
Catastrophic Antiphospholipid Syndrome: A Review

 , ,
, ,
Abstract
:1. Introduction
2. The Complexity of CAPS
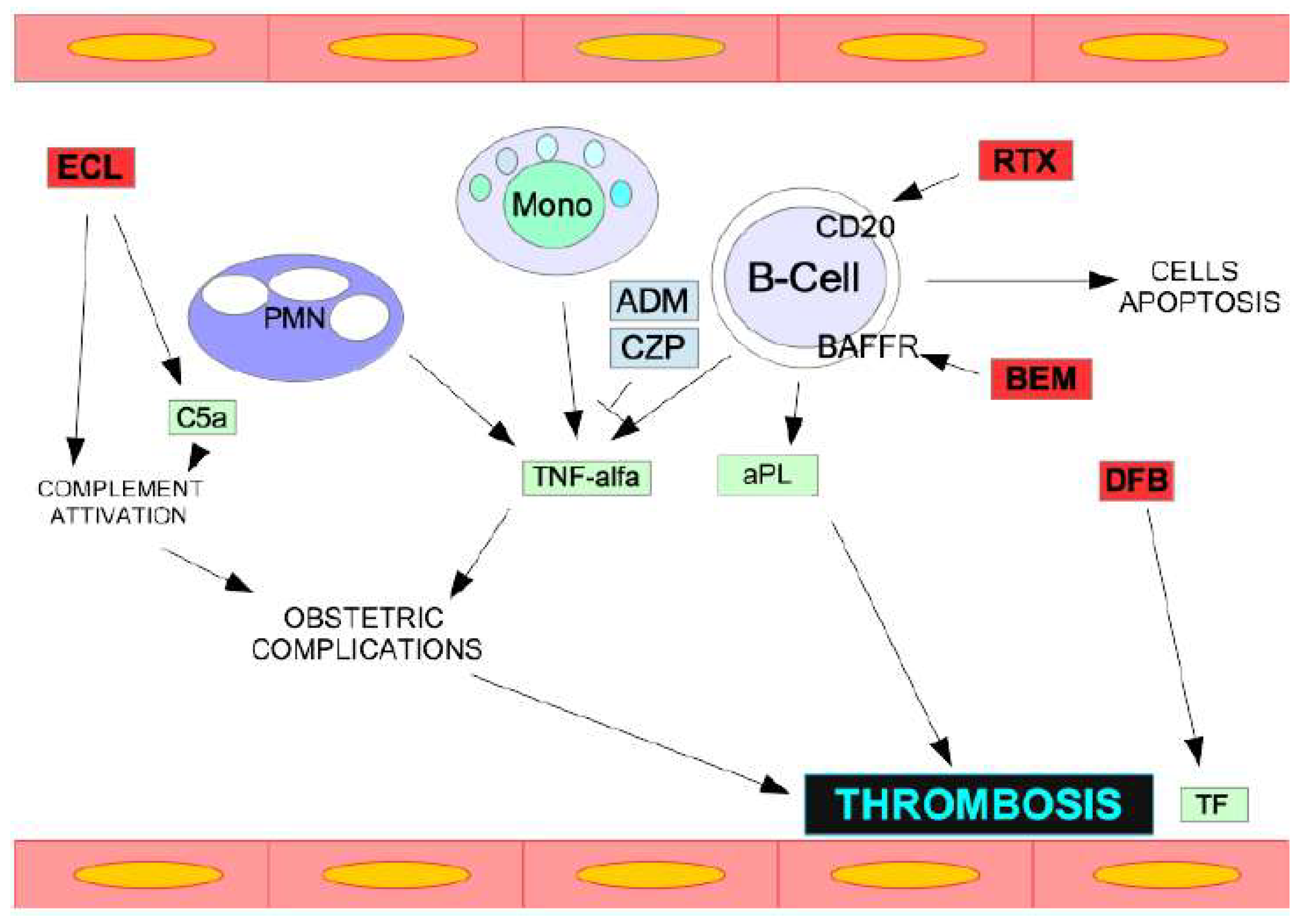
3. Pathophysiology and Mechanisms of CAPS
4. Not a Simple Diagnosis
4.1. Definite CAPS
- Involvement of three organs, systems, or tissues.
- Manifestations appear at the same time or over the course of one week.
- Small vessel occlusion is confirmed histologically in at least one organ or tissue.
- The presence of aPL (anticardiolipin antibodies, anti-beta2-glycoprotein I antibodies, and/or lupus anticoagulant) is documented twice, at least 12 weeks apart.
4.2. Probable CAPS
- Only two organs or tissue are involved.
- Despite anticoagulation, a third event manifests between one week and one month after presentation.
- No histologic evidence.
- No laboratory confirmation of aPL.
5. Differential Diagnosis
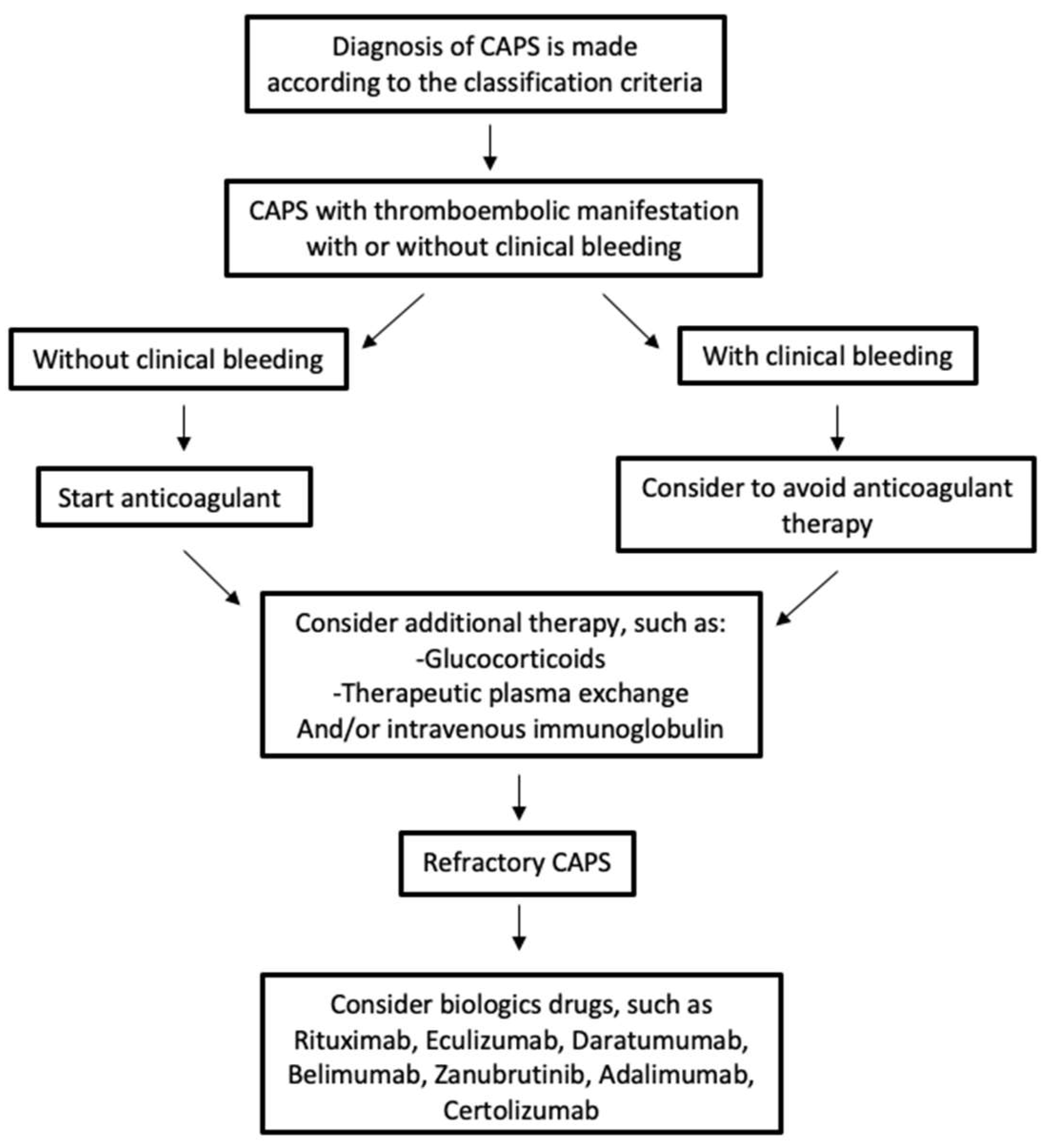
6. Management and Therapy of CAPS
7. Rituximab and Eculizumab in Refractory CAPS
8. CAPS during Pregnancy
9. CAPS in Children
10. Recurrence Risk
11. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.W.M.; De Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.A.; Gharavi, A.E.; Koike, T.; Lockshin, M.D.; Branch, D.W.; Piette, J.-C.; Brey, R.; Derksen, R.; Harris, E.N.; Hughes, G.R.V.; et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: Report of an international workshop. Arthritis Rheum. 1999, 42, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.T.; Triplett, D.A.; Alving, B.; Scharrer, I. Criteria for the diagnosis of lupus anticoagulants: An update. On behalf of the subcommittee on lupus anticoagulant/antiphospholipid antibody of the scientific and standardisation committee of the ISTH. Thromb. Haemost. 1995, 74, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Pengo, V.; Tripodi, A.; Reber, G.; Rand, J.H.; Ortel, T.L.; Galli, M.; De Groot, P.G. Update of the guidelines for lupus anticoagulant detection. Subcommittee on lupus anticoagulant/antiphospholipid antibody of the Scientific and Standardisation committee of the International Society on Thrombosis and Haemostasis. J. Thromb. Haemost. 2009, 7, 1737–1740. [Google Scholar] [CrossRef] [PubMed]
- Barbhaiya, M.; Zuily, S.; Naden, R.; Hendry, A.; Manneville, F.; Amigo, M.-C.; Amoura, Z.; Andrade, D.; Andreoli, L.; Artim-Esen, B.; et al. ACR/EULAR APS Classification Criteria Collaborators. 2023 ACR/EULAR antiphospholipid syndrome classification criteria. Ann. Rheum. Dis. 2023, 82, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Espinosa, G. Update on the catastrophic antiphospholipid syndrome and the “CAPS Registry”. Semin. Thromb. Hemost. 2012, 38, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pintó, I.; Moitinho, M.; Santacreu, I.; Shoenfeld, Y.; Erkan, D.; Espinosa, G.; Cervera, R. Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of 500 patients from the International CAPS Registry. Autoimmun. Rev. 2016, 15, 1120–1124. [Google Scholar] [CrossRef]
- Asherson, R.A. The catastrophic antiphospholipid syndrome, 1998. A review of the clinical features, possible pathogenesis and treatment. Lupus 1998, 7, S55–S62. [Google Scholar] [CrossRef]
- Asherson, R.A.; Cervera, R.; De Groot, P.G.; Erkan, D.; Boffa, M.-C.; Piette, J.-C.; Khamashta, M.A.; Shoenfeld, Y.; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic Antiphospholipid Syndrome Registry Project Group Catastrophic antiphospholipid syndrome: International consensus statement on classification criteria and treatment guidelines. Lupus 2003, 12, 530–534. [Google Scholar] [CrossRef]
- Cervera, R.; Serrano, R.; Pons-Estel, G.J.; Ceberio-Hualde, L.; Shoenfeld, Y.; De Ramón, E.; Buonaiuto, V.; Jacobsen, S.; Zeher, M.M.; Tarr, T.; et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: A multicentre prospective study of 1000 patients. Ann. Rheum. Dis. 2015, 74, 1011–1018. [Google Scholar] [CrossRef]
- Cervera, R.; Bucciarelli, S.; Plasín, M.A.; Gómez-Puerta, J.A.; Plaza, J.; Pons-Estel, G.; Shoenfeld, Y.; Ingelmo, M.; Espinos, G. Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of a series of 280 patients from the “CAPS Registry”. J. Autoimmun. 2009, 32, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Sevim, E.; Zisa, D.; Andrade, D.; Sciascia, S.; Pengo, V.; Tektonidou, M.G.; Ugarte, A.; Gerosa, M.; Belmont, H.M.; Zamorano, M.A.A.; et al. Characteristics of Patients with Antiphospholipid Antibody Positivity in the APS ACTION International Clinical Database and Repository. Arthritis Care Res. 2022, 74, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Kitchens, C.S.; Erkan, D.; Brandão, L.R.; Hahn, S.; James, A.H.; Kulkarni, R.; Pericak-Vance, M.; Vance, J.; Ortel, T.L. Thrombotic Storm Revisited: Preliminary Diagnostic Criteria Suggested by the Thrombotic Storm Study Group. Am. J. Med. 2011, 124, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Serrano, R.; Pons-Estel, G.J.; Espinosa, G.; Quintana, R.M.; Reverter, J.C.; Tassies, D.; Monteagudo, J.; Cervera, R. Long-term follow-up of antiphospholipid syndrome: Real-life experience from a single center. Lupus 2020, 29, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Taraborelli, M.; Reggia, R.; Dall’ara, F.; Fredi, M.; Andreoli, L.; Gerosa, M.; Hoxha, A.; Massaro, L.; Tonello, M.; Costedoat-Chalumeau, N.; et al. Longterm Outcome of Patients with Primary Antiphospholipid Syndrome: A Retrospective Multicenter Study. J. Rheumatol. 2017, 44, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Rodríguez-Pintó, I.; Legault, K.; Erkan, D. 16th International Congress on Antiphospholipid Antibodies Task Force Report on Catastrophic Antiphospholipid Syndrome. Lupus 2020, 29, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Stammler, R.; Nguyen, Y.; Yelnik, C.; Le Guern, V.; Lambert, M.; Paule, R.; Hachulla, E.; Mouthon, L.; Dupré, A.; Ackermann, F.; et al. Precipitating factors of catastrophic antiphospholipid syndrome: The role of anticoagulant treatment in a series of 112 patients. J. Thromb. Haemost. 2023, 21, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Piette, J.; Font, J.; Khamashta, M.A.; Shoenfeld, Y.; Camps, M.T.; Jacobsen, S.; Lakos, G.; Tincani, A.; Kontopoulou-Griva, I.; et al. Antiphospholipid syndrome: Clinical and immunologic manifestations and patterns of disease expression in a cohort of 1000 patients. Arthritis Rheum. 2002, 46, 1019–1027. [Google Scholar] [CrossRef]
- Dupré, A.; Morel, N.; Yelnik, C.; Moguelet, P.; Le Guern, V.; Stammler, R.; Nguyen, Y.; Paule, R.; Dufrost, V.; Ackermann, F.; et al. Cutaneous Involvement in Catastrophic Antiphospholipid Syndrome in a Multicenter Cohort of 65 Patients. JAMA Dermatol. 2023, 159, 62–67. [Google Scholar] [CrossRef]
- Cervera, R.; Rodríguez-Pintó, I.; Espinosa, G. The diagnosis and clinical management of the catastrophic antiphospholipid syndrome: A comprehensive review. J. Autoimmun. 2018, 92, 1–11. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Antiphospholipid syndrome: Complement activation, complement gene mutations, and therapeutic implications. J. Thromb. Haemost. 2020, 19, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Hernandez, O.-D.; Agmon-Levin, N.; Blank, M.; Asherson, R.A.; Shoenfeld, Y. The physiopathology of the catastrophic antiphospholipid (Asherson’s) syndrome: Compelling evidence. J. Autoimmun. 2009, 32, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Agmon-Levin, N.; Rosário, C.; Katz, B.-S.P.; Zandman-Goddard, G.; Meroni, P.; Cervera, R.; Stojanovich, L.; Blank, M.; Pierangeli, S.; Praprotnik, S.; et al. Ferritin in the antiphospholipid syndrome and its catastrophic variant (cAPS). Lupus 2013, 22, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Bontadi, A.; Falcinelli, E.; Giannini, S.; Marturano, A.; Tonello, M.; Hoxha, A.; Pengo, V.; Punzi, L.; Momi, S.; Gresele, P.; et al. Platelet and endothelial activation in catastrophic and quiescent antiphospholipid syndrome. Thromb. Haemost. 2013, 109, 901–908. [Google Scholar] [CrossRef] [PubMed]
- George, D.; Vasanth, L.; Erkan, D.; Bass, A.; Salmon, J.; Lockshin, M.D. Primary antiphospholipid syndrome presenting as HELLP syndrome: A clinical pathology conference held by the Division of Rheumatology at Hospital for Special Surgery. HSS J. 2007, 3, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Erkan, D. Expert Perspective: Management of Microvascular and Catastrophic Antiphospholipid Syndrome. Arthritis Rheumatol. 2021, 73, 1780–1790. [Google Scholar] [CrossRef]
- Legault, K.; Schunemann, H.; Hillis, C.; Yeung, C.; Akl, E.A.; Carrier, M.; Cervera, R.; Crowther, M.; Dentali, F.; Erkan, D.; et al. McMaster RARE-Bestpractices clinical practice guideline on diagnosis and management of the catastrophic antiphospholipid syndrome. J. Thromb. Haemost. 2018, 16, 1656–1664. [Google Scholar] [CrossRef]
- Rodríguez-Pintó, I.; Espinosa, G.; Erkan, D.; Shoenfeld, Y.; Cervera, R. The effect of triple therapy on the mortality of catastrophic anti-phospholipid syndrome patients. Rheumatology 2018, 57, 1264–1270. [Google Scholar] [CrossRef]
- Cervera, R.; Rodríguez-Pintó, I.; Colafrancesco, S.; Conti, F.; Valesini, G.; Rosário, C.; Agmon-Levin, N.; Shoenfeld, Y.; Ferrão, C.; Faria, R.; et al. 14th International Congress on Antiphospholipid Antibodies Task Force Report on Catastrophic Antiphospholipid Syndrome. Autoimmun. Rev. 2014, 13, 699–707. [Google Scholar] [CrossRef]
- Zar, T.; Kaplan, A. Predictable removal of anticardiolipin antibody by therapeutic plasma exchange (TPE) in catastrophic antiphospholipid antibody syndrome (CAPS). Clin. Nephrol. 2008, 70, 77–81. [Google Scholar] [CrossRef]
- Bucciarelli, S.; Espinosa, G.; Cervera, R.; Erkan, D.; Gómez-Puerta, J.A.; Ramos-Casals, M.; Font, J.; Asherson, R.A. Mortality in the catastrophic antiphospholipid syndrome: Causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006, 54, 2568–2576. [Google Scholar] [CrossRef] [PubMed]
- Asherson, R.A.; Cervera, R.; Piette, J.-C.; Font, J.; Lie, J.T.; Burcoglu, A.; Lim, K.; Muñoz-Rodríguez, F.J.; Levy, R.A.; Boué, F.; et al. Catastrophic Antiphospholipid Syndrome: Clinical and Laboratory Features of 50 Patients. Medicine 1998, 77, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, C.; Andronesi, A.G.; Jurcut, C.; Gherghiceanu, M.; Vornicu, A.; Burcea, F.A.; Andronesi, T.D.; Lupusoru, G.E.; Iliuta, L.; Sorohan, B.M.; et al. Successful Treatment of Catastrophic Antiphospholipid Syndrome Using Rituximab: Case Report and Review of the Literature. Medicina 2021, 57, 912. [Google Scholar] [CrossRef] [PubMed]
- Andrew, M.; Schmidt, B. Use of Heparin in Newborn Infants. Semin. Thromb. Hemost. 1988, 14, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Rodríguez-Pintó, I.; Cervera, R.; Morel, N.; Costedoat-Chalumeau, N.; Erkan, D.; Shoenfeld, Y.; Espinosa, G. Rituximab use in the catastrophic antiphospholipid syndrome: Descriptive analysis of the CAPS registry patients receiving rituximab. Autoimmun. Rev. 2013, 12, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, E.; Arkfeld, D.G.; Metyas, S.; Shinada, S.; Ehresmann, S.; A Liebman, H. Rituximab treatment for resistant antiphospholipid syndrome. J. Rheumatol. 2006, 33, 355–357. [Google Scholar] [PubMed]
- Shiber, S.; Yair, M. Catastrophic antiphospholipid syndrome: A case series. Isr. Med. Assoc. J. 2013, 15, 481–484. [Google Scholar]
- López-Benjume, B.; Rodríguez-Pintó, I.; Amigo, M.C.; Erkan, D.; Shoenfeld, Y.; Cervera, R.; Espinosa, G. Eculizumab use in catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis from the “CAPS Registry”. Autoimmun. Rev. 2022, 21, 103055. [Google Scholar] [CrossRef]
- Yelnik, C.M.; Miranda, S.; Mekinian, A.; Lazaro, E.; Quéméneur, T.; Provot, F.; Frimat, M.; Morell-Dubois, S.; Le Guern, V.; Hachulla, E.; et al. Refractory Catastrophic Antiphospholipid Syndrome patients respond inconsistently to Eculizumab. Blood 2020, 136, 2473–2477. [Google Scholar] [CrossRef]
- Kello, N.; El Khoury, L.; Marder, G.; Furie, R.; Zapantis, E.; Horowitz, D.L. Secondary thrombotic microangiopathy in systemic lupus erythematosus and antiphospholipid syndrome, the role of complement and use of eculizumab: Case series and review of literature. Semin. Arthritis Rheum. 2018, 49, 74–83. [Google Scholar] [CrossRef]
- Shapira, I.; Andrade, D.; Allen, S.L.; Salmon, J.E. Brief Report: Induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012, 64, 2719–2723. [Google Scholar] [CrossRef] [PubMed]
- Strakhan, M.; Hurtado-Sbordoni, M.; Galeas, N.; Bakirhan, K.; Alexis, K.; Elrafei, T. 36-Year-Old Female with Catastrophic Antiphospholipid Syndrome Treated with Eculizumab: A Case Report and Review of Literature. Case Rep. Hematol. 2014, 2014, 704371. [Google Scholar] [CrossRef] [PubMed]
- Kronbichler, A.; Frank, R.; Kirschfink, M.; Szilágyi, Á.; Csuka, D.; Prohászka, Z.; Schratzberger, P.; Lhotta, K.; Mayer, G. Efficacy of eculizumab in a patient with immunoadsorption-dependent catastrophic antiphospholipid syndrome: A case report. Medicine 2014, 93, e143. [Google Scholar] [CrossRef] [PubMed]
- Lonze, B.E.; Zachary, A.A.; Magro, C.M.; Desai, N.M.; Orandi, B.J.; Dagher, N.N.; Singer, A.L.; Carter-Monroe, N.; Nazarian, S.M.; Segev, D.L.; et al. Eculizumab Prevents Recurrent Antiphospholipid Antibody Syndrome and Enables Successful Renal Transplantation. Am. J. Transp. 2014, 14, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Chidharla, A.; Syed, S.B.; Chatterjee, T.; Tarantino, M.D. A Case Report of COVID-Associated Catastrophic Antiphospholipid Syndrome Successfully Treated with Eculizumab. J. Blood Med. 2021, 12, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, J.T.; Lim, H.I.; DeSancho, M.T. Successful outcome with eculizumab treatment in a patient with antiphospholipid syndrome presenting with an unusual thrombotic storm. J. Thromb. Thrombolysis 2020, 52, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Gustavsen, A.; Skattum, L.; Bergseth, G.; Lorentzen, B.; Floisand, Y.; Bosnes, V.; Mollnes, T.E.; Barratt-Due, A. Effect on mother and child of eculizumab given before caesarean section in a patient with severe antiphospholipid syndrome: A case report. Medicine 2017, 96, e6338. [Google Scholar] [CrossRef]
- Girish, B.; Gainder, S.; Saha, S.C.; Krishnappa, D. Rare Presentation of Catastrophic Antiphospholipid Syndrome with Myocarditis in Post-Partum Period: Case Report and Review of Literature. J. Obstet. Gynecol. India 2017, 68, 70–72. [Google Scholar] [CrossRef]
- Silver, R.M. Catastrophic antiphospholipid syndrome and pregnancy. Semin. Perinatol. 2018, 42, 26–32. [Google Scholar] [CrossRef]
- Khizroeva, J.; Bitsadze, V.; Makatsariya, A. Catastrophic antiphospholipid syndrome and pregnancy. Clinical report. J. Matern. Neonatal Med. 2018, 32, 2091–2094. [Google Scholar] [CrossRef]
- Hoayek, J.G.; Moussa, H.N.; Rehman, H.A.; Nasab, S.H.; Blackwell, S.C.; Sibai, B.M. Catastrophic antiphospholipid syndrome in pregnancy, a diagnosis that should not be missed. J. Matern. Neonatal Med. 2016, 29, 3950–3955. [Google Scholar] [CrossRef] [PubMed]
- Makatsariyaa, A.D.; Khizroevaa, J.; Bitsadzea, V.O. Catastrophic antiphospholipid syndrome (Ronald Asherson syndrome) and obstetric pathology. J. Perinat. Med. 2018, 46, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Collict, M.; Buhagiar, W.S.; Mercieca, C.; Thake, J. Catastrophic antiphospholipid syndrome in pregnancy: A life-threatening condition. BMJ Case Rep. 2019, 12, e23086. [Google Scholar] [CrossRef] [PubMed]
- Hakman, E.; Mikhael, S. Clinical Report of Probable Catastrophic Antiphospholipid Syndrome in Pregnancy. Case Rep. Obstet. Gynecol. 2018, 2018, 4176456. [Google Scholar]
- Sadick, V.; Lane, S.; Fischer, E.; Seppelt, I.; Shetty, A.; McLean, A. Post-partum catastrophic antiphospholipid syndrome presenting with shock and digital ischaemia—A diagnostic and management challenge. J. Intensive Care Soc. 2018, 19, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Lanier, L.; Jung, E.; Sengupta, S.; Ulrich, V.; Sacharidou, A.; Tarango, C.; Osunbunmi, O.; Shen, Y.-M.; Salmon, J.E.; et al. Identification of a monoclonal antibody that attenuates antiphospholipid syndrome-related pregnancy complications and thrombosis. PLoS ONE 2016, 11, e0158757. [Google Scholar] [CrossRef]
- Defreitas, M.; EdwardsRichards, A.; Raj, V.; Katsoufis, C.; Jeyapalan, A.; McLaughlin, G.; Abitbol, C. Pediatric Catastrophic Antiphospholipid Syndrome: Case Study and Literature Review. Ann. Paediatr. Rheumatol. 2014, 3, 77–87. [Google Scholar] [CrossRef]
- Vieira, A.; Berry, L.; Ofosu, F.; Andrew, M. Heparin sensitivity and resistance in the neonate: An explanation. Thromb. Res. 1991, 63, 85–98. [Google Scholar] [CrossRef]
- Andrew, M.; Vegh, P.; Johnston, M.; Bowker, J.; Ofosu, F.; Mitchell, L. Maturation of the hemostatic system during childhood. Blood 1992, 80, 1998–2005. [Google Scholar] [CrossRef]
- Kuhle, S.; Eulmesekian, P.; Kavanagh, B.; Massicotte, P.; Vegh, P.; Mitchell, L.G. A clinically significant incidence of bleeding in critically ill children receiving therapeutic doses of unfractionated heparin: A prospective cohort study. Haematologica 2007, 92, 244–247. [Google Scholar] [CrossRef]
- Newall, F.; Ignjatovic, V.; Summerhayes, R.; Gan, A.; Butt, W.; Johnston, L.; Monagle, P. In vivo age dependency of unfractionated heparin in infants and children. Thromb. Res. 2009, 123, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Biss, T.T.; Avery, P.J.; Williams, M.D.; Brandão, L.R.; Grainger, J.D.; Kamali, F. The VKORC1 and CYP2C9 genotypes are associated with over-anticoagulation during initiation of warfarin therapy in children. J. Thromb. Haemost. 2012, 11, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Christman, J.W.; Lancaster, L.H.; Blackwell, T.S. Nuclear factor kappa B: A pivotal role in the systemic inflammatory response syndrome and new target for therapy. Intensive Care Med. 1998, 24, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Erkan, D.; Asherson, R.A.; Espinosa, G.; Cervera, R.; Font, J.; Piette, J.C.; Lockshin, M.D. Catastrophic Antiphospholipid Syndrome Registry Project Group. Long term outcome of catastrophic antiphospholipid syndrome survivors. Ann. Rheum. Dis. 2003, 62, 530–533. [Google Scholar] [CrossRef]
- Pleguezuelo, D.E.; Díaz-Simón, R.; Cabrera-Marante, O.; Lalueza, A.; Paz-Artal, E.; Lumbreras, C.; Serrano Hernández, A. Case Report: Resetting the Humoral Immune Response by Targeting Plasma Cells with Daratumumab in Anti-Phospholipid Syndrome. Front. Immunol. 2021, 12, 667515. [Google Scholar] [CrossRef]
- Niznik, S.; Rapoport, M.J.; Avnery, O.; Lubetsky, A.; Haj Yahia, S.; Ellis, M.H.; Agmon-Levin, N. Patterns of Recurrent Thrombosis in Primary Antiphospholipid Syndrome-Multicenter, Real-Life Long-Term Follow-Up. Front. Immunol. 2022, 19, 843718. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Criteria |
|---|
| Domain 1—Macrovascular: venous thromboembolism |
| Domain 2—Macrovascular: arterial thrombosis |
| Domain 3—Microvascular Suspected: Livedo racemosa, Livedoid vasculopathy lesions, Antiphospholipid antibody (aPL) nephropathy, Pulmonary hemorrhage Established: Livedoid vasculopathy, aPL nephropathy, Pulmonary hemorrhage, Myocardial disease, Adrenal hemorrhage or microthrombosis |
| Domain 4—Obstetric: Prefetal death, Fetal death, Preeclampsia with severe features, Central nervous system dysfunction, Placental insufficiency with severe features |
| Domain 5—Cardiac valve: Valve thickening, Valve vegetation, |
| Domain 6—Haematology: Thrombocytopenia, |
| Laboratory Criteria |
| Domain 7—aPL test by coagulation-based functional assay |
| Domain 8—aPL test by solid phase–based assay |
| Current Therapeutics | Drugs and Dose |
|---|---|
| Anticoagulant therapy (direct oral anticoagulants NOT recommended) | vitamin K antagonist with INR target 2–3 |
| Antiaggregation | low-dose aspirin (100 mg/day) |
| Glucocorticoids | methylprednisolone, 0.5 to 1 g intravenously, once daily for three or more days, followed by oral or parenteral therapy with the equivalent of 1 mg/kg prednisone per day with tapering initiated once the patient improves clinically |
| Therapeutic plasmapheresis | exchanges are typically performed once daily for five days (longer in patients with refractory disease) |
| Intravenous immunoglobulin | a typical dose is between 400 mg per kg per day for five days |
| Rituximab (a monoclonal antibody against the B-cell antigen CD20) | 375 mg/m2 once a week for four weeks, or 500 to 1000 mg administered twice, at intervals of 7 or 14 days |
| Drugs | Target Molecules | References |
|---|---|---|
| Rituximab | a monoclonal antibody against the B-cell antigen CD20 | [29] |
| Eculizumab | a monoclonal antibody against the C5 component of complement | [33] |
| Daratumumab | anti-CD38 monoclonal antibody | [34] |
| Belimumab | BAFF/Blys inhibitor | [34] |
| Zanubrutinib | BTK inhibitor | [34] |
| Adalimumab and Certolizumab | anti-TNF-a monoclonal antibody | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siniscalchi, C.; Basaglia, M.; Riva, M.; Meschi, M.; Meschi, T.; Castaldo, G.; Di Micco, P. Catastrophic Antiphospholipid Syndrome: A Review. Immuno 2024, 4, 1-13. https://doi.org/10.3390/immuno4010001
Siniscalchi C, Basaglia M, Riva M, Meschi M, Meschi T, Castaldo G, Di Micco P. Catastrophic Antiphospholipid Syndrome: A Review. Immuno. 2024; 4(1):1-13. https://doi.org/10.3390/immuno4010001
Chicago/Turabian StyleSiniscalchi, Carmine, Manuela Basaglia, Michele Riva, Michele Meschi, Tiziana Meschi, Giampiero Castaldo, and Pierpaolo Di Micco. 2024. "Catastrophic Antiphospholipid Syndrome: A Review" Immuno 4, no. 1: 1-13. https://doi.org/10.3390/immuno4010001