
5-Chlorocoumaranone-Conjugates as Chemiluminescent Protecting Groups (CLPG) and Precursors to Fluorescent Protecting Groups (FPG)

 , , and
, , and 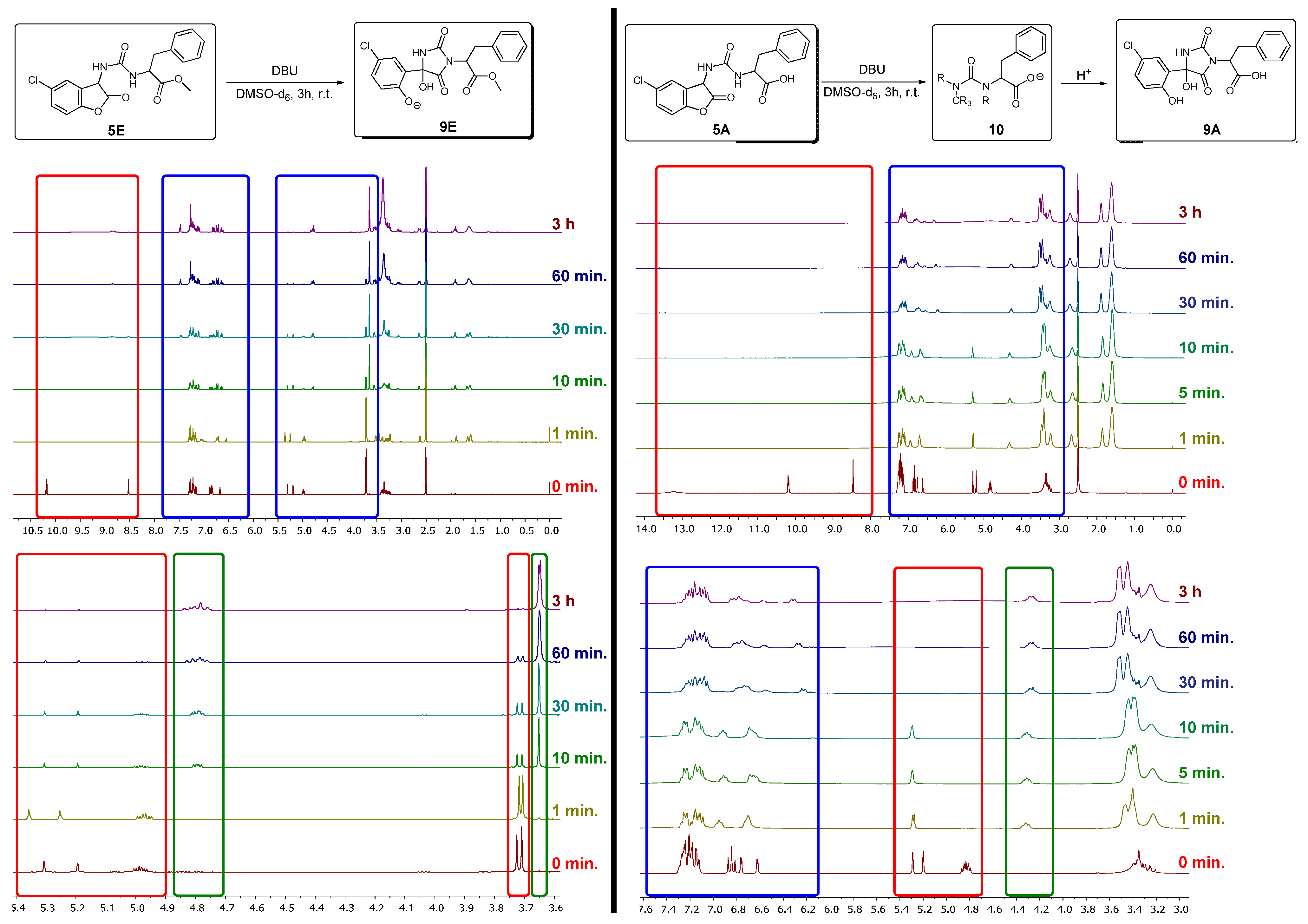{kind=link}
{kind=link}
{kind=link}
{kind=link}
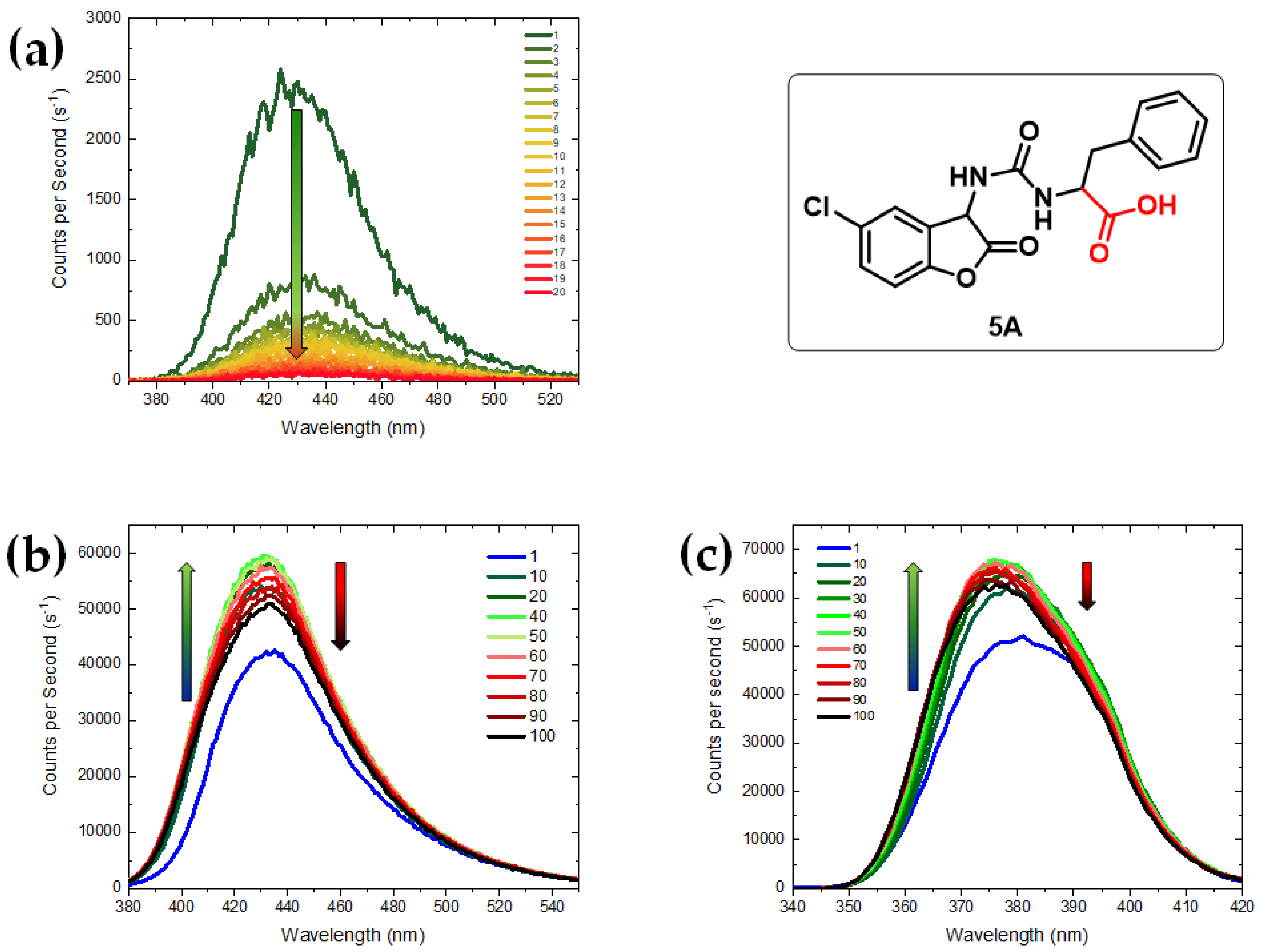{kind=link}
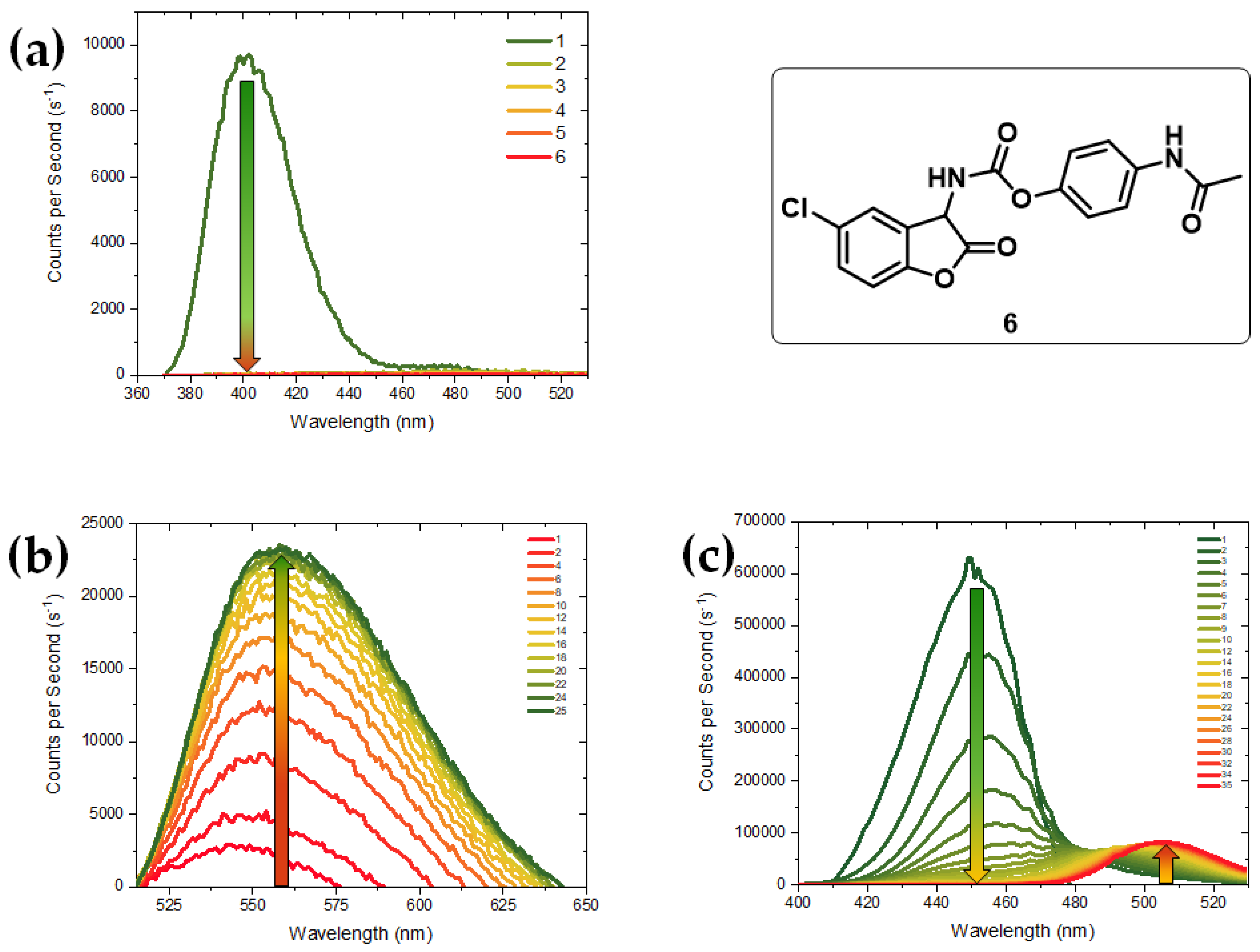{kind=link}
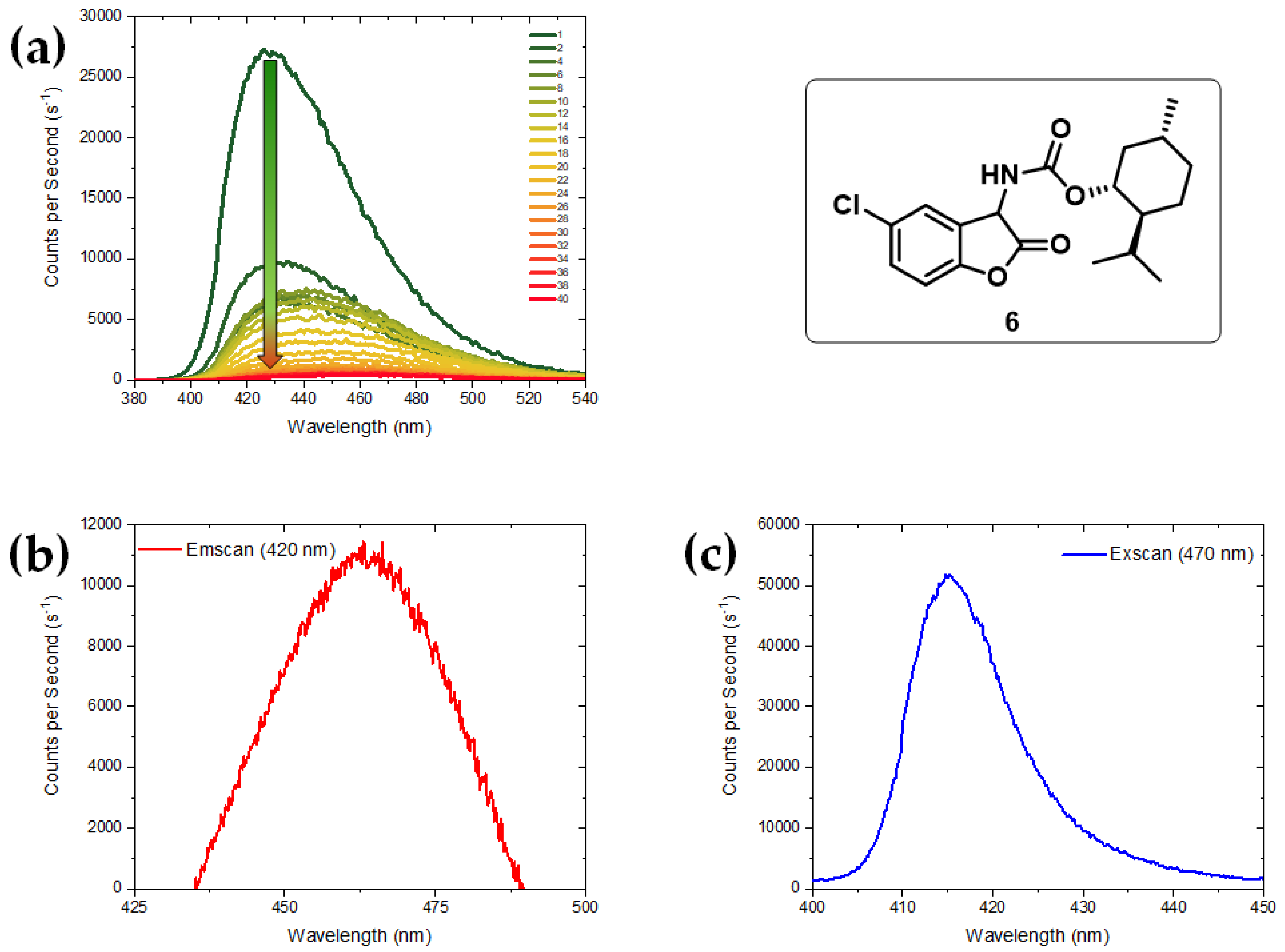{kind=link}
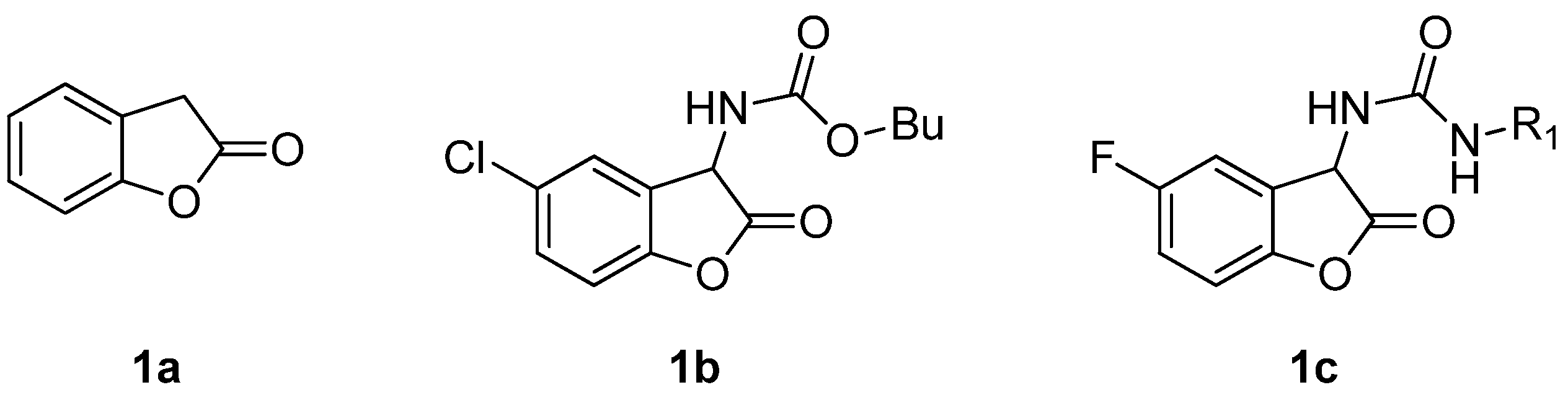{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
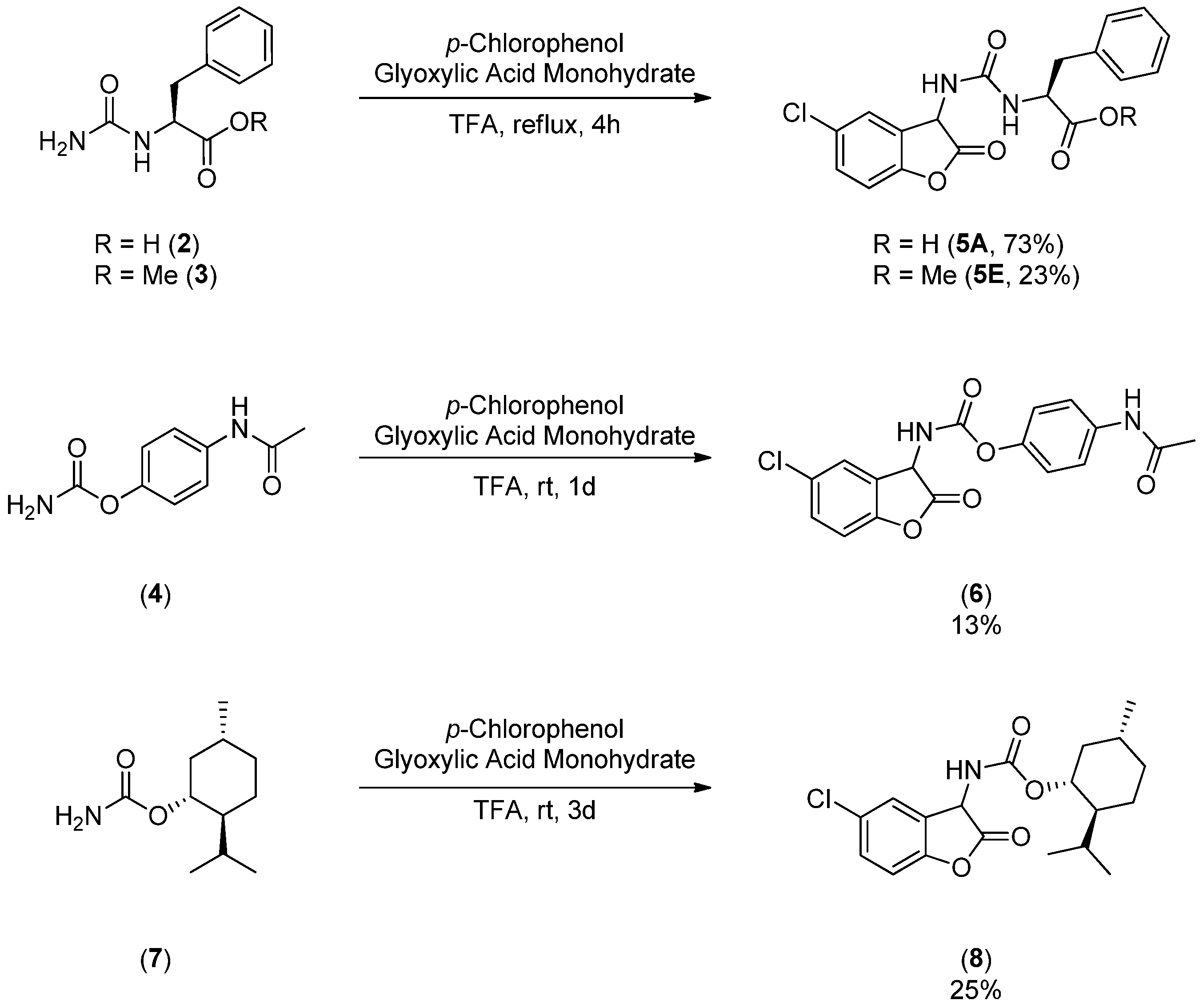
3.1. Syntheses of Coumaranones
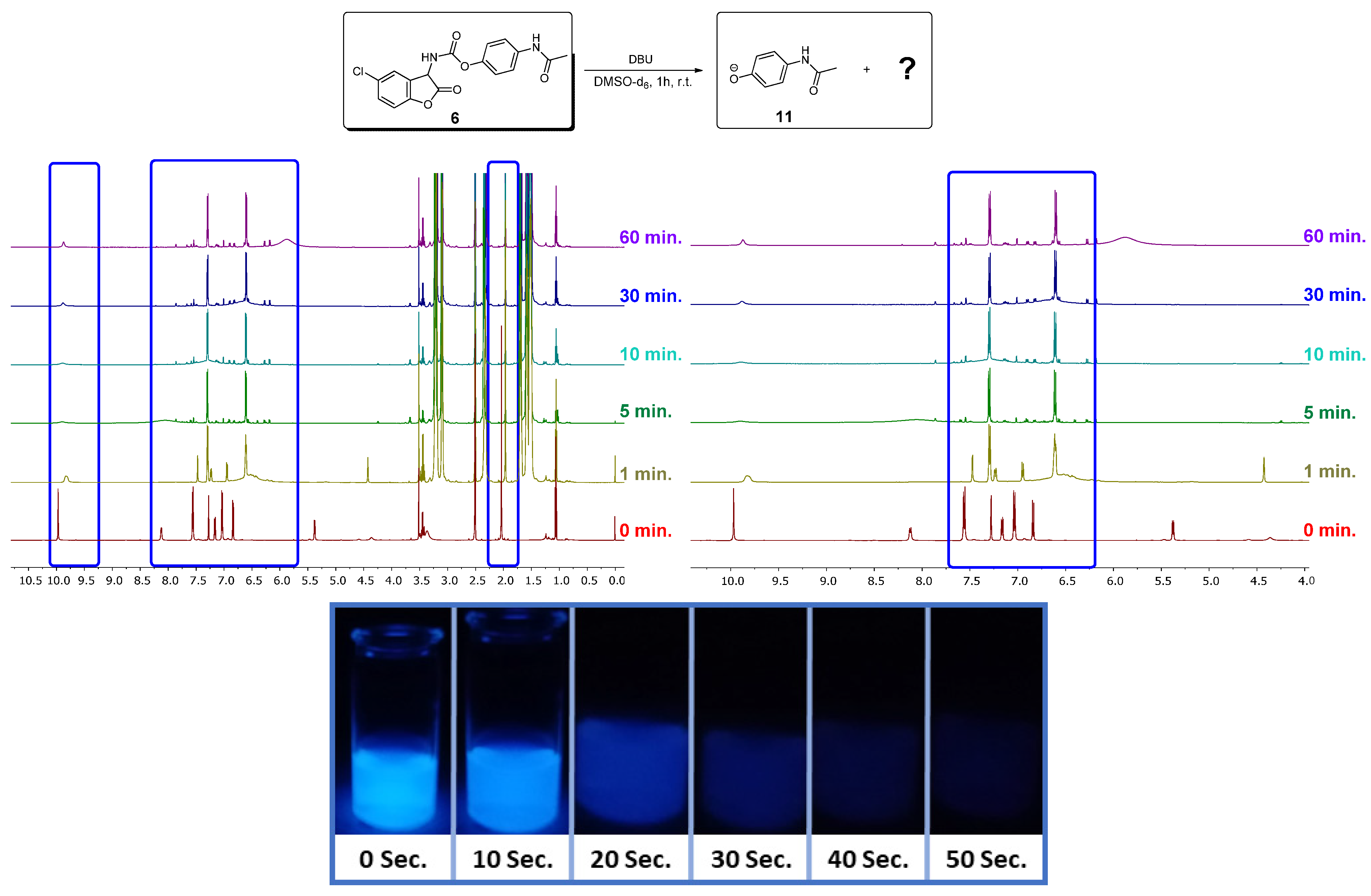
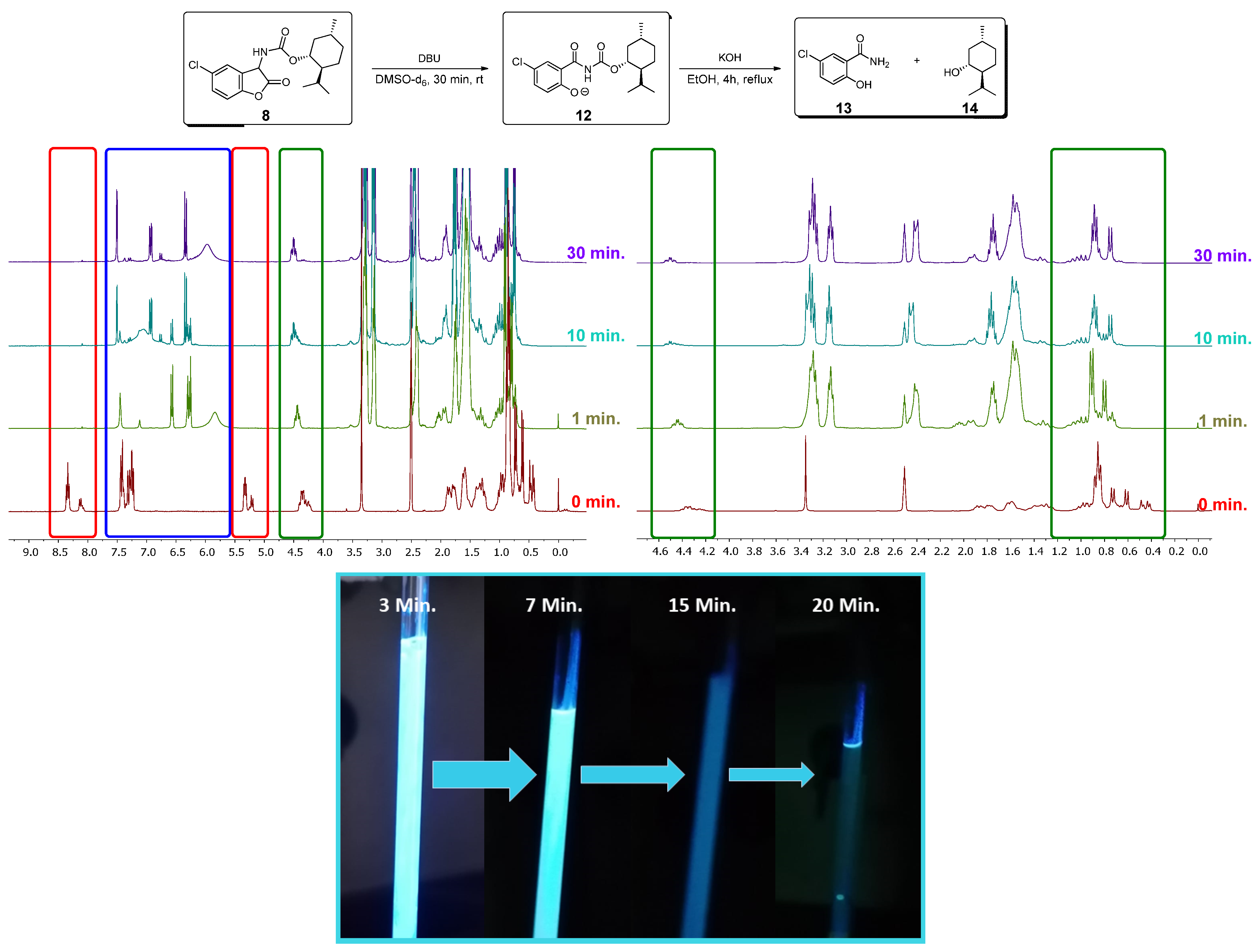
3.2. NMR Investigations of the Cleavage of the Coumaranones
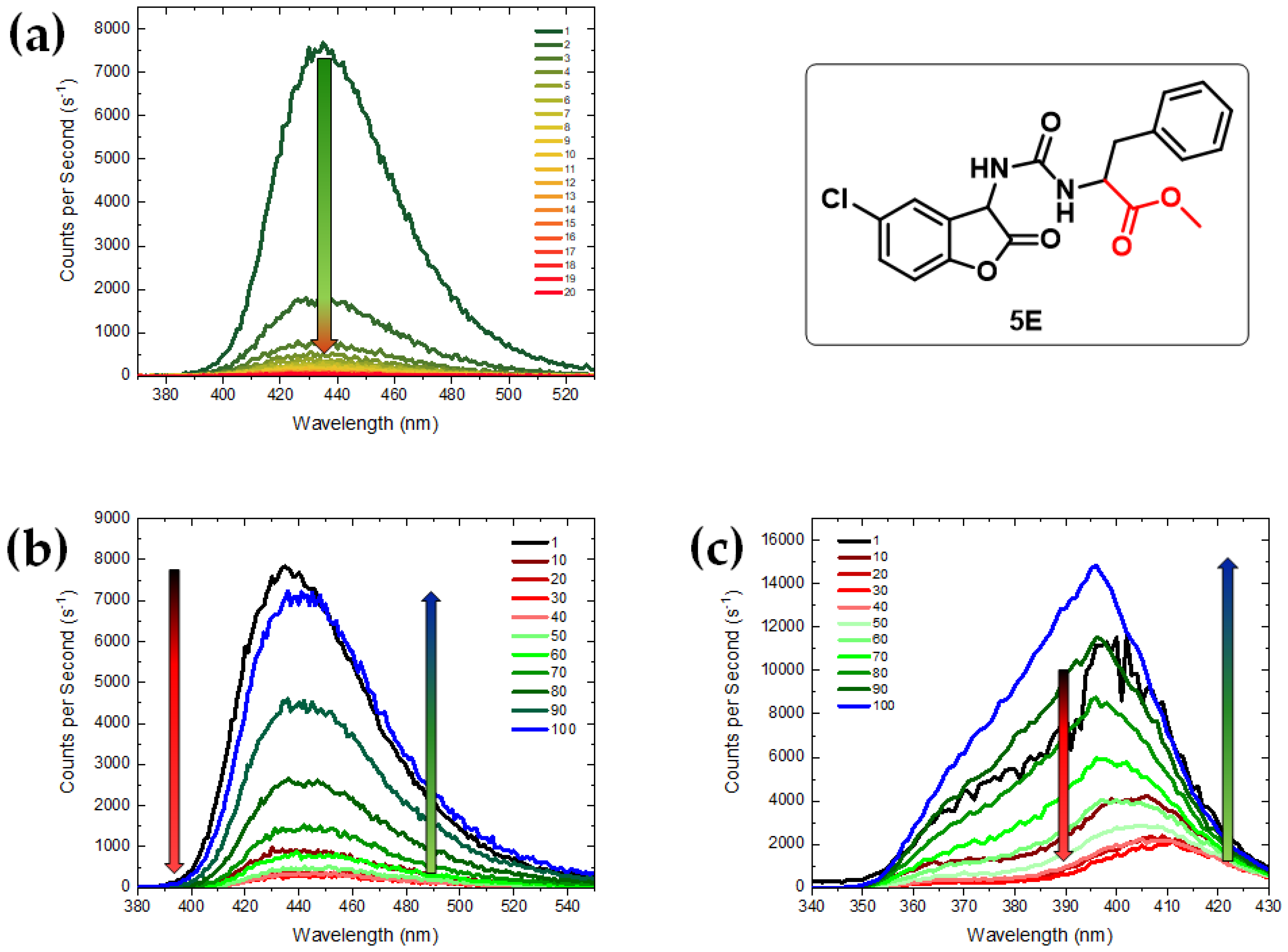
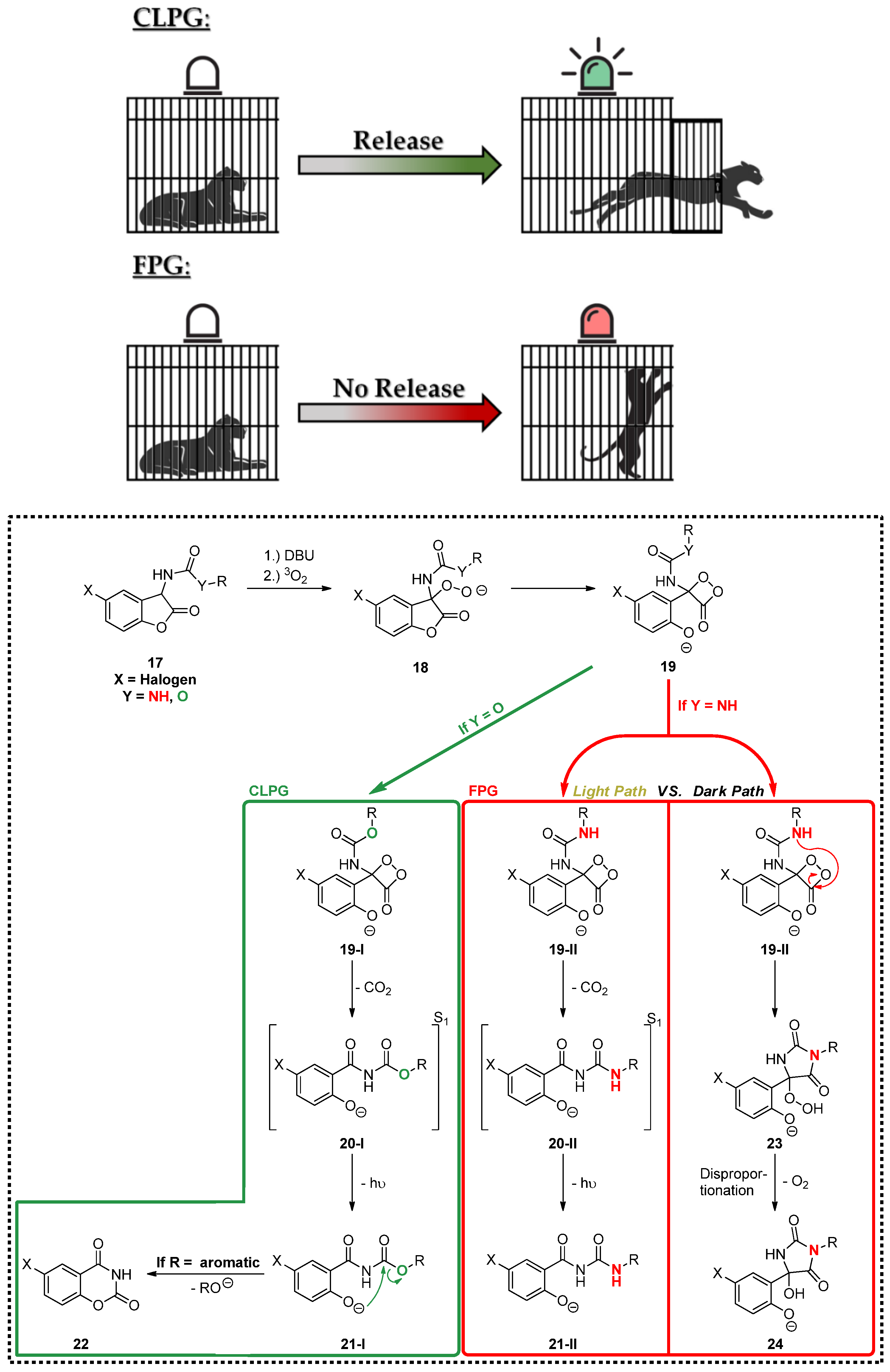
3.3. Chemiluminescence
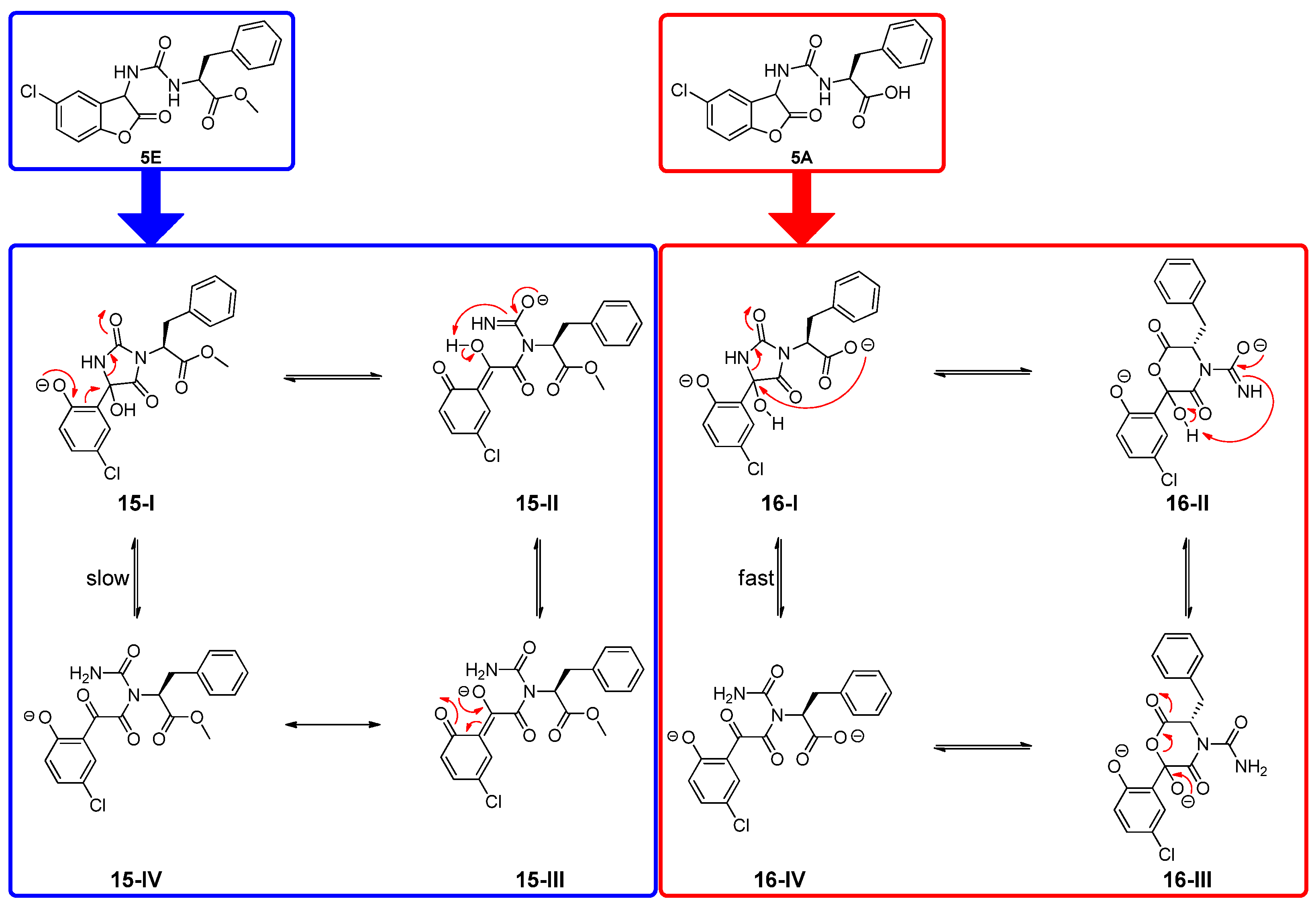
3.4. Mechanistical Considerations for 5A and 5E
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jarowicki, K.; Kocienski, P. Protecting groups. J. Chem. Soc.—Perkin Trans. 1 2001, 2109–2135. [Google Scholar] [CrossRef]
- Kocienski, P. Protecting Groups: Foundations of Organic Chemistry, 3rd ed.; Thieme: Stuttgart, Germany, 2005. [Google Scholar]
- Vidal, S. Protecting Groups: Strategies and Applications in Carbohydrate Chemistry, 1st ed.; Wiley-VCH: Weinheim, Germany; New York, NY, USA, 2019. [Google Scholar]
- Wong, C.H.; Ye, X.S.; Zhang, Z.Y. Assembly of oligosaccharide libraries with a designed building block and an efficient orthogonal protection-deprotection strategy. J. Am. Chem. Soc. 1998, 120, 7137–7138. [Google Scholar] [CrossRef]
- Schelhaas, M.; Waldmann, H. Protecting group strategies in organic synthesis. Angew. Chem. Int. Ed. 1996, 35, 2056–2083. [Google Scholar] [CrossRef]
- Klan, P.; Solomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef]
- Falvey, D.E.; Sundararajan, C. Photoremovable protecting groups based on electron transfer chemistry. Photochem. Photobiol. Sci. 2004, 3, 831–838. [Google Scholar] [CrossRef]
- Pelliccioli, A.P.; Wirz, J. Photoremovable protecting groups: Reaction mechanisms and applications. Photochem. Photobiol. Sci. 2002, 1, 441–458. [Google Scholar] [CrossRef]
- Bochet, C.G. Photolabile protecting groups and linkers. J. Chem. Soc.—Perkin Trans. 1 2002, 125–142. [Google Scholar] [CrossRef]
- Herrmann, A. Using photolabile protecting groups for the controlled release of bioactive volatiles. Photochem. Photobiol. Sci. 2012, 11, 446–459. [Google Scholar] [CrossRef]
- Bochet, C.G. Orthogonal photolysis of protecting groups. Angew. Chem. Int. Ed. 2001, 40, 2071–2073. [Google Scholar] [CrossRef]
- Blanc, A.; Bochet, C.G. Wavelength-controlled orthogonal photolysis of protecting groups. J. Org. Chem. 2002, 67, 5567–5577. [Google Scholar] [CrossRef]
- Josa-Cullere, L.; Llebaria, A. In the search for photocages cleavable with visible light: An overview of recent advances and chemical strategies. ChemPhotoChem 2021, 5, 296–314. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Hinze, O.; Görner, H.; Huchel, U.; Kropf, C.; Sundermeier, U.; Gerke, T. Aromatic aldols and 1,5-diketones as optimized fragrance photocages. Photochem. Photobiol. Sci. 2012, 11, 587–592. [Google Scholar] [CrossRef]
- Soldevilla, A.; Griesbeck, A.G. Chiral photocages based on phthalimide photochemistry. J. Am. Chem. Soc. 2006, 128, 16472–16473. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Schuster, G.B. Chemically-initiated electron exchange luminescence—New chemiluminescent reaction path for organic peroxides. J. Am. Chem. Soc. 1977, 99, 6107–6109. [Google Scholar] [CrossRef]
- Adam, W.; Kazakov, D.V.; Kazakov, V.P. Singlet-oxygen chemiluminescence in peroxide reactions. Chem. Rev. 2005, 105, 3371–3387. [Google Scholar] [CrossRef]
- Rauhut, M.M. Chemiluminescence from concerted peroxide decomposition reactions. Acc. Chem. Res. 1969, 2, 80–87. [Google Scholar] [CrossRef]
- Vacher, M.; Galván, I.F.; Ding, B.-W.; Schramm, S.; Berraud-Pache, R.; Naumov, P.; Ferré, N.; Liu, Y.-J.; Navizet, I.; Roca-Sanjuán, D.; et al. Chemi- and bioluminescence of cyclic peroxides. Chem. Rev. 2018, 118, 6927–6974. [Google Scholar] [CrossRef]
- Augusto, F.A.; Francés-Monerris, A.; Galván, I.F.; Roca-Sanjuán, D.; Bastos, E.L.; Baader, W.J.; Lindh, R. Mechanism of activated chemiluminescence of cyclic peroxides: 1,2-dioxetanes and 1,2-dioxetanones. Phys. Chem. Chem. Phys. 2017, 19, 3955–3962. [Google Scholar] [CrossRef]
- Yue, L.; Yu, L.; Xu, C.; Zhu, C.; Liu, Y. Quantum yields of singlet and triplet chemiexcitation of dimethyl 1,2-dioxetane: Ab initio nonadiabatic molecular dynamic simulations. Phys. Chem. Chem. Phys. 2020, 22, 11440–11451. [Google Scholar] [CrossRef]
- Lofthouse, G.J.; Suschitzky, H.; Wakefield, B.J.; Whittaker, R.A. Synthesis and chemiluminescent reactions of some 3-alkoxy-carbamoylbenzo[b]furan-2(3H)-ones. J. Chem. Soc. Perkin 1 1979, 1634–1639. [Google Scholar] [CrossRef]
- Krieg, R.; Hoffmann, B.; Weiß, D.; Biskup, C. First synthesis of highly chemiluminescent benzo[b]furan-2(3H)-ones bearing a urea substructure. Helv. Chim. Acta 2019, 102, e1800243. [Google Scholar] [CrossRef]
- Schramm, S.; Weiss, D.; Navizet, I.; Roca-Sanjuan, D.; Brandl, H.; Beckert, R.; Gorls, H. Investigations on the synthesis and chemiluminescence of novel 2-coumaranones. ARKIVOC 2013, 4, 174–188. [Google Scholar] [CrossRef]
- Schramm, S.; Navizet, I.; Naumov, P.; Nath, N.K.; Berraud-Pache, R.; Oesau, P.; Weiss, D.; Beckert, R. The light emitter of the 2-coumaranone chemiluminescence: Theoretical and experimental elucidation of a possible model for bioluminescent systems. Eur. J. Org. Chem. 2016, 2016, 678–681. [Google Scholar] [CrossRef]
- Schramm, S.; Navizet, I.; Karothu, D.P.; Oesau, P.; Bensmann, V.; Weiss, D.; Beckert, R.; Naumov, P. Mechanistic investigations of the 2-coumaranone chemiluminescence. Phys. Chem. Chem. Phys. 2017, 19, 22852–22859. [Google Scholar] [CrossRef] [PubMed]
- Schramm, S.; Ciscato, L.F.M.L.; Oesau, P.; Krieg, R.; Richter, J.F.; Navizet, I.; Roca-Sanjuan, D.; Weiss, D.; Beckert, R. Investigations on the synthesis and chemiluminescence of novel 2-coumaranones—II. ARKIVOC 2015, 5, 44–59. [Google Scholar] [CrossRef]
- Schramm, S. Die Chemilumineszenz der 2-Coumaranone: Synthese, Lumineszenzmechanismus und Applikation. Ph.D. Thesis, Friedrich-Schiller-Universität, Jena, Germany, 2016. [Google Scholar]
- Chegaev, K.Y.; Kravchenko, A.N.; Lebedev, O.V.; Strelenko, Y.A. New functional glycoluril derivatives. Mendeleev Commun. 2001, 11, 32–33. [Google Scholar] [CrossRef]
- Nicolas, I.; Jeannin, O.; Pichon, D.; Fourmigué, M. Dibromohydantoins as halogen bond (XB) donors: A route toward the introduction of chirality in halogen bonded systems. CrystEngComm 2016, 18, 9325–9333. [Google Scholar] [CrossRef]
- Graf, R. Umsetzungen mit N-Carbonyl-Sulfonamidsäure-chlorid, II. Alkohole und Phenole. Chem. Ber. 1963, 96, 56–67. [Google Scholar] [CrossRef]
- Konnert, L.; Lamaty, F.; Martinez, J.; Colacino, E. Recent Advances in the Synthesis of Hydantoins: The State of the Art of a Valuable Scaffold. Chem. Rev. 2017, 117, 13757–13809. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lippold, T.; Griesbeck, A.G.; Herzhoff, R.; Wickleder, M.S.; Straub, L.; Flosbach, N.T. 5-Chlorocoumaranone-Conjugates as Chemiluminescent Protecting Groups (CLPG) and Precursors to Fluorescent Protecting Groups (FPG). Photochem 2023, 3, 373-389. https://doi.org/10.3390/photochem3030023
Lippold T, Griesbeck AG, Herzhoff R, Wickleder MS, Straub L, Flosbach NT. 5-Chlorocoumaranone-Conjugates as Chemiluminescent Protecting Groups (CLPG) and Precursors to Fluorescent Protecting Groups (FPG). Photochem. 2023; 3(3):373-389. https://doi.org/10.3390/photochem3030023
Chicago/Turabian StyleLippold, Tim, Axel G. Griesbeck, Robert Herzhoff, Mathias S. Wickleder, Laura Straub, and Niko T. Flosbach. 2023. "5-Chlorocoumaranone-Conjugates as Chemiluminescent Protecting Groups (CLPG) and Precursors to Fluorescent Protecting Groups (FPG)" Photochem 3, no. 3: 373-389. https://doi.org/10.3390/photochem3030023