was then purified on C18 silica to afford the final regio-isomeric mixture of thio-succinamic acid product.
General procedure for the synthesis of (6a–d): Nα-acetyl-L-lysine (38 mg, 0.2 mmol) was dissolved in a solution of methanol (2 mL) and sodium carbonate (128 mg, 1.2 mmol, 6 equiv.), and the solution was allowed to stir for 30 min. Acrylamide (0.2 mmol) was added portion-wise to the solution, and the reaction continued for 8 h. The reaction mixture was then concentrated in vacuo. The crude residue was purified on C18 silica to afford the final product as the sodium carboxylate salt.
3.1. 2-[[(2S)-2-(Acetylamino)-2-carboxyethyl]thio]-4-[(4-pentafluorosulfanylphenyl)amino]-4-oxobutanoic acid (B) & 3-[[(2S)-2-(acetylamino)-2-carboxyethyl]thio]-4-[(4-pentafluorosulfanylphenyl)amino]-4-oxobutanoic Acid (A)
Compound 4a was obtained as a transparent oil (77 mg, 0.16 mmol, 80%). HRMS (EI) Calcd for C15H16F5N2O6S2 [M-H]− 479.0369, [M-H]− Found 479.0372; IR (KBr): 3326, 3206, 2934, 1703, 1597, 1539, 1405, 1373, 1317, 1256, 1230, 1192, 1107, 968, 831 cm−1; 1H-NMR ((CD3)2CO): δ 7.85–7.82 (m, 4H, Ar), 7.79–7.63 (d, 4H, Ar), 4.82–4.74 (m, 2H, CH-S), 3.92–3.88 (m, 2H, CH-N), 3.35–3.25 (m, 2H, CH2-COOH), 3.17–3.04 (m, 4H, CH2-COOH/CH2-S), 2.89–2.81 (m, 2H, CH2-S), 2.03 (s, 3H, CH3), 2.01 (s, 3H, CH3); 13C-NMR ((CD3)2CO): δ 173.26 (C=O), 173.18 (C=O), 171.95 (C=O), 171.90 (C=O), 171.87 (C=O), 171.39 (C=O), 169.96 (C=O), 169.85 (C=O), 148.85 (quin, J = 17 Hz, SF5) 142.92 (Ar-NH), 142.88 (Ar-NH), 127.59 (quin, J = 4 Hz, SF5), 127.59 (Ar-SF5), 127.55 (Ar-SF5), 119.54 (Ar), 119.46 (Ar), 52.81 (CH-S), 52.51 (CH-S), 42.94 (CH-NH), 42.41 (CH-NH), 39.74 (CH2-S), 39.20 (CH2-S), 34.15 (CH2-COOH), 33.97 (CH2-COOH), 22.57 (CH3); 19F-NMR ((CD3)2CO): δ 85.75 (m, 1F), 63.08 (d, J = 150 HZ, 4F).
3.2. 2-[[(2S)-2-(Acetylamino)-2-carboxyethyl]thio]-4-[(4-trifluoromethylphenyl)amino]-4-oxobutanoic acid (B) & 3-[[(2S)-2-(acetylamino)-2-carboxyethyl]thio]-4-[(4-trifluoromethylphenyl)amino]-4-oxobutanoic Acid (A)
Compound 4b was obtained as a transparent oil (79 mg, 0.18 mmol, 94%). HRMS (EI) Calcd for C16H16F3N2O6S [M-H]− 421.0681, [M-H]− Found 421.0684; IR (KBr): 3325, 3213, 3123, 2934, 1716, 1607, 1540, 1411, 1374, 1306, 1258, 1164, 1114, 1067, 1016, 969, 842 cm−1; 1H-NMR ((CD3)2CO): δ 7.87–7.83 (m, 4H, Ar), 7.62–7.60 (m, 4H, Ar), 4.82–4.75 (m, 2H, CH-S), 3.93–3.88 (m, 2H, CH-N), 3.35–3.25 (m, 2H, CH2-COOH), 3.15–3.06 (m, 4H, CH2-COOH/CH2-S), 2.87–2.83 (m, 2H, CH2-S), 2.02 (s, 3H, CH3), 2.00 (s, 3H, CH3); 13C-NMR ((CD3)2CO): δ 173.24 (C=O), 173.15 (C=O), 171.91 (C=O), 171.86 (C=O), 169.84 (C=O), 169.72 (C=O), 143.29 (Ar-NH), 143.25 (Ar-NH), 129.39 (Ar-CF3), 126.77 (q, J = 5 Hz, CF3), 125.44 (Ar-CF3), 125.39 (Ar), 125.12 (Ar), 53.17 (CH-S), 52.82 (CH-S), 43.00 (CH-NH), 42.47 (CH-NH), 39.76 (CH2-S), 39.20 (CH2-S), 34.15 (CH2-COOH), 33.96 (CH2-COOH), 22.54 (CH3); 19F-NMR ((CD3)2CO): δ -62.36 (s, 3F), -62.39 (s, 3F).
3.3. 2-[[(2S)-2-(Acetylamino)-2-carboxyethyl]thio]-4-[(phenyl)amino]-4-oxobutanoic acid (B) & 3-[[(2S)-2-(acetylamino)-2-carboxyethyl]thio]-4-[(phenyl)amino]-4-oxobutanoic Acid (A)
Compound 4c was obtained as a transparent oil (68 mg, 0.19 mmol, 96%). HRMS (EI) Calcd for C15H17N2O6S [M-H]− 353.0807, [M-H]− Found 353.0810; IR (KBr): 3322, 3289, 3079, 1716, 1620, 1597, 1551, 1521, 1445, 1374, 1340, 1317, 1261, 1232m 1177, 1083, 1048, 1000, 915, 869, 811, 754, 738, 688 cm−1; 1H-NMR ((CD3)2CO): δ 7.63 (t, J = 12.4 Hz, 4H, Ar), 7.30–7.25 (m, 4H, Ar), 7.06–7.01 (m, 2H, Ar), 4.84–4.73 (m, 2H, CH-S), 3.92–3.87 (m, 2H, CH-N), 3.33–3.24 (m, 2H, CH2-COOH), 3.17–2.99 (m, 4H, CH2-COOH/CH2-S), 2.83–2.75 (m, 2H, CH2-S), 2.01 (s, 3H, CH3), 1.99 (s, 3H, CH3). 13C-NMR ((CD3)2CO): δ 173.36 (C=O), 173.26 (C=O), 171.99 (C=O), 171.16 (C=O), 169.31 (C=O), 169.22 (C=O), 169.19 (C=O), 169.10 (C=O), 139.96 (Ar-NH), 139.89 (Ar-NH), 139.87 (Ar), 139.80 (Ar), 129.48 (Ar), 124.29 (Ar), 124.21 (Ar), 120.20 (Ar), 53.22 (CH-S), 43.24 (CH-NH), 42.77 (CH-NH), 39.70 (CH2-S), 39.65 (CH2-S), 39.10 (CH2-COOH), 39.05 (CH2-COOH), 22.63 (CH3), 22.59 (CH3).
3.4. 2-[[(2S)-2-(Acetylamino)-2-carboxyethyl]thio]-4-[(4-methoxyphenyl)amino]-4-oxobutanoic acid (B) & 3-[[(2S)-2-(acetylamino)-2-carboxyethyl]thio]-4-[(4-methoxyphenyl)amino]-4-oxobutanoic Acid (A)
Compound 4d was obtained as a transparent oil (71 mg, 0.18 mmol, 92%). HRMS (EI) Calcd for C16H19N2O7S [M-H]− 383.0913, [M-H]− Found 383.0912; IR (KBr): 3308, 3079, 2935, 2838, 1716, 1651, 1541, 1521, 1464, 1415, 1374, 1301, 1241, 1178, 1029, 969, 830 cm−1; 1H-NMR ((CD3)2CO): δ 7.55–7.52 (m, 4H, Ar), 6.86–6.83 (m, 4H, Ar), 4.84–4.72 (m, 2H, CH-S), 3.92–3.86 (m, 2H, CH-N), 3.75 (s, 3H, O-CH3), 3.74 (s, 3H, O-CH3), 3.29–3.23 (m, 2H, CH2-COOH), 3.17–2.96 (m, 4H, CH2-COOH/CH2-S), 2.80–2.72 (m, 2H, CH2-S), 2.02 (s, 3H, CH3), 1.99 (s, 3H, CH3): 13C-NMR ((CD3)2CO): δ 173.40 (C=O), 173.29 (C=O), 171.96 (C=O), 171.32 (C=O), 171.24 (C=O), 168.86 (C=O), 168.82 (C=O), 168.72 (C=O), 156.85 (Ar-OCH3), 156.79 (Ar-OCH3), 132.95 (Ar-NH), 132.87 (Ar-NH), 121.89 (Ar), 121.79 (Ar), 121.76 (Ar), 121.66 (Ar), 55.59 (O-CH3), 53.14 (CH-S), 52.87 (CH-S), 43.36 (CH-NH), 42.92 (CH-NH), 39.45 (CH2-S), 38.84 (CH2-S), 34.19 (CH2-COOH), 34.05 (CH2-COOH), 22.58 (CH3).
3.5. 2-[[(2S)-2-(Acetylamino)-6-((2-carboxyethyl)amino)-[(4-pentafluorosulfanylphenyl)amino]-4-hexanoic Acid (6a)
Compound 6a was obtained as a transparent oil (76 mg, 0.166 mmol, 83%). HRMS (EI) Calcd for C17H23F5N3O4S [M-H]− 460.1329, [M-H]− Found 460.1329; IR (KBr): 3263, 2932, 1693, 1625, 1503, 1438, 1399, 1314, 1260, 1188, 1100, 1033, 812, 649, 578, 541, 493 cm−1; 1H-NMR (D2O): δ 7.72 (d, J = 8 Hz, 2H, Ar), 7.52 (d, J = 8.4 Hz, 2H, Ar), 4.07 (dd, J = 5, 8 Hz, 1H, CH-N), 3.36–3.28 (m, 2H, CH2-N), 3.05–2.96 (m, 2H, CH2-CONH), 2.91 (t, J = 8 Hz, 2H, CH2-N), 1.93 (s, 3H, CH3), 1.78–1.18 (m, 8H, CH2); 13C-NMR (D2O): δ 179.06 (C=O), 173.53 (C=O), 170.50 (C=O), 148.81 (q, J = 18 Hz, SF5), 142.44 (Ar-NH), 140.66 (Ar-SF5), 131.17 (Ar), 126.88 (quin, J = 4 Hz, SF5), 120.16 (Ar), 54.89 (CH-N), 48.84 (CH2-N), 47.44 (CH2-N), 42.92 (CH2-CONH), 31.01 (CH2), 26.29 (CH2), 24.97 (CH2), 22.07 (CH3); 19F-NMR (D2O): δ 85.65 (m, 1F), 63.10 (d, J = 160 Hz, 4F).
3.6. 2-[[(2S)-2-(Acetylamino)-6-((2-carboxyethyl)amino)-[(4-trifluoromethylphenyl)amino]-4-hexanoic Acid (6b)
Compound 6b was obtained as a transparent oil (71 mg, 0.178 mmol, 89%). HRMS (EI) Calcd for C18H23F3N3O4 [M-H]− 402.1640, [M-H]− Found 402.1641; IR (KBr): 3273, 1606, 1548, 1409, 1324, 1162, 1112, 1067, 1016, 841, 510 cm−1; 1H-NMR (MeOD): δ 7.56–7.49 (m, 4H, Ar), 4.15 (dd, J = 5, 8 Hz, 1H, CH-N), 3.39–3.35 (m, 2H, CH2-N), 3.07 (m, 2H, CH2-CONH), 2.88 (t, J = 8 Hz, 2H, CH2-N), 1.90 (s, 3H, CH3), 1.74–1.06 (m, 8H, CH2). 13C-NMR (D6-DMSO): 13C-NMR (D6-DMSO): δ 174.49 (C=O), 169.62 (C=O), 168.74 (C=O), 142.62 (Ar-NH), 127.95 (q, J = 5 Hz, CF3), 119.35 (Ar), 119.08 (Ar), 47.41 (CH-N), 43.30 (CH2-N), 33.87 (CH2-N), 31.67 (CH2-CONH), 31.50 (CH2), 26.83 (CH2), 26.49 (CH2), 22.71 (CH3); 19F-NMR (D6-DMSO): δ -59.78 (s, 3F).
3.7. 2-[[(2S)-2-(Acetylamino)-6-((2-carboxyethyl)amino)-[(phenyl)amino]-4-hexanoic Acid (6c)
Compound 6c was obtained as a transparent oil (42 mg, 0.128 mmol, 64%). HRMS (EI) Calcd for C17H24N3O4 [M-H]− 334.1766, [M-H]− Found 334.1770; IR (KBr): 3295, 2945, 1560, 1499, 1444, 1399, 1311, 757, 693, 626, 560, 460 cm−1; 1H-NMR (D6-DMSO): δ 7.59 (d, J = 7.6 Hz, 2H, Ar), 7.29 (t, J = 8 Hz, 2H, Ar), 7.04 (t, J = 7.2 Hz, Ar), 4.13–4.08 (m, 1H, CH-N), 2.87 (t, J = 7.2, 2H, CH2-N), 2.81 (t, J = 7.2, 2H, CH2-CONH), 1.93 (s, 3H, CH3), 1.68–1.29 (m, 8H, CH2); 13C-NMR (D6-DMSO): δ 173.83 (C=O), 169.31 (C=O), 168.21 (C=O), 138.92 (Ar-NH), 128.75 (Ar), 123.36 (Ar), 119.18 (Ar), 51.90 (CH-N), 48.60 (CH2-N), 46.77 (CH2-N), 42.77 (CH2-CONH), 30.62 (CH2), 26.56 (CH2), 25.21 (CH2), 22.57 (CH3)
3.8. 2-[[(2S)-2-(Acetylamino)-6-((2-carboxyethyl)amino)-[(4-methoxyphenyl)amino]-4-hexanoic Acid (6d)
Compound 6d was obtained as a transparent oil (42 mg, 0.116 mmol, 58%). HRMS (EI) Calcd for C18H26N3O5 [M-H]− 364.1872, [M-H]− Found 364.1872; IR (KBr): 3275, 1607, 1551, 1409, 1324, 1113, 1068, 1016, 842, 596, 511 cm−1; 1H-NMR (D6-DMSO): δ 7.49 (d, J = 8 Hz, 2H, Ar), 6.85 (d, J = 8 Hz, 2H, Ar), 3.91–3.86 (m, 1H, CH-N), 3.70 (s, 3H, O-CH3), 2.90 (t, J = 8 Hz, 2H, CH2-N), 2.71 (m, 2H, CH2-CONH), 2.59 (t, J = 8 Hz, CH2-N), 1.81 (s, 3H, CH3) 1.64–1.22 (m, 8H, CH2); 13C-NMR (D2O): δ 167.98 (C=O), 162.44 (C=O), 159.36 (C=O), 145.44 (Ar-OCH3), 118.71 (Ar-NH), 112.75 (Ar), 103.33 (Ar), 44.44 (O-CH3), 43.83 (CH-N), 43.78 (CH2-N), 37.76 (CH2-N), 36.31 (CH2), 32.13 (CH2), 28.14 (CH2), 20.50 (CH3).


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
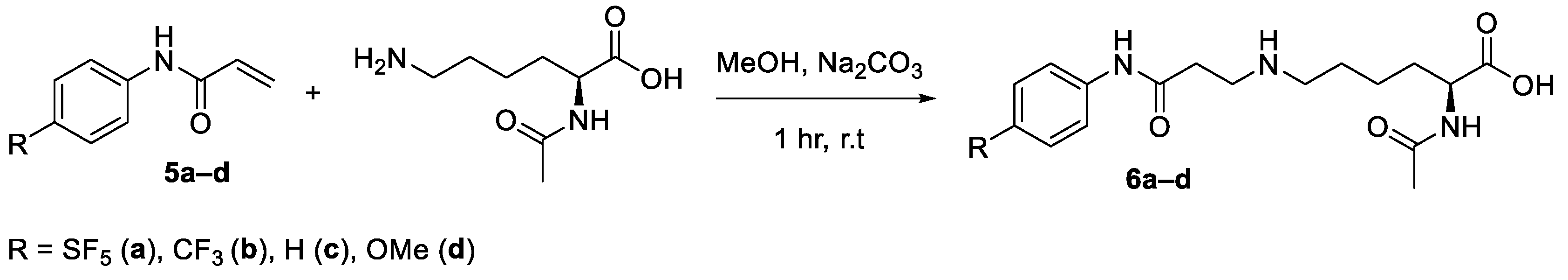{kind=link}
{kind=link}