
Inflammatory Network of Liver Fibrosis and How It Can Be Targeted Therapeutically
 ,
,
Abstract
:1. Introduction
1.1. Complications of Liver Regeneration Therapeutics
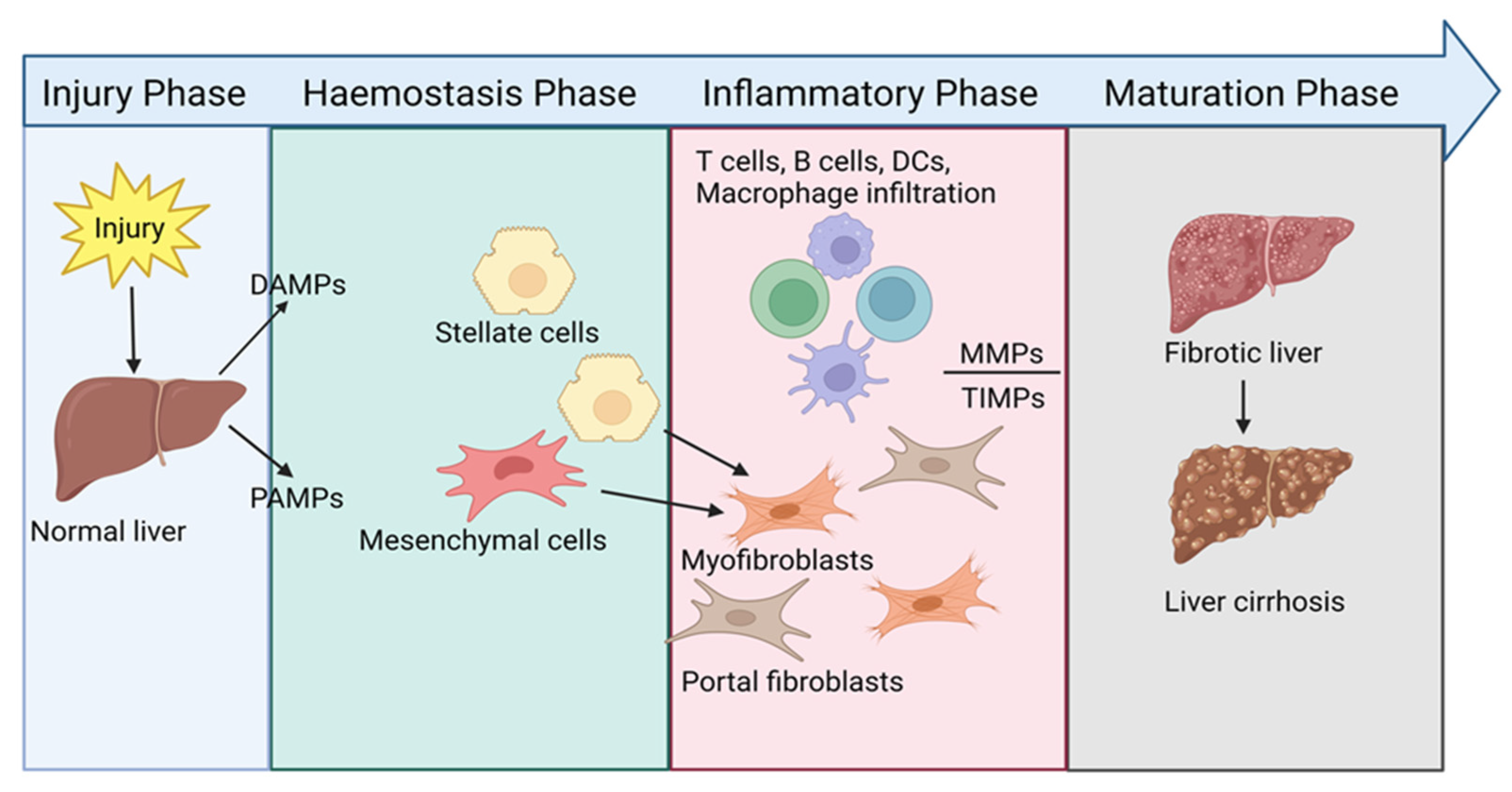
1.2. Initiation of Liver Fibrosis
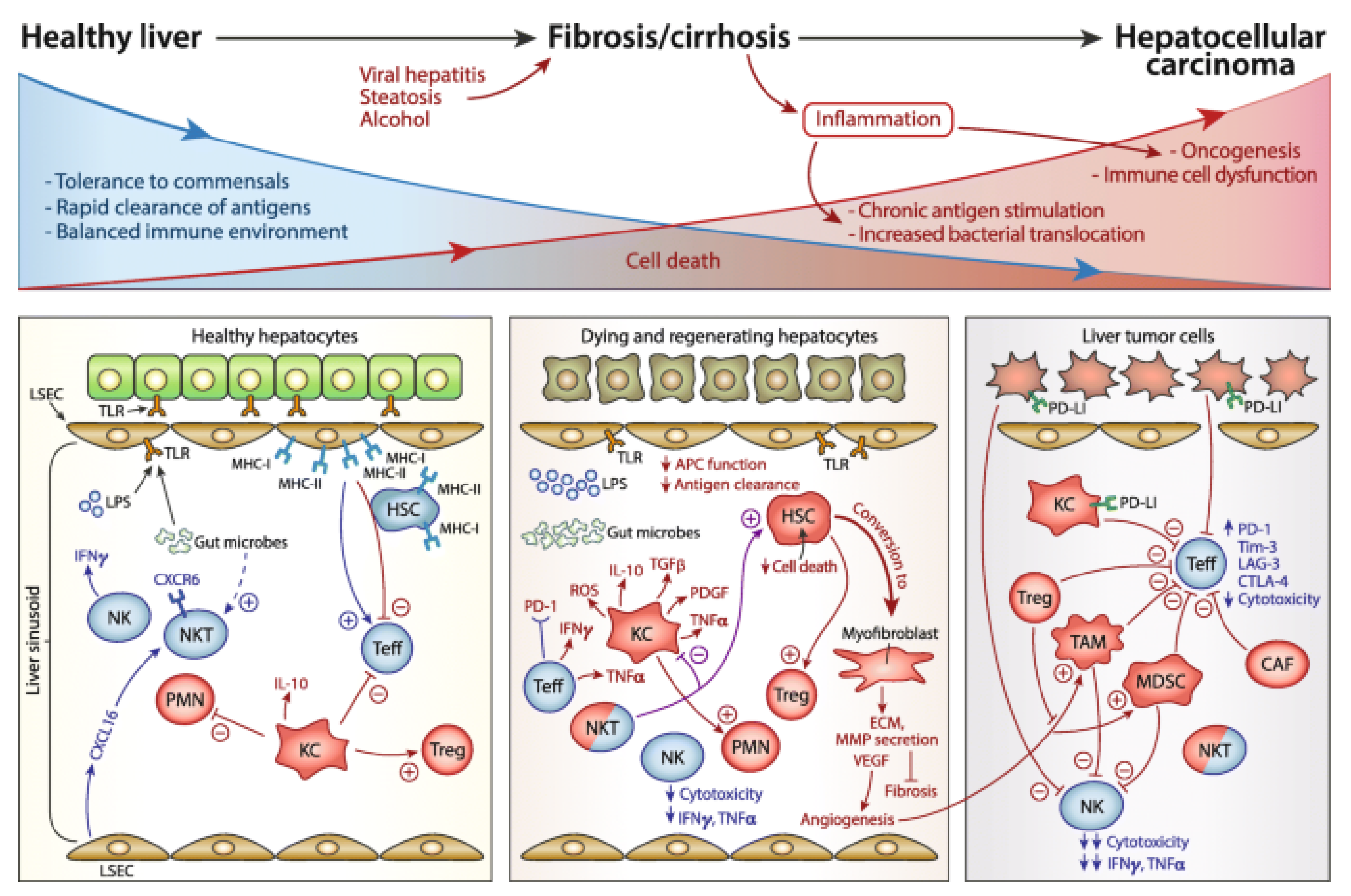
2. Immunopathology of Liver Fibrosis
2.1. Innate Immune Cells
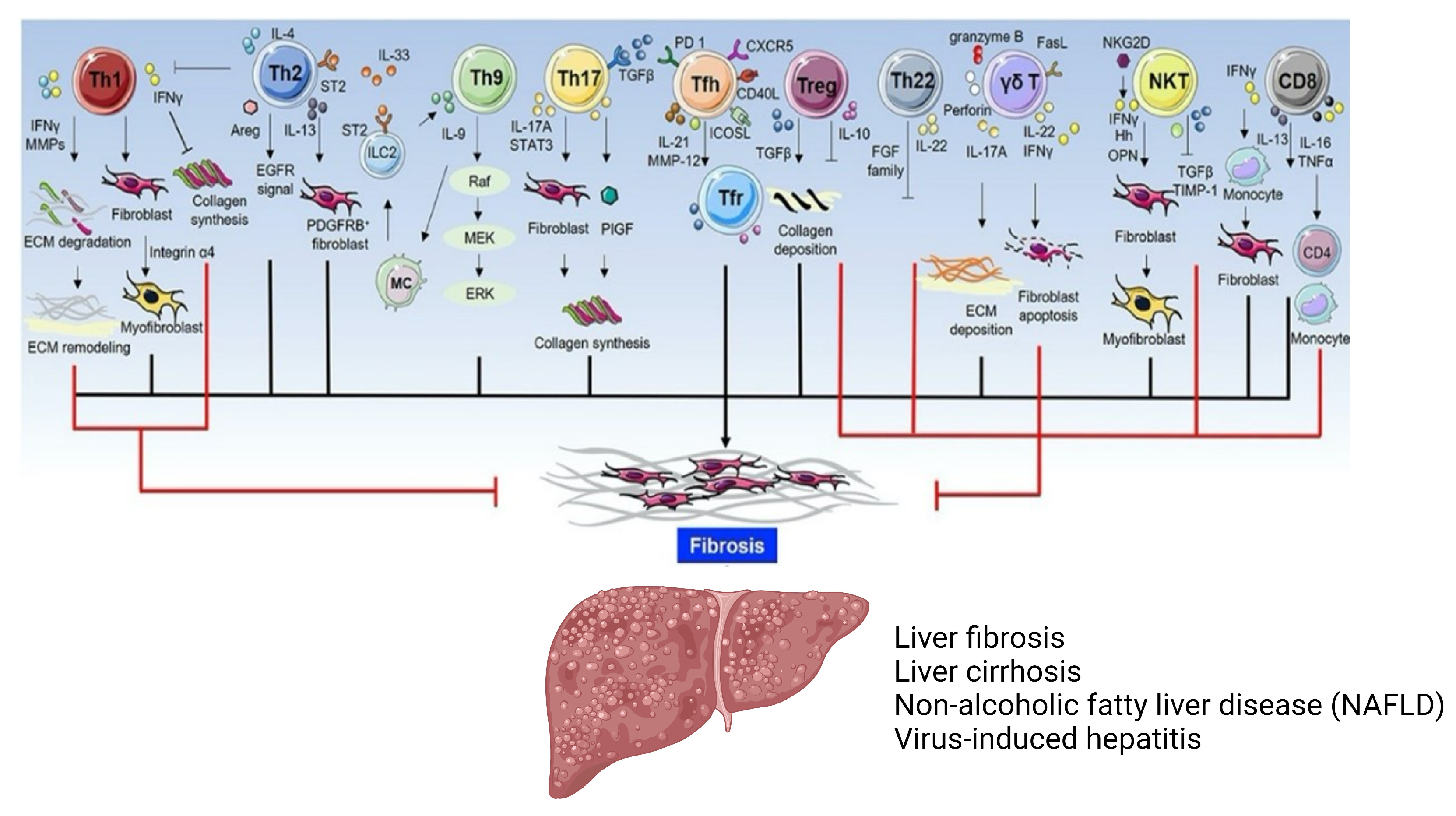
2.2. Adaptive Immune Cells
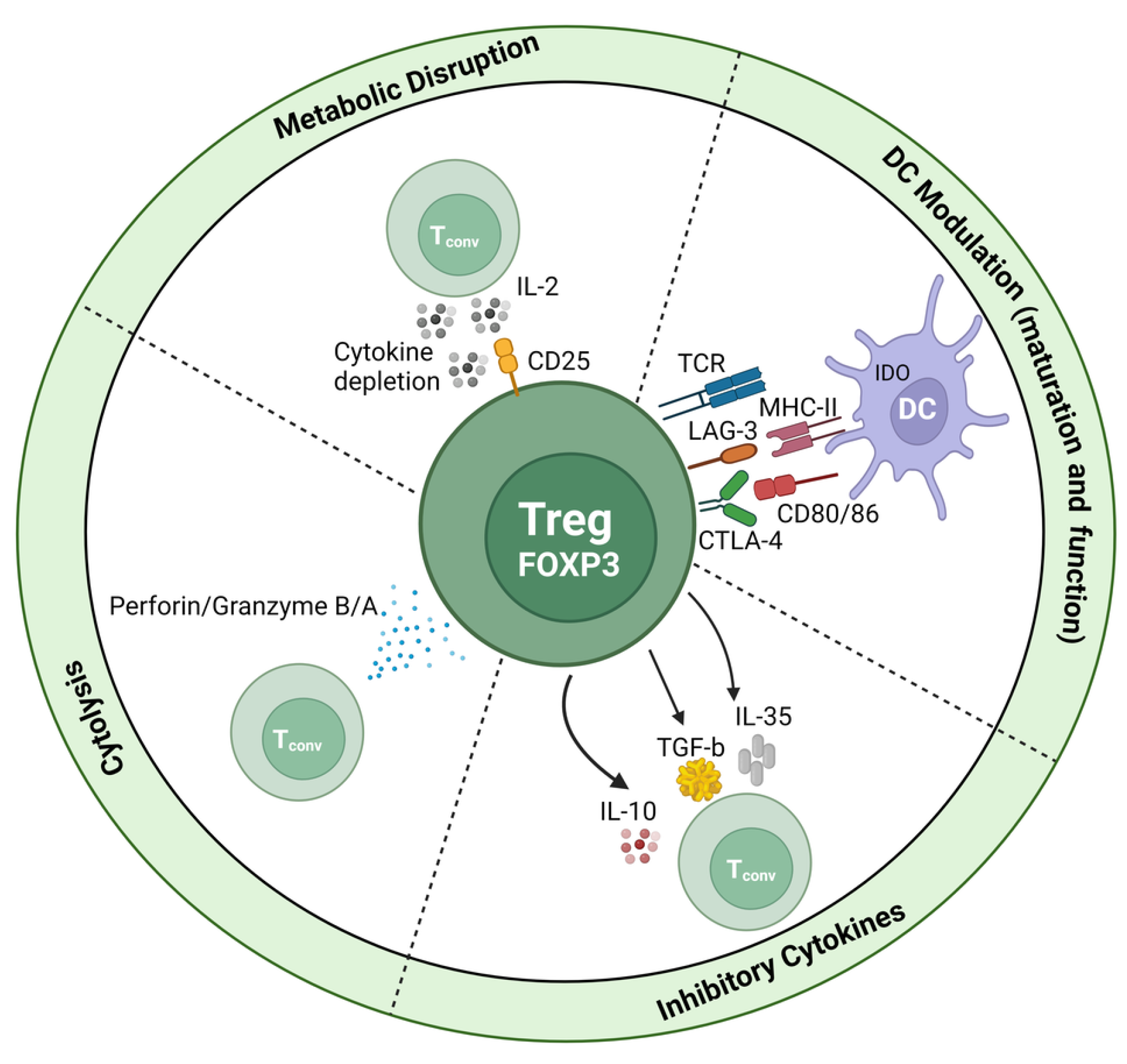
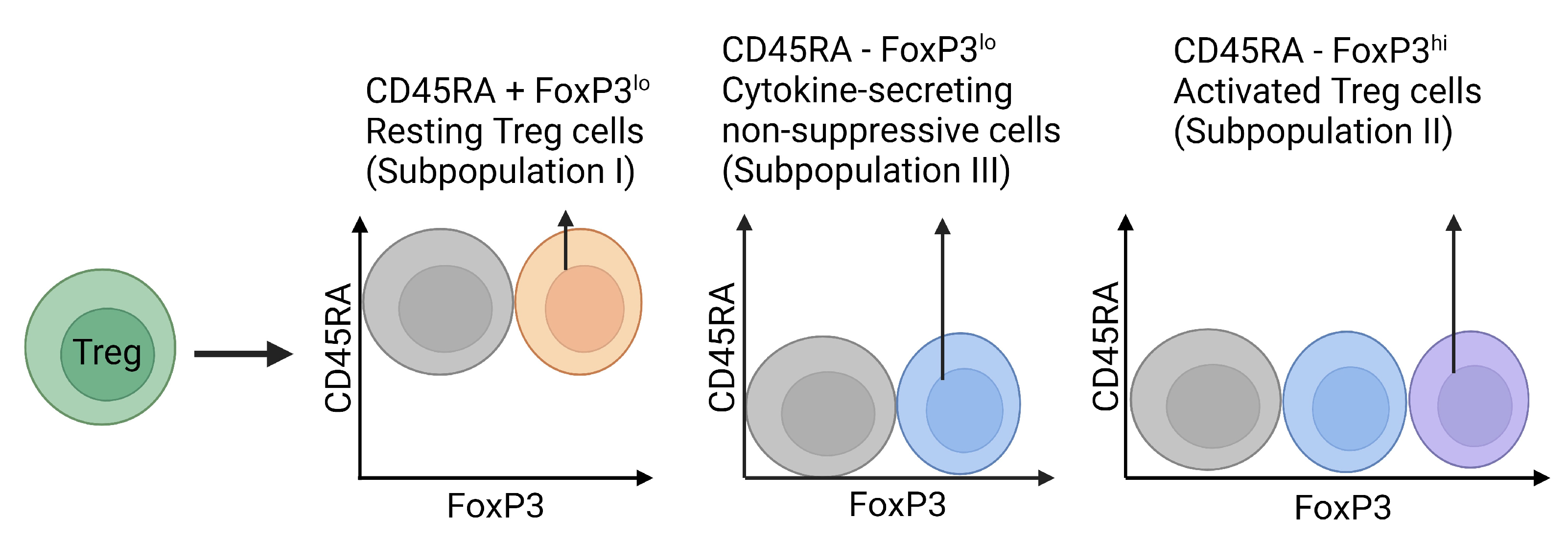
3. Regulatory T Cells
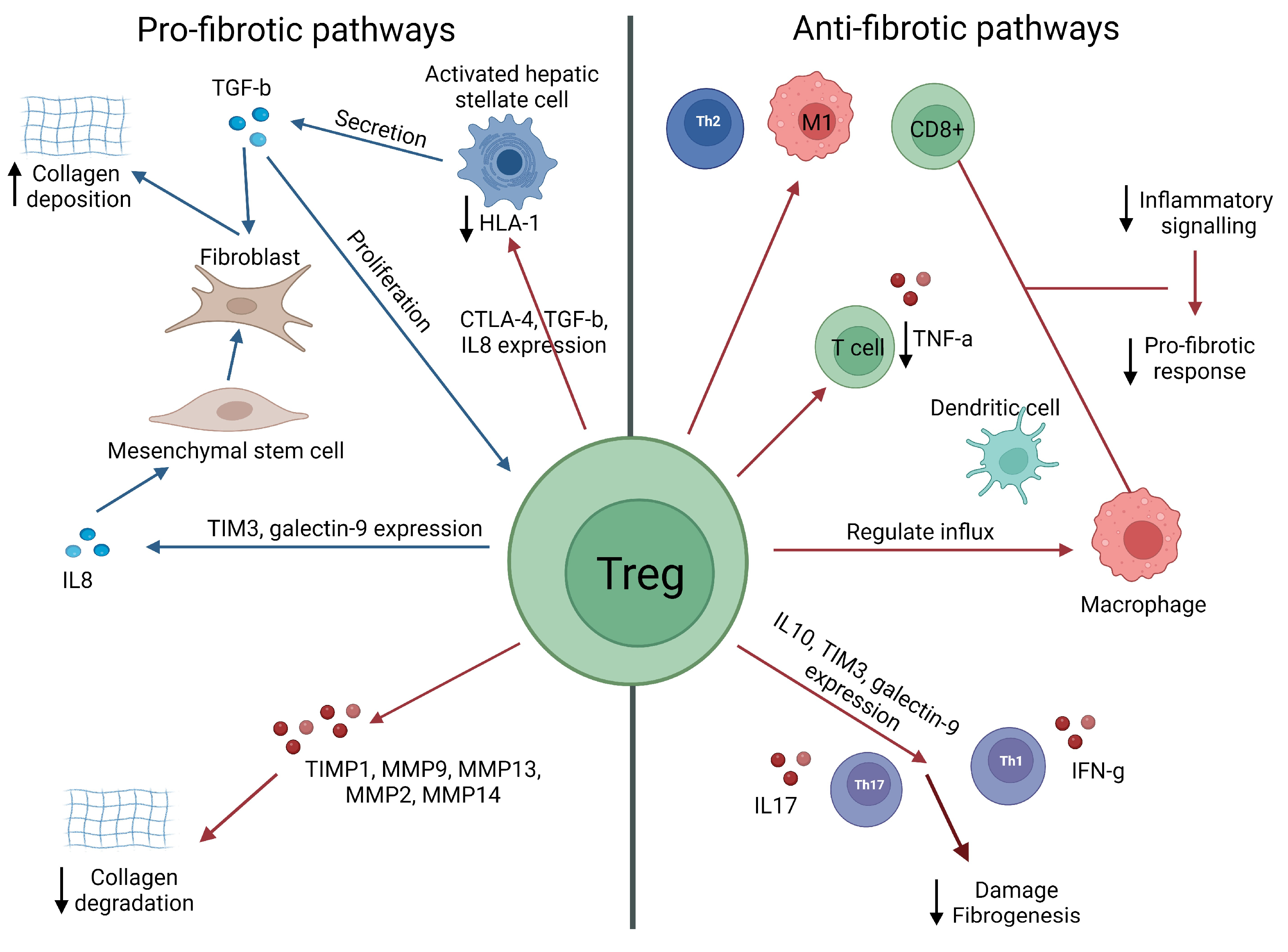
3.1. Tregs as Potential Inducers of Liver Fibrosis
3.2. Potential Role of Tregs in the Attenuation of Liver Fibrosis
3.3. Tregs: Pro-Fibrotic or Protective?
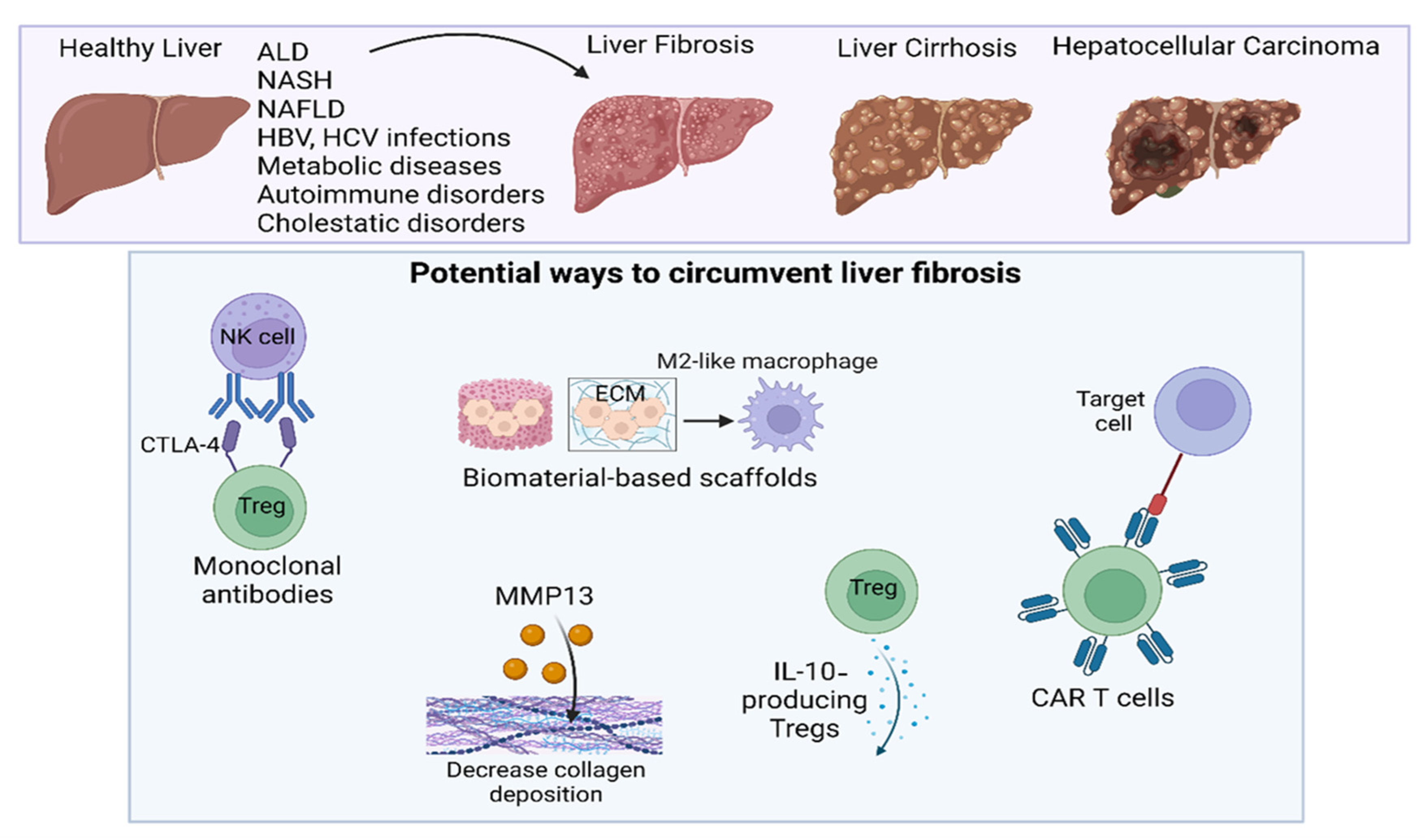
4. Immunotherapy of Liver Fibrosis
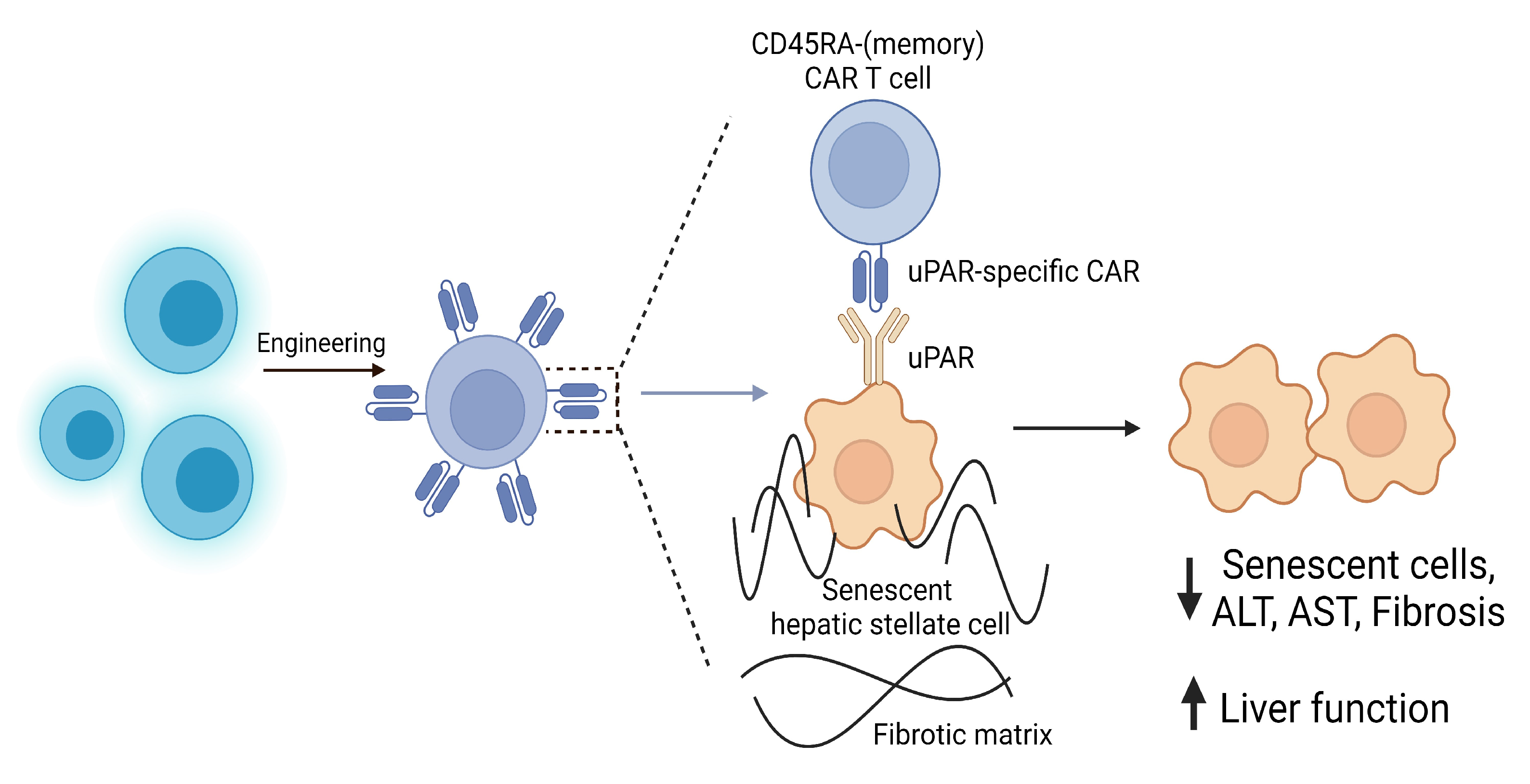
4.1. Clinical Potential of CAR T Cells
4.2. Treg Manipulation as a Next-Generation Therapy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan, X.; Ma, X.; Jiang, Y.; Wen, J.; Yang, L.; Chen, D.; Cao, X.; Peng, C. A Comprehensive Review of Natural Products against Liver Fibrosis: Flavonoids, Quinones, Lignans, Phenols, and Acids. Evid.-Based Complement. Altern. Med. 2020, 2020, 7171498. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Keenan, B.P.; Fong, L.; Kelley, R.K. Immunotherapy in hepatocellular carcinoma: The complex interface between inflammation, fibrosis, and the immune response. J. Immunother. Cancer 2019, 7, 267. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47, 2–6. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Sharma, A.; Nagalli, S. Chronic Liver Disease; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef]
- Xia, M.-F.; Bian, H.; Gao, X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef]
- Fattahi, M.R.; Niknam, R.; Safarpour, A.; Sepehrimanesh, M.; Lotfi, M. The Prevalence of Metabolic Syndrome in Non-alcoholic Fatty Liver Disease; A Population-Based Study. Middle East J. Dig. Dis. 2016, 8, 131–137. [Google Scholar] [CrossRef]
- Elferink, R.O. Cholestasis. Gut 2003, 52, ii42. [Google Scholar] [CrossRef]
- Nallagangula, K.S.; Nagaraj, S.K.; Venkataswamy, L.; Chandrappa, M. Liver fibrosis: A compilation on the biomarkers status and their significance during disease progression. Future Sci. OA 2017, 4, FSO250. [Google Scholar] [CrossRef]
- Yeom, S.K.; Lee, C.H.; Cha, S.H.; Park, C.M. Prediction of liver cirrhosis, using diagnostic imaging tools. World J. Hepatol. 2015, 7, 2069–2079. [Google Scholar] [CrossRef]
- Ginès, P.; Graupera, I.; Lammert, F.; Angeli, P.; Caballeria, L.; Krag, A.; Guha, I.N.; Murad, S.D.; Castera, L. Screening for liver fibrosis in the general population: A call for action. Lancet Gastroenterol. Hepatol. 2016, 1, 256–260. [Google Scholar] [CrossRef]
- Lucero, C.; Brown, R.S., Jr. Noninvasive Measures of Liver Fibrosis and Severity of Liver Disease. Gastroenterol. Hepatol. 2016, 12, 33–40. [Google Scholar]
- Wang, X.; Wu, B. Critical issues in the diagnosis and treatment of liver cirrhosis. Gastroenterol. Rep. 2019, 7, 227–230. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef]
- Bessone, F. Non-steroidal anti-inflammatory drugs: What is the actual risk of liver damage? World J. Gastroenterol. 2010, 16, 5651–5661. [Google Scholar] [CrossRef]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef]
- da Silva Morais, A.; Vieira, S.; Zhao, X.; Mao, Z.; Gao, C.; Oliveira, J.M.; Reis, R.L. Advanced Biomaterials and Processing Methods for Liver Regeneration: State-of-the-Art and Future Trends. Adv. Healthc. Mater. 2020, 9, 1901435. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, Z.; Wang, F.-S. Liver fibrosis: Mechanisms of immune-mediated liver injury. Cell. Mol. Immunol. 2012, 9, 296–301. [Google Scholar] [CrossRef]
- Yang, L.; Seki, E. Toll-Like Receptors in Liver Fibrosis: Cellular Crosstalk and Mechanisms. Front. Physiol. 2012, 3, 138. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Dobie, R.; Henderson, N.C. Homing in on the hepatic scar: Recent advances in cell-specific targeting of liver fibrosis. F1000Research 2016, 5, 1749. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Recent advances in understanding liver fibrosis: Bridging basic science and individualized treatment concepts. F1000Research 2018, 7, 921. [Google Scholar] [CrossRef]
- Xu, J.; Liu, X.; Koyama, Y.; Wang, P.; Lan, T.; Kim, I.-G.; Kim, I.H.; Ma, H.-Y.; Kisseleva, T. The types of hepatic myofibroblasts contributing to liver fibrosis of different etiologies. Front. Pharmacol. 2014, 5, 167. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, B.G.; Pestana, R.C.; Abugabal, Y.I.; Krishnan, S.; Chen, J.; Hassan, M.M.; Wolff, R.A.; Rashid, A.; Amin, H.M.; Kaseb, A.O. Origin and role of hepatic myofibroblasts in hepatocellular carcinoma. Oncotarget 2020, 11, 1186–1201. [Google Scholar] [CrossRef]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J. Histochem. Cytochem. 2018, 66, 213–227. [Google Scholar] [CrossRef]
- Naim, A.; Pan, Q.; Baig, M.S. Matrix Metalloproteinases (MMPs) in Liver Diseases. J. Clin. Exp. Hepatol. 2017, 7, 367–372. [Google Scholar] [CrossRef]
- Roeb, E. Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol. 2018, 68–69, 463–473. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Dou, L.; Ono, Y.; Chen, Y.F.; Thomson, A.W.; Chen, X.P. Hepatic Dendritic Cells, the Tolerogenic Liver Environment, and Liver Disease. Semin. Liver Dis. 2018, 38, 170–180. [Google Scholar] [CrossRef]
- Bartneck, M.; Wang, J. Therapeutic Targeting of Neutrophil Granulocytes in Inflammatory Liver Disease. Front. Immunol. 2019, 10, 2257. [Google Scholar] [CrossRef]
- Pulli, B.; Ali, M.; Iwamoto, Y.; Zeller, M.W.G.; Schob, S.; Linnoila, J.J.; Chen, J.W. Myeloperoxidase-Hepatocyte-Stellate Cell Cross Talk Promotes Hepatocyte Injury and Fibrosis in Experimental Nonalcoholic Steatohepatitis. Antioxid. Redox Signal. 2015, 23, 1255–1269. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef]
- van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef]
- Cardoso, C.C.; Matiollo, C.; Pereira, C.H.J.; Fonseca, J.S.; Alves, H.E.L.; da Silva, O.M.; de Souza Menegassi, V.; dos Santos, C.R.; de Moraes, A.C.R.; de Lucca Schiavon, L.; et al. Patterns of dendritic cell and monocyte subsets are associated with disease severity and mortality in liver cirrhosis patients. Sci. Rep. 2021, 11, 5923. [Google Scholar] [CrossRef]
- Ploeger, D.T.; Hosper, N.A.; Schipper, M.; Koerts, J.A.; de Rond, S.; Bank, R.A. Cell plasticity in wound healing: Paracrine factors of M1/M2 polarized macrophages influence the phenotypical state of dermal fibroblasts. Cell Commun. Signal. 2013, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-Y.; Li, X.-F.; Meng, X.-M.; Huang, C.; Zhang, L.; Li, J. Macrophage Phenotype in Liver Injury and Repair. Scand. J. Immunol. 2017, 85, 166–174. [Google Scholar] [CrossRef]
- Witherel, C.E.; Abebayehu, D.; Barker, T.H.; Spiller, K.L. Macrophage and Fibroblast Interactions in Biomaterial-Mediated Fibrosis. Adv. Healthc. Mater. 2019, 8, 1801451. [Google Scholar] [CrossRef]
- Li, M.; Hou, Q.; Zhong, L.; Zhao, Y.; Fu, X. Macrophage Related Chronic Inflammation in Non-Healing Wounds. Front. Immunol. 2021, 12, 681710. [Google Scholar] [CrossRef]
- Chung, L.; Maestas, D.R.; Housseau, F.; Elisseeff, J.H. Key players in the immune response to biomaterial scaffolds for regenerative medicine. Adv. Drug Deliv. Rev. 2017, 114, 184–192. [Google Scholar] [CrossRef]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef]
- Liu, Y.; Munker, S.; Müllenbach, R.; Weng, H.-L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S.; Park, O. Liver natural killer and natural killer T cells: Immunobiology and emerging roles in liver diseases. J. Leukoc. Biol. 2009, 86, 513–528. [Google Scholar] [CrossRef]
- Fasbender, F.; Widera, A.; Hengstler, J.G.; Watzl, C. Natural Killer Cells and Liver Fibrosis. Front. Immunol. 2016, 7, 19. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S.; Jeong, W.-I.L. Activation of natural killer cells inhibits liver fibrosis: A novel strategy to treat liver fibrosis. Expert Rev. Gastroenterol. Hepatol. 2007, 1, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, J.; Wang, J.; Zhou, Q.; Yang, B.; He, Q.; Weng, Q. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharmacol. Res. 2020, 155, 104720. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Mertens, P.R.; Gressner, A.M.; Dooley, S. IFN-gamma abrogates profibrogenic TGF-beta signaling in liver by targeting expression of inhibitory and receptor Smads. J. Hepatol. 2007, 46, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C. Current and future anti-fibrotic therapies for chronic liver disease. Clin. Liver Dis. 2008, 12, 939–962. [Google Scholar] [CrossRef]
- Rahman, A.H.; Aloman, C. Dendritic cells and liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Almeda-Valdes, P.; Aguilar Olivos, N.E.; Barranco-Fragoso, B.; Uribe, M.; Méndez-Sánchez, N. The Role of Dendritic Cells in Fibrosis Progression in Nonalcoholic Fatty Liver Disease. BioMed Res. Int. 2015, 2015, 768071. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.-Y.; Wang, N.; Feng, Y.; Wang, X.; Feng, Y. Recent Insights Into the Role of Immune Cells in Alcoholic Liver Disease. Front. Immunol. 2019, 10, 1328. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N.; Valencia-Rodríguez, A.; Coronel-Castillo, C.; Vera-Barajas, A.; Contreras-Carmona, J.; Ponciano-Rodríguez, G.; Zamora-Valdés, D. The cellular pathways of liver fibrosis in non-alcoholic steatohepatitis. Ann. Transl. Med. 2020, 8, 400. [Google Scholar] [CrossRef]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef]
- Bhogal, R.K.; Bona, C.A. B cells: No longer bystanders in liver fibrosis. J. Clin. Investig. 2005, 115, 2962–2965. [Google Scholar] [CrossRef]
- Faggioli, F.; Palagano, E.; Di Tommaso, L.; Donadon, M.; Marrella, V.; Recordati, C.; Mantero, S.; Villa, A.; Vezzoni, P.; Cassani, B. B lymphocytes limit senescence-driven fibrosis resolution and favor hepatocarcinogenesis in mouse liver injury. Hepatology 2018, 67, 1970–1985. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Ricci, O.E.; Paoletti, F.; Surrenti, C. Immune mechanisms for hepatic fibrogenesis. T-lymphocyte-mediated stimulation of fibroblast collagen production in chronic active hepatitis. Liver 1985, 5, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Koda, Y.; Teratani, T.; Chu, P.-S.; Hagihara, Y.; Mikami, Y.; Harada, Y.; Tsujikawa, H.; Miyamoto, K.; Suzuki, T.; Taniki, N.; et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat. Commun. 2021, 12, 4474. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, P.; Wu, Y.; Wang, L. Metabolic tissue-resident CD8+ T cells: A key player in obesity-related diseases. Obes. Rev. 2021, 22, 13133. [Google Scholar] [CrossRef] [PubMed]
- Breuer, D.A.; Pacheco, M.C.; Washington, M.K.; Montgomery, S.A.; Hasty, A.H.; Kennedy, A.J. CD8+ T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2020, 318, 211–224. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Wang, X.-Z. Interleukin-10 and chronic liver disease. World J. Gastroenterol. 2006, 12, 1681–1685. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef]
- Li, H.; You, H.; Fan, X.; Jia, J. Hepatic macrophages in liver fibrosis: Pathogenesis and potential therapeutic targets. BMJ Open Gastroenterol. 2016, 3, e000079. [Google Scholar] [CrossRef]
- Barron, L.; Wynn, T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 723–728. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhu, J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int. J. Mol. Sci. 2020, 21, 8011. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.; Gottschalk, S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 2021, 12, 684642. [Google Scholar] [CrossRef]
- Rainard, P.; Cunha, P.; Martins, R.P.; Gilbert, F.B.; Germon, P.; Foucras, G. Type 3 immunity: A perspective for the defense of the mammary gland against infections. Vet. Res. 2020, 51, 129. [Google Scholar] [CrossRef] [PubMed]
- Fabre, T.; Molina, M.F.; Soucy, G.; Goulet, J.-P.; Willems, B.; Villeneuve, J.-P.; Bilodeau, M.; Shoukry, N.H. Type 3 cytokines IL-17A and IL-22 drive TGF-β–dependent liver fibrosis. Sci. Immunol. 2018, 3, 7754. [Google Scholar] [CrossRef] [PubMed]
- Shoukry, N.H.; Fabre, T.; Molina, M.F.; Soucy, G.; Willems, B.; Villeneuve, J.-P.; Bilodeau, M. Th17 Cytokines Drive Liver Fibrosis Progression by Regulating TGF-β Signaling through Activation of MAPKs. J. Immunol. 2017, 198, 197.12. [Google Scholar] [CrossRef]
- Lafdil, F.; Miller, A.M.; Ki, S.H.; Gao, B. Th17 cells and their associated cytokines in liver diseases. Cell. Mol. Immunol. 2010, 7, 250–254. [Google Scholar] [CrossRef]
- Mason, G.M.; Lowe, K.; Melchiotti, R.; Ellis, R.; de Rinaldis, E.; Peakman, M.; Heck, S.; Lombardi, G.; Tree, T.I.M. Phenotypic Complexity of the Human Regulatory T Cell Compartment Revealed by Mass Cytometry. J. Immunol. 2015, 195, 2030. [Google Scholar] [CrossRef]
- Wan, M.; Han, J.; Ding, L.; Hu, F.; Gao, P. Novel Immune Subsets and Related Cytokines: Emerging Players in the Progression of Liver Fibrosis. Front. Med. 2021, 8, 604894. [Google Scholar] [CrossRef]
- Kitz, A.; Singer, E.; Hafler, D. Regulatory T Cells: From Discovery to Autoimmunity. Cold Spring Harb. Perspect. Med. 2018, 8, a029041. [Google Scholar] [CrossRef]
- Devaud, C.; Darcy, P.K.; Kershaw, M.H. Foxp3 expression in T regulatory cells and other cell lineages. Cancer Immunol. Immunother. 2014, 63, 869–876. [Google Scholar] [CrossRef]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10, 43. [Google Scholar] [CrossRef]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; de St. Groth, B.F.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Wallet, M.A.; Sen, P.; Tisch, R. Immunoregulation of dendritic cells. Clin. Med. Res. 2005, 3, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, 22236. [Google Scholar] [CrossRef] [PubMed]
- Rana, J.; Biswas, M. Regulatory T cell therapy: Current and future design perspectives. Cell. Immunol. 2020, 356, 104193. [Google Scholar] [CrossRef]
- Curotto de Lafaille, M.A.; Lafaille, J.J. Natural and Adaptive Foxp3+ Regulatory T Cells: More of the Same or a Division of Labor? Immunity 2009, 30, 626–635. [Google Scholar] [CrossRef]
- Lin, X.; Chen, M.; Liu, Y.; Guo, Z.; He, X.; Brand, D.; Zheng, S.G. Advances in distinguishing natural from induced Foxp3(+) regulatory T cells. Int. J. Clin. Exp. Pathol. 2013, 6, 116–123. [Google Scholar]
- Freudenberg, K.; Lindner, N.; Dohnke, S.; Garbe, A.I.; Schallenberg, S.; Kretschmer, K. Critical Role of TGF-β and IL-2 Receptor Signaling in Foxp3 Induction by an Inhibitor of DNA Methylation. Front. Immunol. 2018, 9, 125. [Google Scholar] [CrossRef]
- Tischner, D.; Wiegers, G.J.; Fiegl, H.; Drach, M.; Villunger, A. Mutual antagonism of TGF-beta and Interleukin-2 in cell survival and lineage commitment of induced regulatory T cells. Cell Death Differ. 2012, 19, 1277–1287. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X. Foxp3 Instability Helps tTregs Distinguish Self and Non-self. Front. Immunol. 2019, 10, 2226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liao, X.; Kang, Y. Tregs: Where We Are and What Comes Next? Front. Immunol. 2017, 8, 1578. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ma, X.; Gong, R.; Zhu, J.; Wei, L.; Yao, J. Recent advances in CD8+ regulatory T cell research. Oncol. Lett. 2018, 15, 8187–8194. [Google Scholar] [CrossRef]
- Wawman, R.E.; Bartlett, H.; Oo, Y.H. Regulatory T Cell Metabolism in the Hepatic Microenvironment. Front. Immunol. 2018, 8, 1889. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G.; Lohse, A.W. Immune tolerance: What is unique about the liver. J. Autoimmun. 2010, 34, 1–6. [Google Scholar] [CrossRef]
- Jeffery, H.C.; Braitch, M.K.; Brown, S.; Oo, Y.H. Clinical Potential of Regulatory T Cell Therapy in Liver Diseases: An Overview and Current Perspectives. Front. Immunol. 2016, 7, 334. [Google Scholar] [CrossRef]
- Devi, M.; Vijayalakshmi, D.; Dhivya, K.; Janane, M. Memory T Cells (CD45RO) Role and Evaluation in Pathogenesis of Lichen Planus and Lichenoid Mucositis. J. Clin. Diagn. Res. 2017, 11, 84–86. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Way, S.S.; Abbas, A.K. Regulatory T cell memory. Nat. Rev. Immunol. 2016, 16, 90–101. [Google Scholar] [CrossRef]
- Hu, C.-C.; Jeng, W.-J.; Chen, Y.-C.; Fang, J.-H.; Huang, C.-H.; Teng, W.; Hsieh, Y.-C.; Lin, Y.-C.; Chien, R.-N.; Sheen, I.S.; et al. Memory Regulatory T cells Increase Only In Inflammatory Phase of Chronic Hepatitis B Infection and Related to Galectin-9/Tim-3 interaction. Sci. Rep. 2017, 7, 15280. [Google Scholar] [CrossRef]
- Jung, M.K.; Shin, E.-C. Regulatory T Cells in Hepatitis B and C Virus Infections. Immune Netw. 2016, 16, 330–336. [Google Scholar] [CrossRef]
- Scharte, M.; Han, X.; Bertges, D.J.; Fink, M.P.; Delude, R.L. Cytokines induce HIF-1 DNA binding and the expression of HIF-1-dependent genes in cultured rat enterocytes. Am. J. Physiol.-Gastrointest. Liver Physiol. 2003, 284, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Colgan, S.P.; Eltzschig, H.K. Hypoxia-inducible factors as molecular targets for liver diseases. J. Mol. Med. 2016, 94, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef] [PubMed]
- Atif, M.; Warner, S.; Oo, Y.H. Linking the gut and liver: Crosstalk between regulatory T cells and mucosa-associated invariant T cells. Hepatol. Int. 2018, 12, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Jeffery, H.C.; Hunter, S.; Bhogal, R.; Birtwistle, J.; Braitch, M.K.; Roberts, S.; Ming, M.; Hannah, J.; Thomas, C.; et al. Human intrahepatic regulatory T cells are functional, require IL-2 from effector cells for survival, and are susceptible to Fas ligand-mediated apoptosis. Hepatology 2016, 64, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Z.-q.; Zhang, L.; Zheng, H.; Zhou, M.-g.; Liu, D.-w. Burden of viral hepatitis caused by specific aetiologies in China, 1990–2016: Findings from the GBD 2016. BMC Public Health 2020, 20, 1461. [Google Scholar] [CrossRef] [PubMed]
- Mailer, R.K.W.; Gisterå, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia Induces Differentiation of Regulatory T Cells in the Liver. Circ. Res. 2017, 120, 1740–1753. [Google Scholar] [CrossRef]
- Zhai, N.; Chi, X.; Li, T.; Song, H.; Li, H.; Jin, X.; Crispe, I.N.; Su, L.; Niu, J.; Tu, Z. Hepatitis C virus core protein triggers expansion and activation of CD4+CD25+ regulatory T cells in chronic hepatitis C patients. Cell. Mol. Immunol. 2015, 12, 743–749. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Feng, K.; Fan, D.; Xue, W.; Lu, J. ‘Repair’ Treg Cells in Tissue Injury. Cell. Physiol. Biochem. 2017, 43, 2155–2169. [Google Scholar] [CrossRef]
- Xu, D.; Fu, J.; Jin, L.; Zhang, H.; Zhou, C.; Zou, Z.; Zhao, J.-M.; Zhang, B.; Shi, M.; Ding, X.; et al. Circulating and Liver Resident CD4+CD25+ Regulatory T Cells Actively Influence the Antiviral Immune Response and Disease Progression in Patients with Hepatitis B. J. Immunol. 2006, 177, 739. [Google Scholar] [CrossRef]
- Peng, G.; Li, S.; Wu, W.; Sun, Z.; Chen, Y.; Chen, Z. Circulating CD4+ CD25+ regulatory T cells correlate with chronic hepatitis B infection. Immunology 2008, 123, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Stoop, J.N.; van der Molen, R.G.; Baan, C.C.; van der Laan, L.J.W.; Kuipers, E.J.; Kusters, J.G.; Janssen, H.L.A. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology 2005, 41, 771–778. [Google Scholar] [CrossRef] [PubMed]
- El-Badawy, O.; Sayed, D.; Badary, M.S.; Abd-Alrahman, M.E.; El-Feky, M.A.; Thabit, A.G. Relations of regulatory T cells with hepatitis markers in chronic hepatitis B virus infection. Hum. Immunol. 2012, 73, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Lei, Z.; Wang, X.; Qi, Q.; He, J.; Liu, D.; Wang, X.; Chen, X.; Zhu, J.; Li, Y.; et al. Hepatitis B envelope antigen increases Tregs by converting CD4+CD25- T cells into CD4+CD25+Foxp3+ Tregs. Exp. Ther. Med. 2020, 20, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Mucida, D.; Tyznik, A.J.; Kronenberg, M.; Cheroutre, H. Hepatic stellate cells function as regulatory bystanders. J. Immunol. 2011, 186, 5549–5555. [Google Scholar] [CrossRef] [PubMed]
- Khanam, A.; Chua, J.V.; Kottilil, S. Immunopathology of Chronic Hepatitis B Infection: Role of Innate and Adaptive Immune Response in Disease Progression. Int. J. Mol. Sci. 2021, 22, 5497. [Google Scholar] [CrossRef] [PubMed]
- Ebinuma, H.; Nakamoto, N.; Li, Y.; Price, D.A.; Gostick, E.; Levine, B.L.; Tobias, J.; Kwok, W.W.; Chang, K.-M. Identification and in vitro expansion of functional antigen-specific CD25+ FoxP3+ regulatory T cells in hepatitis C virus infection. J. Virol. 2008, 82, 5043–5053. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ikeda, F.; Stadanlick, J.; Nunes, F.A.; Alter, H.J.; Chang, K.-M. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 2003, 38, 1437–1448. [Google Scholar] [CrossRef]
- Ward, S.M.; Fox, B.C.; Brown, P.J.; Worthington, J.; Fox, S.B.; Chapman, R.W.; Fleming, K.A.; Banham, A.H.; Klenerman, P. Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J. Hepatol. 2007, 47, 316–324. [Google Scholar] [CrossRef]
- Rushbrook, S.M.; Ward, S.M.; Unitt, E.; Vowler, S.L.; Lucas, M.; Klenerman, P.; Alexander, G.J.M. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J. Virol. 2005, 79, 7852–7859. [Google Scholar] [CrossRef]
- Boettler, T.; Spangenberg, H.C.; Neumann-Haefelin, C.; Panther, E.; Urbani, S.; Ferrari, C.; Blum, H.E.; von Weizsäcker, F.; Thimme, R. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J. Virol. 2005, 79, 7860–7867. [Google Scholar] [CrossRef] [PubMed]
- Langhans, B.; Krämer, B.; Louis, M.; Nischalke, H.D.; Hüneburg, R.; Staratschek-Jox, A.; Odenthal, M.; Manekeller, S.; Schepke, M.; Kalff, J.; et al. Intrahepatic IL-8 producing Foxp3+CD4+ regulatory T cells and fibrogenesis in chronic hepatitis C. J. Hepatol. 2013, 59, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Kotsiou, O.S.; Gourgoulianis, K.I.; Zarogiannis, S.G. IL-33/ST2 Axis in Organ Fibrosis. Front. Immunol. 2018, 9, 2432. [Google Scholar] [CrossRef] [PubMed]
- She, Y.X.; Yu, Q.Y.; Tang, X.X. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.W.; Seidler, S.; Gassler, N.; Nattermann, J.; Luedde, T.; Trautwein, C.; Tacke, F. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS ONE 2011, 6, 21381. [Google Scholar] [CrossRef]
- Wang, H.; Lafdil, F.; Wang, L.; Yin, S.; Feng, D.; Gao, B. Tissue inhibitor of metalloproteinase 1 (TIMP-1) deficiency exacerbates carbon tetrachloride-induced liver injury and fibrosis in mice: Involvement of hepatocyte STAT3 in TIMP-1 production. Cell Biosci. 2011, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Himmel, M.E.; Crome, S.Q.; Ivison, S.; Piccirillo, C.; Steiner, T.S.; Levings, M.K. Human CD4+FOXP3+ regulatory T cells produce CXCL8 and recruit neutrophils. Eur. J. Immunol. 2011, 41, 306–312. [Google Scholar] [CrossRef]
- Kryczek, I.; Wang, L.; Wu, K.; Li, W.; Zhao, E.; Cui, T.; Wei, S.; Liu, Y.; Wang, Y.; Vatan, L.; et al. Inflammatory regulatory T cells in the microenvironments of ulcerative colitis and colon carcinoma. OncoImmunology 2016, 5, 1105430. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Frangogiannis, N.G. Chemokines and cardiac fibrosis. Front Biosci (Sch. Ed.) 2009, 1, 391–405. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, W.; Xing, D.; Li, P.; Fu, J.; Gong, K.; Hage, F.G.; Oparil, S.; Chen, Y.-F. Endothelial cells overexpressing IL-8 receptor reduce cardiac remodeling and dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H590–H598. [Google Scholar] [CrossRef]
- Claassen, M.A.A.; Janssen, H.L.A.; de Knegt, R.J.; Boonstra, A. Controversy on the role of FoxP3+ regulatory T cells in fibrogenesis in chronic hepatitis C virus infections. J. Hepatol. 2014, 60, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Kamimura, K.; Kobayashi, Y.; Ohtsuka, M.; Miura, H.; Ohashi, R.; Yokoo, T.; Kanefuji, T.; Suda, T.; Tsuchida, M.; et al. Effective Prevention of Liver Fibrosis by Liver-targeted Hydrodynamic Gene Delivery of Matrix Metalloproteinase-13 in a Rat Liver Fibrosis Model. Mol. Ther. Nucleic Acids 2016, 5, 276. [Google Scholar] [CrossRef]
- Kim, E.-J.; Cho, H.-J.; Park, D.; Kim, J.Y.; Kim, Y.B.; Park, T.G.; Shim, C.-K.; Oh, Y.-K. Antifibrotic effect of MMP13-encoding plasmid DNA delivered using polyethylenimine shielded with hyaluronic acid. Mol. Ther. 2011, 19, 355–361. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wu, L.; Xie, W.; Shao, Y.; Jiang, J.; Zhao, Z.; Yan, M.; Chen, Z.; Cui, D. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol. 2017, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tang, X.; Yang, M.; Zhang, S.; Li, S.; Chen, Y.; Liu, M.; Guo, Y.; Lu, M. Interleukin 10 Gene-Modified Bone Marrow-Derived Dendritic Cells Attenuate Liver Fibrosis in Mice by Inducing Regulatory T Cells and Inhibiting the TGF-β/Smad Signaling Pathway. Mediat. Inflamm. 2019, 2019, 4652596. [Google Scholar] [CrossRef]
- Starkey Lewis, P.; Campana, L.; Aleksieva, N.; Cartwright, J.A.; Mackinnon, A.; O’Duibhir, E.; Kendall, T.; Vermeren, M.; Thomson, A.; Gadd, V.; et al. Alternatively activated macrophages promote resolution of necrosis following acute liver injury. J. Hepatol. 2020, 73, 349–360. [Google Scholar] [CrossRef]
- Watanabe, Y.; Tsuchiya, A.; Seino, S.; Kawata, Y.; Kojima, Y.; Ikarashi, S.; Starkey Lewis, P.J.; Lu, W.-Y.; Kikuta, J.; Kawai, H.; et al. Mesenchymal Stem Cells and Induced Bone Marrow-Derived Macrophages Synergistically Improve Liver Fibrosis in Mice. Stem Cells Transl. Med. 2019, 8, 271–284. [Google Scholar] [CrossRef]
- Bird, T.G.; Müller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.-Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, 1230. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Oo, Y.H.; Ackrill, S.; Cole, R.; Jenkins, L.; Anderson, P.; Jeffery, H.C.; Jones, N.; Jeffery, L.E.; Lutz, P.; Wawman, R.E.; et al. Liver homing of clinical grade Tregs after therapeutic infusion in patients with autoimmune hepatitis. JHEP Rep. 2019, 1, 286–296. [Google Scholar] [CrossRef]
- Sánchez-Fueyo, A.; Whitehouse, G.; Grageda, N.; Cramp, M.E.; Lim, T.Y.; Romano, M.; Thirkell, S.; Lowe, K.; Fry, L.; Heward, J.; et al. Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. Am. J. Transpl. 2020, 20, 1125–1136. [Google Scholar] [CrossRef]
- Rhodes, K.R.; Meyer, R.A.; Wang, J.; Tzeng, S.Y.; Green, J.J. Biomimetic tolerogenic artificial antigen presenting cells for regulatory T cell induction. Acta Biomater. 2020, 112, 136–148. [Google Scholar] [CrossRef]
- Gu, L.; Deng, W.S.; Sun, X.F.; Zhou, H.; Xu, Q. Rapamycin ameliorates CCl4-induced liver fibrosis in mice through reciprocal regulation of the Th17/Treg cell balance. Mol. Med. Rep. 2016, 14, 1153–1161. [Google Scholar] [CrossRef]
- Anthony, B.; Allen, J.T.; Li, Y.S.; McManus, D.P. Hepatic stellate cells and parasite-induced liver fibrosis. Parasites Vectors 2010, 3, 60. [Google Scholar] [CrossRef]
- Silva Pereira, S.; Trindade, S.; De Niz, M.; Figueiredo, L.M. Tissue tropism in parasitic diseases. Open Biol. 2019, 9, 190036. [Google Scholar] [CrossRef]
- Tang, Z.-L.; Huang, Y.; Yu, X.-B. Current status and perspectives of Clonorchis sinensis and clonorchiasis: Epidemiology, pathogenesis, omics, prevention and control. Infect. Dis. Poverty 2016, 5, 71. [Google Scholar] [CrossRef]
- Allen, J.E.; Sutherland, T.E. Host protective roles of type 2 immunity: Parasite killing and tissue repair, flip sides of the same coin. Semin. Immunol. 2014, 26, 329–340. [Google Scholar] [CrossRef]
- Zhang, B.-B.; Yan, C.; Fang, F.; Du, Y.; Ma, R.; Li, X.-Y.; Yu, Q.; Meng, D.; Tang, R.-X.; Zheng, K.-Y. Increased hepatic Th2 and Treg subsets are associated with biliary fibrosis in different strains of mice caused by Clonorchis sinensis. PLoS ONE 2017, 12, 171005. [Google Scholar] [CrossRef]
- Xu, S.; Gu, M.; Wu, K.; Li, G. Unraveling the Role of Hydroxyproline in Maintaining the Thermal Stability of the Collagen Triple Helix Structure Using Simulation. J. Phys. Chem. B 2019, 123, 7754–7763. [Google Scholar] [CrossRef]
- Yan, C.; Zhang, B.-B.; Hua, H.; Li, B.; Zhang, B.; Yu, Q.; Li, X.-Y.; Liu, Y.; Pan, W.; Liu, X.-Y.; et al. The Dynamics of Treg/Th17 and the Imbalance of Treg/Th17 in Clonorchis sinensis-Infected Mice. PLoS ONE 2015, 10, 143217. [Google Scholar] [CrossRef]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab. Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef]
- Ikeno, Y.; Ohara, D.; Takeuchi, Y.; Watanabe, H.; Kondoh, G.; Taura, K.; Uemoto, S.; Hirota, K. Foxp3+ Regulatory T Cells Inhibit CCl4-Induced Liver Inflammation and Fibrosis by Regulating Tissue Cellular Immunity. Front. Immunol. 2020, 11, 584048. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, M.; Liu, X.; Bai, L.; Kong, M.; Chen, Y.; Zheng, S.; Liu, S.; Wan, Y.-J.Y.; Duan, Z.; et al. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Transl. Res. 2016, 169, 67–79. [Google Scholar] [CrossRef]
- Zhou, X.; Hovell, C.J.; Pawley, S.; Hutchings, M.I.; Arthur, M.J.; Iredale, J.P.; Benyon, R.C. Expression of matrix metalloproteinase-2 and -14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004, 24, 492–501. [Google Scholar] [CrossRef]
- Zhang, X.; Lou, J.; Bai, L.; Chen, Y.; Zheng, S.; Duan, Z. Immune Regulation of Intrahepatic Regulatory T Cells in Fibrotic Livers of Mice. Med. Sci. Monit. 2017, 23, 1009–1016. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S. Natural killer and natural killer T cells in liver fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1061–1069. [Google Scholar] [CrossRef]
- Langhans, B.; Alwan, A.W.; Krämer, B.; Glässner, A.; Lutz, P.; Strassburg, C.P.; Nattermann, J.; Spengler, U. Regulatory CD4+ T cells modulate the interaction between NK cells and hepatic stellate cells by acting on either cell type. J. Hepatol. 2015, 62, 398–404. [Google Scholar] [CrossRef]
- Hesse, M.; Piccirillo, C.A.; Belkaid, Y.; Prufer, J.; Mentink-Kane, M.; Leusink, M.; Cheever, A.W.; Shevach, E.M.; Wynn, T.A. The Pathogenesis of Schistosomiasis Is Controlled by Cooperating IL-10-Producing Innate Effector and Regulatory T Cells. J. Immunol. 2004, 172, 3157. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Rosen, H.R. Galectin-9: Diverse roles in hepatic immune homeostasis and inflammation. Hepatology 2017, 66, 271–279. [Google Scholar] [CrossRef]
- Fujita, K.; Niki, T.; Nomura, T.; Oura, K.; Tadokoro, T.; Sakamoto, T.; Tani, J.; Yoneyama, H.; Morishita, A.; Kuroda, N.; et al. Correlation between serum galectin-9 levels and liver fibrosis. J. Gastroenterol. Hepatol. 2018, 33, 492–499. [Google Scholar] [CrossRef]
- Claassen, M.A.; de Knegt, R.J.; Tilanus, H.W.; Janssen, H.L.; Boonstra, A. Abundant numbers of regulatory T cells localize to the liver of chronic hepatitis C infected patients and limit the extent of fibrosis. J. Hepatol. 2010, 52, 315–321. [Google Scholar] [CrossRef]
- McGill, M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Stross, L.; Günther, J.; Gasteiger, G.; Asen, T.; Graf, S.; Aichler, M.; Esposito, I.; Busch, D.H.; Knolle, P.; Sparwasser, T.; et al. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute experimental hepatitis B virus infection in mice. Hepatology 2012, 56, 873–883. [Google Scholar] [CrossRef]
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Teo Hansen Selnø, A.; Teuscher Abeleira, A.M.; Benlaouer, O.; Gonçalves Silva, I.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594. [Google Scholar] [CrossRef]
- Banerjee, H.; Nieves-Rosado, H.; Kulkarni, A.; Murter, B.; McGrath, K.V.; Chandran, U.R.; Chang, A.; Szymczak-Workman, A.L.; Vujanovic, L.; Delgoffe, G.M.; et al. Expression of Tim-3 drives phenotypic and functional changes in Treg cells in secondary lymphoid organs and the tumor microenvironment. Cell Rep. 2021, 36, 109699. [Google Scholar] [CrossRef]
- Lan, Y.-T.; Wang, Z.-l.; Tian, P.; Gong, X.-N.; Fan, Y.-C.; Wang, K. Treg/Th17 imbalance and its clinical significance in patients with hepatitis B-associated liver cirrhosis. Diagn. Pathol. 2019, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, H.; Guo, T. Th17/Treg imbalance is an indicator of liver cirrhosis process and a risk factor for HCC occurrence in HBV patients. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 399–407. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.-K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef]
- Katz, S.C.; Ryan, K.; Ahmed, N.; Plitas, G.; Chaudhry, U.I.; Kingham, T.P.; Naheed, S.; Nguyen, C.; Somasundar, P.; Espat, N.J.; et al. Obstructive Jaundice Expands Intrahepatic Regulatory T Cells, Which Impair Liver T Lymphocyte Function but Modulate Liver Cholestasis and Fibrosis. J. Immunol. 2011, 187, 1150. [Google Scholar] [CrossRef]
- Sa’dyah, N.A.C.; Putra, A.; Dirja, B.T.; Hidayah, N.; Yasmine Azzahara, S.; Candra Satria Irawan, R. Suppression of transforming growth factor-β by mesenchymal stem-cells accelerates liver regeneration in liver fibrosis animal model. Universa Med. 2021, 40, 29–35. [Google Scholar] [CrossRef]
- Steen, E.H.; Wang, X.; Balaji, S.; Butte, M.J.; Bollyky, P.L.; Keswani, S.G. The Role of the Anti-Inflammatory Cytokine Interleukin-10 in Tissue Fibrosis. Adv. Wound Care 2020, 9, 184–198. [Google Scholar] [CrossRef]
- Sziksz, E.; Pap, D.; Lippai, R.; Béres, N.J.; Fekete, A.; Szabó, A.J.; Vannay, Á. Fibrosis Related Inflammatory Mediators: Role of the IL-10 Cytokine Family. Mediat. Inflamm. 2015, 2015, 764641. [Google Scholar] [CrossRef]
- Ostroukhova, M.; Qi, Z.; Oriss, T.B.; Dixon-McCarthy, B.; Ray, r.; Ray, A. Treg-mediated immunosuppression involves activation of the Notch-HES1 axis by membrane-bound TGF-β. J. Clin. Investig. 2006, 116, 996–1004. [Google Scholar] [CrossRef]
- Oh, S.A.; Li, M.O. TGF-β: Guardian of T Cell Function. J. Immunol. 2013, 191, 3973. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Maher, T.M.; Strek, M.E. Antifibrotic therapy for idiopathic pulmonary fibrosis: Time to treat. Respir. Res. 2019, 20, 205. [Google Scholar] [CrossRef]
- Lim, B.J.; Lee, W.-K.; Lee, H.W.; Lee, K.S.; Kim, J.K.; Chang, H.Y.; Lee, J.I. Selective deletion of hepatocyte platelet-derived growth factor receptor α and development of liver fibrosis in mice. Cell Commun. Signal. 2018, 16, 93. [Google Scholar] [CrossRef]
- Zhao, Z.; Lin, C.Y.; Cheng, K. siRNA- and miRNA-based therapeutics for liver fibrosis. Transl. Res. 2019, 214, 17–29. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Wu, H.M.; Kim, T.H.; Kim, A.; Koo, J.H.; Joo, M.S.; Kim, S.G. Liver X Receptor α–Induced Cannabinoid Receptor 2 Inhibits Ubiquitin-Specific Peptidase 4 Through miR-27b, Protecting Hepatocytes From TGF-β. Hepatol. Commun. 2019, 3, 1373–1387. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Johansen, A.K.Z.; Molkentin, J.D. CARdiac Immunotherapy: T Cells Engineered to Treat the Fibrotic Heart. Mol. Ther. 2019, 27, 1869–1871. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Yu, J.; Liu, Z.; Li, C.; Wei, Q.; Zheng, S.; Saeb-Parsy, K.; Xu, X. Regulatory T Cell Therapy Following Liver Transplantation. Liver Transplant. 2021, 27, 264–280. [Google Scholar] [CrossRef]
- Bai, W.; Wang, P.; Yu, F. CAR-T cells shed light on the treatments of fatal liver diseases. Biotarget 2018, 2, 6. [Google Scholar] [CrossRef]
- Akdoğan, Ö.; Atak Yücel, A.; Gök Sargin, Z.; Sönmez, C.; Esendağli Yilmaz, G.; Özenirler, S. Evaluation of Plasma Urokinase-Type Plasminogen Activator Receptor (UPAR) in Patients With Chronic Hepatitis B, C and Non-Alcoholic Fatty Liver Disease (NAFLD) as Serological Fibrosis Marker. J. Clin. Exp. Hepatol. 2019, 9, 29–33. [Google Scholar] [CrossRef]
- Li, G.; Lin, J.; Peng, Y.; Qin, K.; Wen, L.; Zhao, T.; Feng, Q. Curcumol may reverse early and advanced liver fibrogenesis through downregulating the uPA/uPAR pathway. Phytother. Res. 2020, 34, 1421–1435. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.H.; Zhu, Q.; Li, H.H.; Ra, H.J.; Majumdar, S.; Gulick, D.L.; Jerome, J.A.; Madsen, D.H.; Christofidou-Solomidou, M.; Speicher, D.W.; et al. Fibroblast Activation Protein (FAP) Accelerates Collagen Degradation and Clearance from Lungs in Mice. J. Biol. Chem. 2016, 291, 8070–8089. [Google Scholar] [CrossRef] [PubMed]
- Lay, A.J.; Zhang, H.E.; McCaughan, G.W.; Gorrell, M.D. Fibroblast activation protein in liver fibrosis. Front. Biosci. (Landmark Ed) 2019, 24, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Monslow, J.; Klampatsa, A.; Leibowitz, M.; Sun, J.; Liousia, M.; Woodruff, P.; Moon, E.; Todd, L.; Puré, E.; et al. Loss of cells expressing fibroblast activation protein has variable effects in models of TGF-β and chronic bleomycin-induced fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 317, 271–282. [Google Scholar] [CrossRef]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef]
- Pang, N.; Zhang, F.; Ma, X.; Zhu, Y.; Zhao, H.; Xin, Y.; Wang, S.; Chen, Z.; Wen, H.; Ding, J. TGF-β/Smad signaling pathway regulates Th17/Treg balance during Echinococcus multilocularis infection. Int. Immunopharmacol. 2014, 20, 248–257. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Raffin, C.; Vo, L.T.; Bluestone, J.A. Treg cell-based therapies: Challenges and perspectives. Nat. Rev. Immunol. 2020, 20, 158–172. [Google Scholar] [CrossRef]
- Chen, X.; Oppenheim, J.J. Resolving the identity myth: Key markers of functional CD4+FoxP3+ regulatory T cells. Int. Immunopharmacol. 2011, 11, 1489–1496. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authorship, Year, Title | Model | Study Findings | Ref. |
|---|---|---|---|
| Zhao, X. et al. (2013) Endothelial cells overexpressing IL-8 receptor reduce cardiac remodelling following myocardial infarction. | Myocardial infarction model induced by left anterior descending coronary artery ligation in male Sprague-Dawley rats. | Cell-based therapy was able to indirectly reduce fibrosis by decreasing inflammatory cell infiltration by reducing IL-8-mediated pro-inflammatory responses. This may be possible to apply to liver fibrosis models. | [132] |
| Abe, H. et al. (2016) Effective prevention of liver fibrosis by liver-targeted hydrodynamic gene delivery of matrix metalloproteinase-13 in a rat liver fibrosis model. | Liver fibrosis model induced by bile duct ligation in female Wistar rats. | Liver-targeted delivery of MMP13 gene led to decreased levels of hyaluronic acid and a decreased volume of fibrotic tissue, thus may be an effective antifibrotic genetic therapeutic for preventing liver fibrosis progression. | [134] |
| Kim, E.-J. et al. (2011) Antifibrotic effect of MMP13-encoding plasmid DNA delivered using polyethylenimine shielded with hyaluronic acid. | CCl4-induced murine model of liver fibrosis. | Administration of plasmid DNA encoding MMP13 (pMMP13) resulted in increased expression of MMP13 and reduced collagen I deposition in liver tissue with the potential to ameliorate collagen deposition and support fibrosis recovery. | [135] |
| He, B. et al. (2017) The imbalance of Th17/Treg cells is involved in the progression of non-alcoholic fatty liver disease in mice. | Murine model (C57BL/6 mice) of NAFLD. | High-fat-fed mice had a higher frequency of intrahepatic Th17 cells, and lower frequencies of Tregs compared to a normal diet. Use of polyene phosphatidylcholine capsules demonstrated the ability to restore the Treg/Th17 ratio cell balance to reduce inflammation and thus fibrosis within the liver. | [136] |
| Xu, Y. et al. (2019) Interleukin 10 gene-modified bone marrow-derived dendritic cells attenuate liver fibrosis in mice by inducing regulatory T cells and inhibiting the TGF-β/Smad signaling pathway. | CCl4-induced murine model (BALB/c mice) of liver fibrosis. | Lentiviral transduction of IL-10 gene in DCs could expand Tregs and inhibit the TGF-b/smad signalling pathway, offering an exciting therapeutic option for liver fibrosis via production of Tregs. | [137] |
| Starkey Lewis, P. et al. (2020) Alternatively activated macrophages promote the resolution of necrosis following acute liver injury. | Murine model (APAP-ALI mice) of acetaminophen-induced acute livery injury. | Macrophage-based cell therapy demonstrated the ability to reduce hepatocellular necrosis and inflammation and promote liver regeneration. | [138] |
| Watanabe, Y. et al. (2019) Mesenchymal stem cells and induced bone-marrow-derived macrophages synergistically improve liver fibrosis in mice. | CCl4-induced murine model (C57BL/6 mice) of cirrhosis. | Cell therapies using MSCs could be an effective method of treating liver fibrosis, most likely by supporting an M2 phenotypic switch in macrophages, facilitating high phagocytic activity for hepatocyte debris clearance, whilst also increasing hepatocyte proliferation. | [139] |
| Bird, T.G. et al. (2018) TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. | Murine model (C57BL/6J mice) of acetaminophen poisoning), and post-transplant human liver tissue. | Using a novel TGFβ inhibitor (SB525334), treatment was able to improve the survival of hepatocytes via a reduction in senescence and enhance liver regeneration. This likely ameliorates liver fibrosis progression by removing the risk of liver injury and hepatic senescence associated with acetaminophen poisoning. | [140] |
| Amor, C. et al. (2020) Senolytic CAR T cells reverse senescence-associated pathologies. | Murine models of CCl4-induced liver fibrosis and diet-induced NASH, and human liver biopsy samples from patients with liver fibrosis. | uPAR-targeted CAR T cells were shown to eliminate senescent cells both in vitro and in vivo, with no notable toxicity in mice, and were also shown to eliminate senescent HSCs which instigated the resolution of hepatic fibrosis and improved liver functionality. | [141] |
| Aghajanian, H. et al. (2019) Targeting cardiac fibrosis with engineered T cells. | Murine model of myocardial infarction model via transverse aortic constriction. | Adoptive transfer of FAP-specific CAR T cells were shown to target quiescent cardiac fibroblasts and reduced the level of fibrosis within the heart. Restoration of cardiac function was also seen post-treatment. | [142] |
| Oo, Y.H. et al. (2019) Liver homing of clinical-grade Tregs after therapeutic infusion in patients with autoimmune hepatitis. | Proof-of-concept study in four patients with autoimmune hepatitis. | Adoptive transfer of autologous polyclonal GMP-grade Tregs showed a suppressive effector phenotype with homing to the inflamed liver, with off-target migration only occurring to the spleen and bone marrow. | [143] |
| Sánchez-Fueyo, A. et al. (2020) Applicability, safety, and biological activity of regulatory T cell therapy in liver transplantation. | Two-site, open-label, dose escalation, Phase I clinical trial in patients undergoing adult cadaveric liver transplantation. | The trial demonstrated that adoptive transfer of autologous polyclonal GMP-grade Tregs was safe and increased the levels of circulatory Tregs. This presents the opportunity of a Treg-based immunotherapy to reduce the dependence of immunosuppression after liver transplantation. | [144] |
| Rhodes, K.R. et al. (2020) Biomimetic tolerogenic artificial antigen presenting cells for regulatory T cell induction. | Single dose of intravenous administration of APCs into wild-type C57BL/6 mice. | APCs were able to induce Tregs and instigate a potent suppressive phenotype with a high concentration of TGFβ within the spleen and lymph nodes. The therapy represents an exciting “off-the-shelf” therapeutic in comparison to adoptive transfer, but its capacity to treat liver diseases is still unclear. | [145] |
| Gu, L. et al. (2016) Rapamycin ameliorates CCl4-induced liver fibrosis in mice via reciprocal regulation of the Th17/Treg cell balance. | CCl4-induced murine model (C57BL/6 mice) of liver fibrosis. | Intraperitoneal administration of rapamycin was shown to correct the Th17/Treg imbalance by reducing Th17 cells and increasing Treg cell levels, with a high suppressive ability leading to suppression in hepatic fibrogenesis. | [146] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lowe, K.O.; Tanase, C.E.; Maghami, S.; Fisher, L.E.; Ghaemmaghami, A.M. Inflammatory Network of Liver Fibrosis and How It Can Be Targeted Therapeutically. Immuno 2023, 3, 375-408. https://doi.org/10.3390/immuno3040023
Lowe KO, Tanase CE, Maghami S, Fisher LE, Ghaemmaghami AM. Inflammatory Network of Liver Fibrosis and How It Can Be Targeted Therapeutically. Immuno. 2023; 3(4):375-408. https://doi.org/10.3390/immuno3040023
Chicago/Turabian StyleLowe, Kirstin O., Constantin E. Tanase, Susan Maghami, Leanne E. Fisher, and Amir M. Ghaemmaghami. 2023. "Inflammatory Network of Liver Fibrosis and How It Can Be Targeted Therapeutically" Immuno 3, no. 4: 375-408. https://doi.org/10.3390/immuno3040023