

OutSplice: A Novel Tool for the Identification of Tumor-Specific Alternative Splicing Events
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
2.2. Computational Resources
2.3. Genome Index Building and Alignment
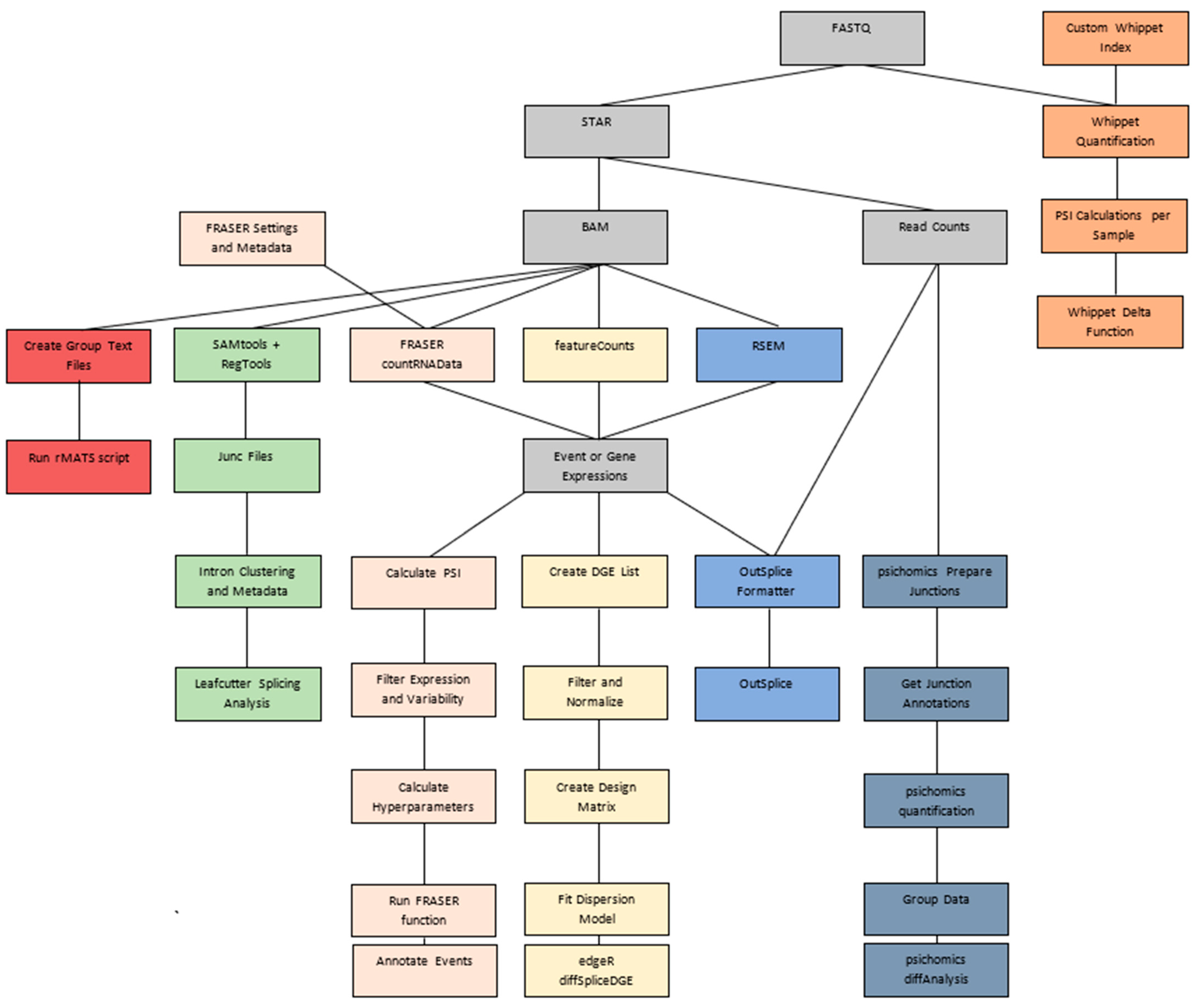
2.4. Algorithm Data Formatting Pipelines
2.4.1. OutSplice Pipeline
2.4.2. edgeR Pipeline
2.4.3. LeafCutter/LeafCutterMD Pipeline
2.4.4. Psichomics Pipeline
2.4.5. rMATs Pipeline
2.4.6. Whippet Pipeline
2.4.7. FRASER Pipeline
2.4.8. Simulated Data Creation
3. Results and Discussion
3.1. OutSplice
3.2. edgeR
3.3. LeafCutter
3.4. Psichomics
3.5. rMATS
3.6. Whippet
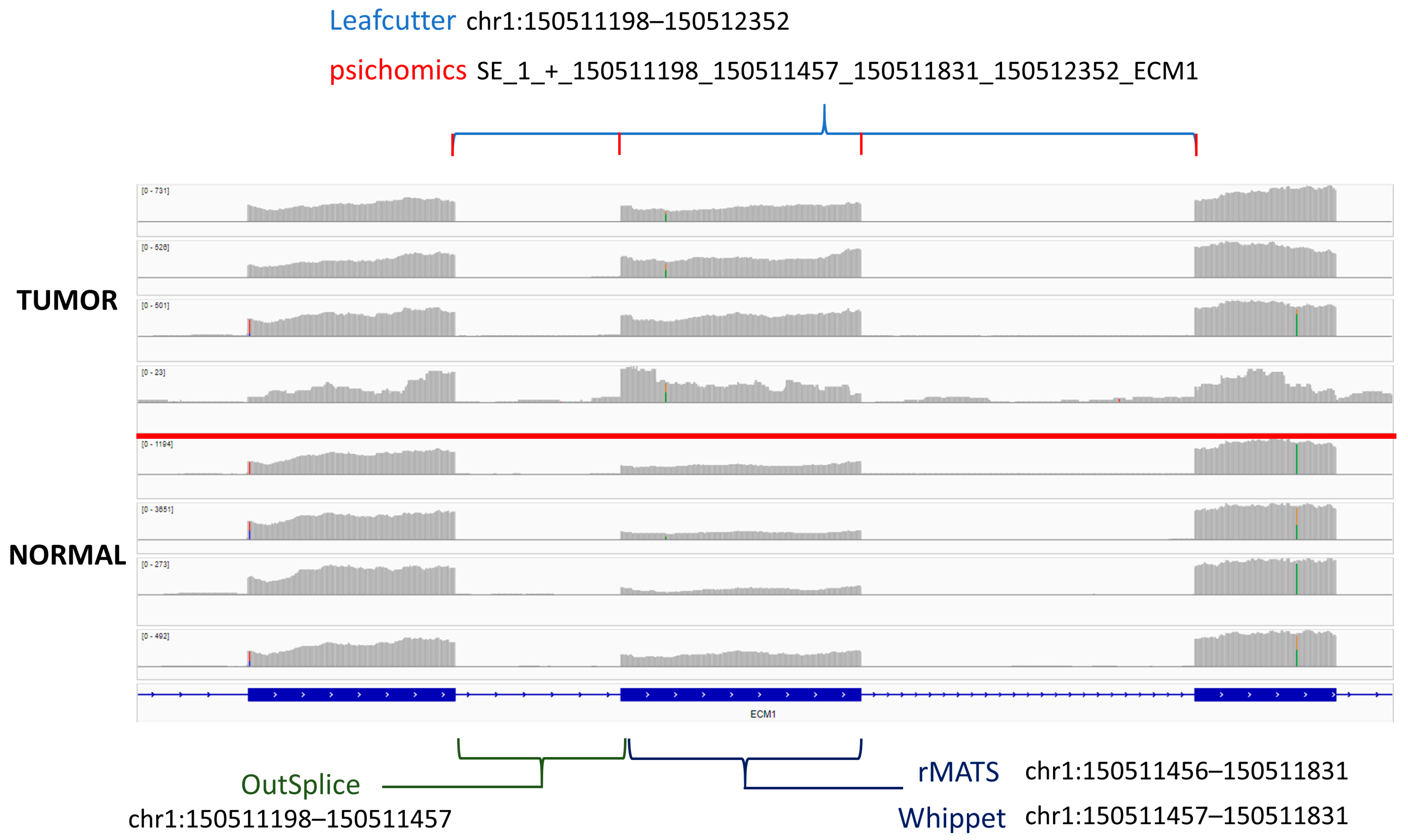
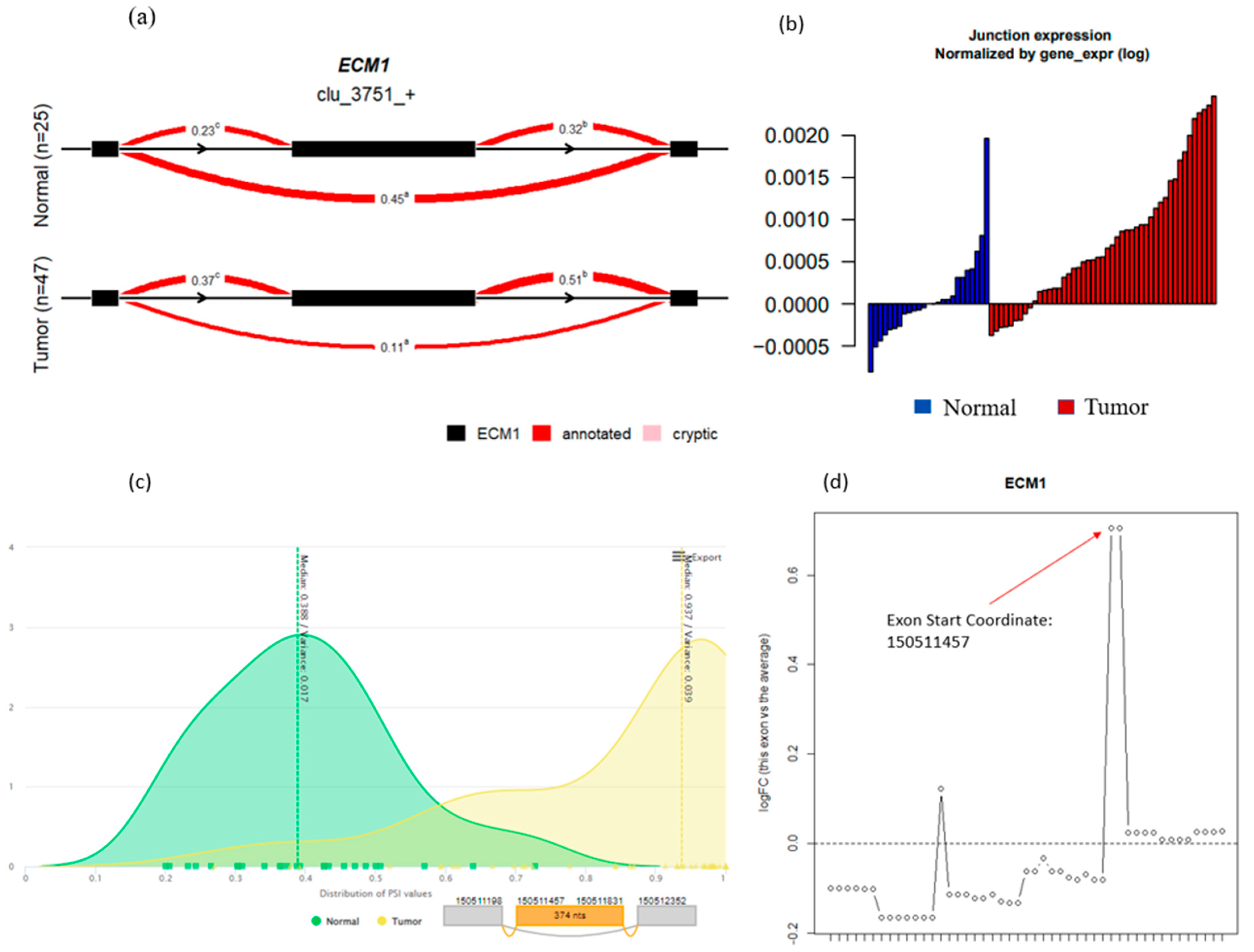
3.7. Gene Overlap and Algorithm Comparisons
3.8. Outlier Analysis and Comparison
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Screaton, G.R.; Bell, M.V.; Jackson, D.G.; Cornelis, F.B.; Gerth, U.; Bell, J.I. Genomic Structure of DNA Encoding the Lymphocyte Homing Receptor Cd44 Reveals at Least 12 Alternatively Spliced Exons. Proc. Natl. Acad. Sci. USA 1992, 89, 12160–12164. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Tarin, D. Significance of Cd44 Gene-Products for Cancer-Diagnosis and Disease Evaluation. Lancet 1992, 340, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef]
- Venkataramany, A.S.; Schieffer, K.M.; Lee, K.; Cottrell, C.E.; Wang, P.Y.; Mardis, E.R.; Cripe, T.P.; Chandler, D.S. Alternative RNA splicing defects in pediatric cancers: New insights in tumorigenesis and potential therapeutic vulnerabilities. Ann. Oncol. 2022, 33, 578–592. [Google Scholar] [CrossRef]
- Guo, T.; Sakai, A.; Afsari, B.; Considine, M.; Danilova, L.; Favorov, A.V.; Yegnasubramanian, S.; Kelley, D.Z.; Flam, E.; Ha, P.K.; et al. A Novel Functional Splice Variant of AKT3 Defined by Analysis of Alternative Splice Expression in HPV-Positive Oropharyngeal Cancers. Cancer Res. 2017, 77, 5248–5258. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; The Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef]
- Pan, Y.; Phillips, J.W.; Zhang, B.D.; Noguchi, M.; Kutschera, E.; McLaughlin, J.; Nesterenko, P.A.; Mao, Z.; Bangayan, N.J.; Wang, R.; et al. IRIS: Discovery of cancer immunotherapy targets arising from pre-mRNA alternative splicing. Proc. Natl. Acad. Sci. USA 2023, 120, e2221116120. [Google Scholar] [CrossRef]
- Wood, M.A.; Weeder, B.R.; David, J.K.; Nellore, A.; Thompson, R.F. Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival. Genome Med. 2020, 12, 33. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.T.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.D.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma (vol 547, pg 217, 2017). Nature 2018, 555, 402. [Google Scholar] [CrossRef]
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Huang, Y.; Du, Y.; Orellana, C.F.; Singh, D.; Johnson, A.R.; Monroy, A.; Kuan, P.F.; Hammond, S.M.; Makowski, L.; et al. DiffSplice: The genome-wide detection of differential splicing events with RNA-seq. Nucleic. Acids Res. 2013, 41, e39. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Hartley, S.W.; Mullikin, J.C. Detection and visualization of differential splicing in RNA-Seq data with JunctionSeq. Nucleic. Acids Res. 2016, 44, e127. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic. Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Li, Y.I.; Knowles, D.A.; Humphrey, J.; Barbeira, A.N.; Dickinson, S.P.; Im, H.K.; Pritchard, J.K. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 2018, 50, 151–158. [Google Scholar] [CrossRef]
- Wang, Q.; Rio, D.C. JUM is a computational method for comprehensive annotation-free analysis of alternative pre-mRNA splicing patterns. Proc. Natl. Acad. Sci. USA 2018, 115, E8181–E8190. [Google Scholar] [CrossRef]
- Saraiva-Agostinho, N.; Barbosa-Morais, N.L. psichomics: Graphical application for alternative splicing quantification and analysis. Nucleic. Acids Res. 2019, 47, e7. [Google Scholar] [CrossRef]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef]
- Sterne-Weiler, T.; Weatheritt, R.J.; Best, A.J.; Ha, K.C.H.; Blencowe, B.J. Efficient and Accurate Quantitative Profiling of Alternative Splicing Patterns of Any Complexity on a Laptop. Mol. Cell 2018, 72, 187–200.e6. [Google Scholar] [CrossRef]
- Vaquero-Garcia, J.; Barrera, A.; Gazzara, M.R.; Gonzalez-Vallinas, J.; Lahens, N.F.; Hogenesch, J.B.; Lynch, K.W.; Barash, Y. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 2016, 5, e11752. [Google Scholar] [CrossRef]
- Denti, L.; Rizzi, R.; Beretta, S.; Vedova, G.D.; Previtali, M.; Bonizzoni, P. ASGAL: Aligning RNA-Seq data to a splicing graph to detect novel alternative splicing events. BMC Bioinform. 2018, 19, 444. [Google Scholar] [CrossRef]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Ratsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar] [CrossRef]
- Ryan, M.C.; Cleland, J.; Kim, R.; Wong, W.C.; Weinstein, J.N. SpliceSeq: A resource for analysis and visualization of RNA-Seq data on alternative splicing and its functional impacts. Bioinformatics 2012, 28, 2385–2387. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Guo, T.; Xu, G.; Sakai, A.; Ren, S.; Fukusumi, T.; Ando, M.; Sadat, S.; Saito, Y.; Khan, Z.; et al. Characterization of Alternative Splicing Events in HPV-Negative Head and Neck Squamous Cell Carcinoma Identifies an Oncogenic DOCK5 Variant. Clin. Cancer Res. 2018, 24, 5123–5132. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Guo, T.; Sakai, A.; Ren, S.; Fukusumi, T.; Ando, M.; Sadat, S.; Saito, Y.; Califano, J.A. A novel splice variant of LOXL2 promotes progression of human papillomavirus-negative head and neck squamous cell carcinoma. Cancer 2020, 126, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, A.; Laiho, A.; Venalainen, M.S.; McGlinchey, A.J.; Wang, N.; Elo, L.L. Systematic evaluation of differential splicing tools for RNA-seq studies. Brief. Bioinform. 2020, 21, 2052–2065. [Google Scholar] [CrossRef]
- Jiang, M.H.; Zhang, S.Y.; Yin, H.X.; Zhuo, Z.Y.; Meng, G.Y. A comprehensive benchmarking of differential splicing tools for RNA-seq analysis at the event level. Brief. Bioinform. 2023, 24, bbad121. [Google Scholar] [CrossRef]
- Mertes, C.; Scheller, I.F.; Yepez, V.A.; Celik, M.H.; Liang, Y.; Kremer, L.S.; Gusic, M.; Prokisch, H.; Gagneur, J. Detection of aberrant splicing events in RNA-seq data using FRASER. Nat. Commun. 2021, 12, 529. [Google Scholar] [CrossRef]
- Jenkinson, G.; Li, Y.I.; Basu, S.; Cousin, M.A.; Oliver, G.R.; Klee, E.W. LeafCutterMD: An algorithm for outlier splicing detection in rare diseases. Bioinformatics 2020, 36, 4609–4615. [Google Scholar] [CrossRef]
- Guo, T.; Gaykalova, D.A.; Considine, M.; Wheelan, S.; Pallavajjala, A.; Bishop, J.A.; Westra, W.H.; Ideker, T.; Koch, W.M.; Khan, Z.; et al. Characterization of functionally active gene fusions in human papillomavirus related oropharyngeal squamous cell carcinoma. Int. J. Cancer 2016, 139, 373–382. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. Gencode 2021. Nucleic. Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Veeneman, B.A.; Shukla, S.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Nesvizhskii, A.I. Two-pass alignment improves novel splice junction quantification. Bioinformatics 2016, 32, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Ghosh, D. Discrete nonparametric algorithms for outlier detection with genomic data. J. Biopharm. Stat. 2010, 20, 193–208. [Google Scholar] [CrossRef]
- Ochs, M.F.; Farrar, J.E.; Considine, M.; Wei, Y.; Meshinchi, S.; Arceci, R.J. Outlier Analysis and Top Scoring Pair for Integrated Data Analysis and Biomarker Discovery. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 520–532. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic. Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Cotto, K.C.; Feng, Y.-Y.; Ramu, A.; Richters, M.; Freshour, S.L.; Skidmore, Z.L.; Xia, H.; McMichael, J.F.; Kunisaki, J.; Campbell, K.M.; et al. RegTools: Integrated analysis of genomic and transcriptomic data for the discovery of splice-associated variants in cancer. Nat. Commun. 2023, 14, 1589. [Google Scholar] [CrossRef]
- Frazee, A.C.; Jaffe, A.E.; Langmead, B.; Leek, J.T. Polyester: Simulating RNA-seq datasets with differential transcript expression. Bioinformatics 2015, 31, 2778–2784. [Google Scholar] [CrossRef]
- Tapial, J.; Ha, K.C.H.; Sterne-Weiler, T.; Gohr, A.; Braunschweig, U.; Hermoso-Pulido, A.; Quesnel-Vallieres, M.; Permanyer, J.; Sodaei, R.; Marquez, Y.; et al. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017, 27, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; pp. 1–212. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zambo, K.D.A.; Zamuner, F.T.; Ou, T.; Hopkins, C.; Kelley, D.Z.; Wulf, H.A.; Winkler, E.; Erbe, R.; Danilova, L.; et al. Chromatin structure regulates cancer-specific alternative splicing events in primary HPV-related oropharyngeal squamous cell carcinoma. Epigenetics 2020, 15, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.Z.; Flam, E.L.; Guo, T.; Danilova, L.V.; Zamuner, F.T.; Bohrson, C.; Considine, M.; Windsor, E.J.; Bishop, J.A.; Zhang, C.; et al. Functional characterization of alternatively spliced GSN in head and neck squamous cell carcinoma. Transl. Res. 2018, 202, 109–119. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene with Significant Splicing Event(s) | Description | Event Types Found | edgeR | LeafCutter | OutSplice | Psichomics | rMATS | Whippet |
|---|---|---|---|---|---|---|---|---|
| ECM1 | Extracellular Matrix Protein 1 | Skipping and Alternative 5′ Splice Sites | X | X | X | X | X | |
| COL6A3 | Collagen type VI alpha-3 chain | Skipping and Mutually Exclusive Exons | X | X | X | X | X | |
| KIAA1217 | Embryonic skeletal system development | Skipping, Alternative First Exon, Retained Intron, and Mutually Exclusive Exons | X | X | X | X | X | |
| HDAC9 | Histone deacetylase 9 | Skipping, Alternative First Exon, Tandam Start Site, and Mutually Exclusive Exons | X | X | X | X | X | |
| MBNL1 | Muscleblind-like splicing regulator 1 | Skipping and Mutually Exclusive Exons | X | X | X | X | X | |
| VPS39 | VPS39 subunit of the HOPS complex | Skipping and Tandam Start Site | X | X | X | X | X | |
| PLEKHG1 | Pleckstrin homology and RhoGEF domain containing G1 | Skipping, Insertion, Tandam Start Site, and Mutually Exclusive Exons | X | X | X | X | X | X |
| ITGB4 | Integrin subunit beta 4 | Skipping | X | X | X | X | X | X |
| PTPN6 | Protein tyrosine phosphatase non-receptor type 6 | Skipping, Alternative Acceptor, Alternative First Exon, Alternative 3′ Start Site, Mutually Exclusive Exons, and Retained Introns | X | X | X | X | X | |
| MTMR1 | Myotubularin-related protein 1 | Skipping and Mutually Exclusive Exons | X | X | X | X | X | |
| PARD3 | Par-3 family cell polarity regulator | Skipping, Alternative 5′ Splice Site, Alternative First Exon, and Mutually Exclusive Exons | X | X | X | X | X | |
| NUMA1 | Nuclear mitotic apparatus protein 1 | Alternative First Exon, Tandem Transcription Start Site, Retained Intron, and Mutually Exclusive Exons | X | X | X | X | X | |
| RABGAP1L | RAB GTPase-activating protein 1 | Skipping and Insertion | X | X | X | X | X | |
| MDM2 | Proto-oncogene | Skipping, Alternative First Exon, and Mutually Exclusive Exons | X | X | X | X | X | |
| MCM7 | Minichromosome maintenance complex component 7 | Skipping, Alternative First Exon, and Retained Intron | X | X | X | X | X | |
| MEI1 | Meiotic double-stranded break formation protein 1 | Skipping, Insertion, and Deletion Events | X | X | X | X | X | |
| FCGR2B | FC gamma receptor IIb | Skipping and Retained Intron | X | X | X | X | X |
| Identified Genes | Identified + Significant Genes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| True Positive | Total Identified | Sensitivity | Specificity | FDR | True Positive | Total Identified + Significant | Sensitivity | Specificity | FDR | |
| edgeR | 200 | 1014 | 1 | 0.03 | 0.8 | 192 | 265 | 0.96 | 0.91 | 0.28 |
| LeafCutter | 185 | 213 | 0.93 | 0.97 | 0.13 | 181 | 194 | 0.91 | 0.98 | 0.07 |
| OutSplice | 91 | 366 | 0.46 | 0.67 | 0.75 | 78 | 336 | 0.39 | 0.69 | 0.77 |
| psichomics | 108 | 123 | 0.54 | 0.98 | 0.12 | 102 | 115 | 0.51 | 0.98 | 0.11 |
| rMATS | 149 | 392 | 0.75 | 0.70 | 0.62 | 115 | 120 | 0.58 | 0.99 | 0.04 |
| Whippet | 200 | 972 | 1 | 0.07 | 0.79 | 77 | 79 | 0.39 | 1 | 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bendik, J.; Kalavacherla, S.; Webster, N.; Califano, J.; Fertig, E.J.; Ochs, M.F.; Carter, H.; Guo, T. OutSplice: A Novel Tool for the Identification of Tumor-Specific Alternative Splicing Events. BioMedInformatics 2023, 3, 853-868. https://doi.org/10.3390/biomedinformatics3040053
Bendik J, Kalavacherla S, Webster N, Califano J, Fertig EJ, Ochs MF, Carter H, Guo T. OutSplice: A Novel Tool for the Identification of Tumor-Specific Alternative Splicing Events. BioMedInformatics. 2023; 3(4):853-868. https://doi.org/10.3390/biomedinformatics3040053
Chicago/Turabian StyleBendik, Joseph, Sandhya Kalavacherla, Nicholas Webster, Joseph Califano, Elana J. Fertig, Michael F. Ochs, Hannah Carter, and Theresa Guo. 2023. "OutSplice: A Novel Tool for the Identification of Tumor-Specific Alternative Splicing Events" BioMedInformatics 3, no. 4: 853-868. https://doi.org/10.3390/biomedinformatics3040053