
The Bitter Side of Sugar Consumption: A Mitochondrial Perspective on Diabetes Development


Abstract
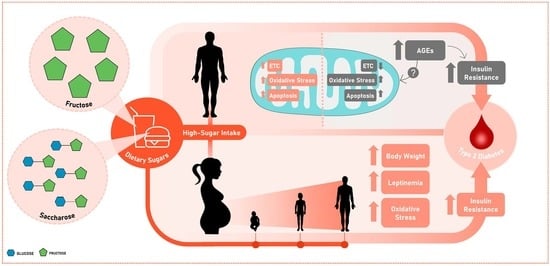
:
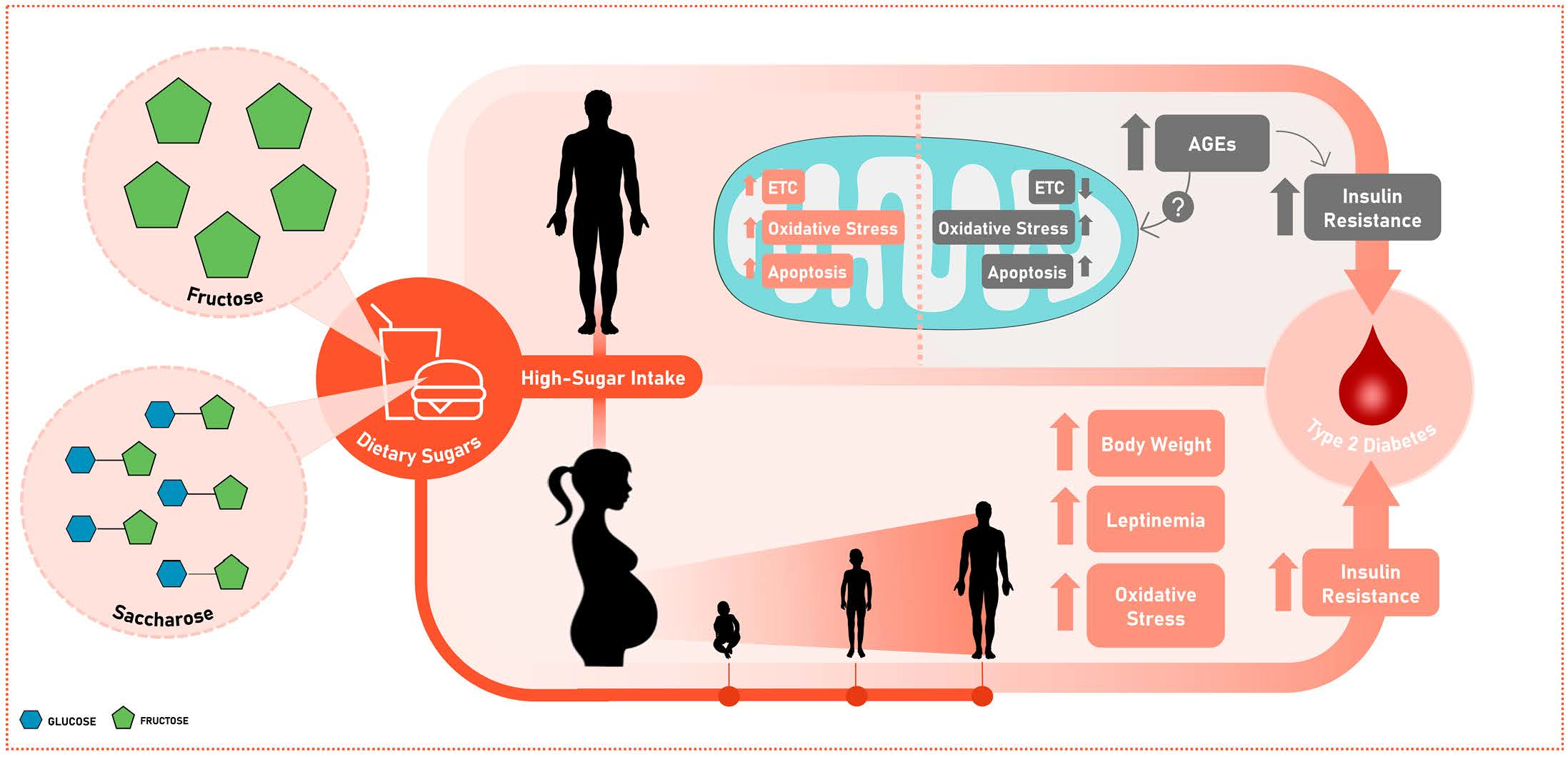{kind=link}
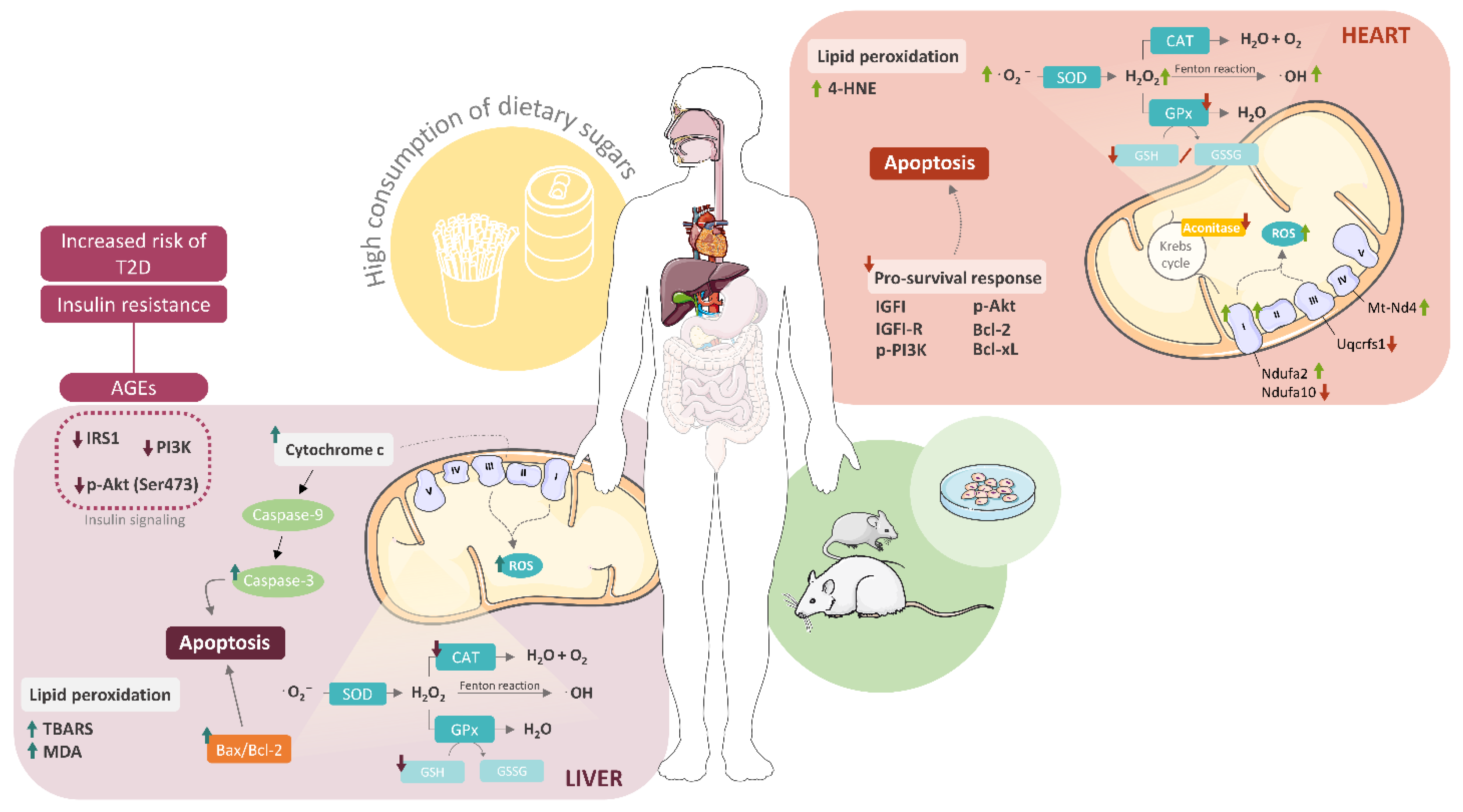{kind=link}
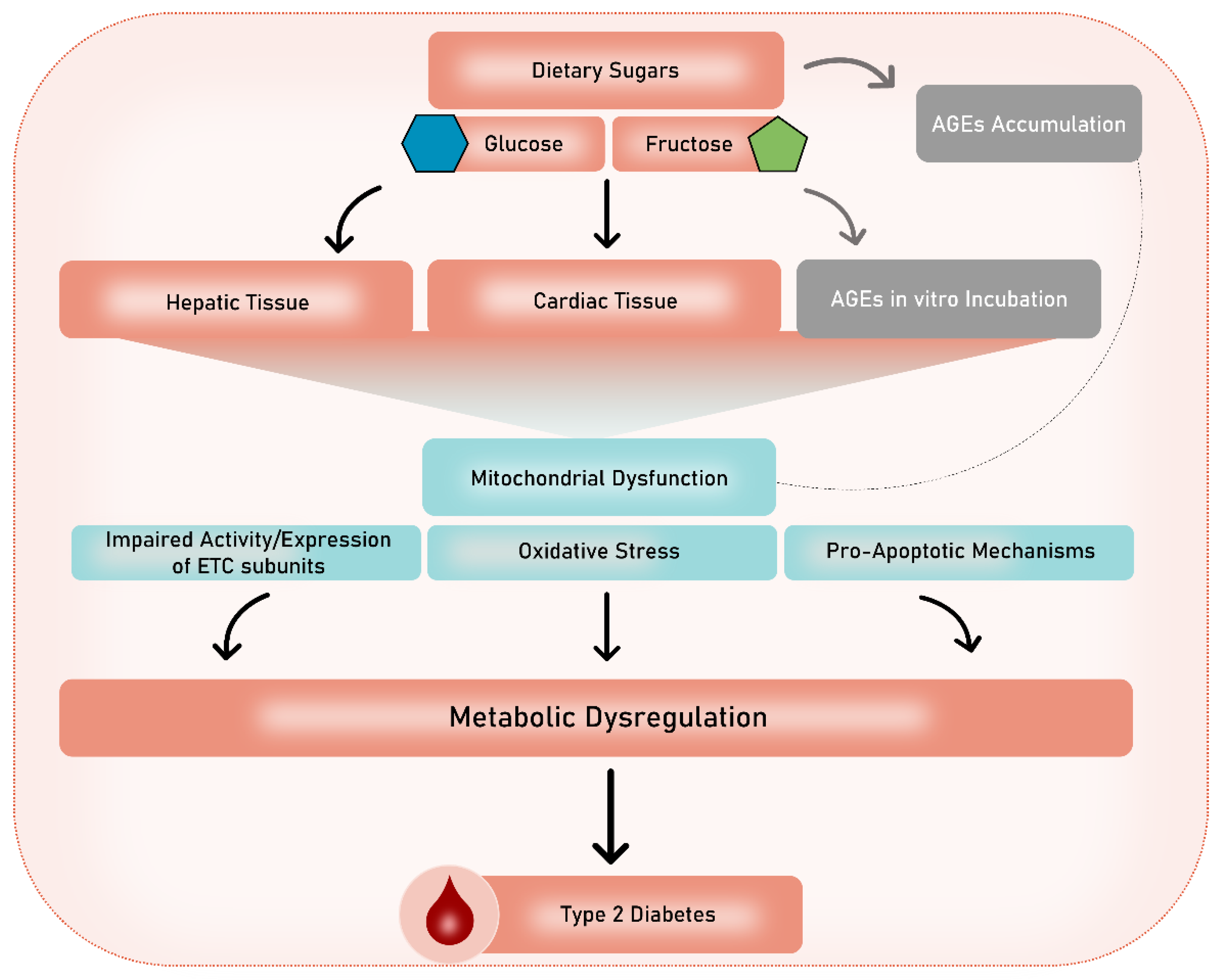{kind=link}
1. Introduction
1.1. Metabolic Syndrome Development: The Case of Type 2 Diabetes
1.2. The Bitter Risks of Increased Dietary Sugars Consumption
2. The Impact of Dietary Sugars on Mitochondrial Function and Promotion of Type 2 Diabetes
2.1. High-Fructose Intake and Mitochondrial Function Modulation
2.2. Advanced Glycation End-Products Accumulation Induce Mitochondrial Dysfunction: A Potential Mechanism in Type 2 Diabetes
3. Type 2 Diabetes and Its Origin in the Womb: The Consequences of Maternal High-Sugar Diet in the Offspring’s Metabolic Function
4. Mitochondrial-Targeted Therapies for T2D Management
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the Metabolic Syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Lemieux, I.; Després, J.P. Metabolic Syndrome: Past, Present and Future. Nutrients 2020, 12, 3501. [Google Scholar] [CrossRef] [PubMed]
- Bovolini, A.; Garcia, J.; Andrade, M.A.; Duarte, J.A. Metabolic Syndrome Pathophysiology and Predisposing Factors. Int. J. Sports Med. 2021, 42, 199–214. [Google Scholar] [CrossRef]
- Gonzalez-Chávez, A.; Chávez-Fernández, J.A.; Elizondo-Argueta, S.; González-Tapia, A.; León-Pedroza, J.I.; Ochoa, C. Metabolic Syndrome and Cardiovascular Disease: A Health Challenge. Arch. Med. Res. 2018, 49, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-B.; Kim, D.H.; Kim, S.M.; Kim, N.H.; Choi, K.M.; Baik, S.H.; Park, Y.G.; Han, K.; Yoo, H.J. Risk of Type 2 Diabetes According to the Cumulative Exposure to Metabolic Syndrome or Obesity: A Nationwide Population-based Study. J. Diabetes Investig. 2020, 11, 1583. [Google Scholar] [CrossRef]
- Li, X.; Zhai, Y.; Zhao, J.; He, H.; Li, Y.; Liu, Y.; Feng, A.; Li, L.; Huang, T.; Xu, A.; et al. Impact of Metabolic Syndrome and It’s Components on Prognosis in Patients with Cardiovascular Diseases: A Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 704145. [Google Scholar] [CrossRef]
- Marott, S.C.W.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; Benn, M. Components of the Metabolic Syndrome and Risk of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3212–3221. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Monforte, M.; Sánchez, E.; Barrio, F.; Costa, B.; Flores-Mateo, G. Metabolic Syndrome and Dietary Patterns: A Systematic Review and Meta-Analysis of Observational Studies. Eur. J. Nutr. 2017, 56, 925–947. [Google Scholar] [CrossRef]
- Redruello-Requejo, M.; Samaniego-Vaesken, M.D.L.; Partearroyo, T.; Rodríguez-Alonso, P.; Soto-Méndez, M.J.; Hernández-Ruiz, Á.; Villoslada, F.L.; Leis, R.; de Victoria, E.M.; Moreno, J.M.; et al. Dietary Intake of Individual (Intrinsic and Added) Sugars and Food Sources from Spanish Children Aged One to <10 Years-Results from the EsNuPI Study. Nutrients 2022, 14, 1667. [Google Scholar] [CrossRef]
- Prinz, P. The Role of Dietary Sugars in Health: Molecular Composition or Just Calories? Eur. J. Clin. Nutr. 2019, 73, 1216–1223. [Google Scholar] [CrossRef]
- Fidler Mis, N.; Braegger, C.; Bronsky, J.; Campoy, C.; Domellöf, M.; Embleton, N.D.; Hojsak, I.; Hulst, J.; Indrio, F.; Lapillonne, A.; et al. Sugar in Infants, Children and Adolescents: A Position Paper of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition Committee on Nutrition. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 681–696. [Google Scholar] [CrossRef] [Green Version]
- Newens, K.J.; Walton, J. A Review of Sugar Consumption from Nationally Representative Dietary Surveys across the World. J. Hum. Nutr. Diet. 2016, 29, 225–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taskinen, M.R.; Packard, C.J.; Borén, J. Dietary Fructose and the Metabolic Syndrome. Nutrients 2019, 11, 1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, V.S.; Hu, F.B. Sugar-Sweetened Beverages and Cardiometabolic Health: An Update of the Evidence. Nutrients 2019, 11, 1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Keim, N.L.; et al. Circulating Concentrations of Monocyte Chemoattractant Protein-1, Plasminogen Activator Inhibitor-1, and Soluble Leukocyte Adhesion Molecule-1 in Overweight/Obese Men and Women Consuming Fructose- or Glucose-Sweetened Beverages for 10 Weeks. J. Clin. Endocrinol. Metab. 2011, 96, E2034–E2038. [Google Scholar] [CrossRef] [Green Version]
- Della Corte, K.W.; Perrar, I.; Penczynski, K.J.; Schwingshackl, L.; Herder, C.; Buyken, A.E. Effect of Dietary Sugar Intake on Biomarkers of Subclinical Inflammation: A Systematic Review and Meta-Analysis of Intervention Studies. Nutrients 2018, 10, 606. [Google Scholar] [CrossRef] [Green Version]
- Della Corte, K.A.; Penczynski, K.; Kuhnle, G.; Perrar, I.; Herder, C.; Roden, M.; Wudy, S.A.; Remer, T.; Alexy, U.; Buyken, A.E. The Prospective Association of Dietary Sugar Intake in Adolescence with Risk Markers of Type 2 Diabetes in Young Adulthood. Front. Nutr. 2021, 7, 362. [Google Scholar] [CrossRef]
- Gill, J.M.R.; Sattar, N. Fruit Juice: Just Another Sugary Drink? Lancet Diabetes Endocrinol. 2014, 2, 444–446. [Google Scholar] [CrossRef]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of Sugar Sweetened Beverages, Artificially Sweetened Beverages, and Fruit Juice and Incidence of Type 2 Diabetes: Systematic Review, Meta-Analysis, and Estimation of Population Attributable Fraction. BMJ 2015, 351, h3576. [Google Scholar] [CrossRef] [Green Version]
- Choo, V.L.; Viguiliouk, E.; Blanco Mejia, S.; Cozma, A.I.; Khan, T.A.; Ha, V.; Wolever, T.M.S.; Leiter, L.A.; Vuksan, V.; Kendall, C.W.C.; et al. Food Sources of Fructose-Containing Sugars and Glycaemic Control: Systematic Review and Meta-Analysis of Controlled Intervention Studies. BMJ 2018, 363, k4644. [Google Scholar] [CrossRef]
- Paglia, L. The Sweet Danger of Added Sugars. Eur. J. Paediatr. Dent. 2019, 20, 89. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.v.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M.; Mv, P.; Gk, F.; Qa, H.; Aj, D.; et al. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. J. Physiol. Endocrinol. Metab. 2019, 316, 268–285. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased Fructose Consumption Is Associated with Fibrosis Severity in Patients with Nonalcoholic Fatty Liver Disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sil, R.; Chakraborti, A.S. Oxidative Inactivation of Liver Mitochondria in High Fructose Diet-Induced Metabolic Syndrome in Rats: Effect of Glycyrrhizin Treatment. Phytother. Res. 2016, 30, 1503–1512. [Google Scholar] [CrossRef]
- Grilo, L.F.; Tocantins, C.; Diniz, M.S.; Gomes, R.M.; Oliveira, P.J.; Matafome, P.; Pereira, S.P. Metabolic Disease Programming: From Mitochondria to Epigenetics, Glucocorticoid Signalling and Beyond. Eur. J. Clin. Investig. 2021, 51, e13625. [Google Scholar] [CrossRef]
- Lou, P.H.; Lucchinetti, E.; Scott, K.Y.; Huang, Y.; Gandhi, M.; Hersberger, M.; Clanachan, A.S.; Lemieux, H.; Zaugg, M. Alterations in Fatty Acid Metabolism and Sirtuin Signaling Characterize Early Type-2 Diabetic Hearts of Fructose-Fed Rats. Physiol. Rep. 2017, 5, e13388. [Google Scholar] [CrossRef]
- Zhang, Y.B.; Meng, Y.H.; Chang, S.; Zhang, R.Y.; Shi, C. High Fructose Causes Cardiac Hypertrophy via Mitochondrial Signaling Pathway. Am. J. Transl. Res. 2016, 8, 4869. [Google Scholar]
- Szűcs, G.; Sója, A.; Péter, M.; Sárközy, M.; Bruszel, B.; Siska, A.; Földesi, I.; Szabó, Z.; Janáky, T.; Vígh, L.; et al. Prediabetes Induced by Fructose-Enriched Diet Influences Cardiac Lipidome and Proteome and Leads to Deterioration of Cardiac Function Prior to the Development of Excessive Oxidative Stress and Cell Damage. Oxidative Med. Cell. Longev. 2019, 2019, 3218275. [Google Scholar] [CrossRef]
- García-Berumen, C.I.; Ortiz-Avila, O.; Vargas-Vargas, M.A.; del Rosario-Tamayo, B.A.; Guajardo-López, C.; Saavedra-Molina, A.; Rodríguez-Orozco, A.R.; Cortés-Rojo, C. The Severity of Rat Liver Injury by Fructose and High Fat Depends on the Degree of Respiratory Dysfunction and Oxidative Stress Induced in Mitochondria. Lipids Health Dis. 2019, 18, 78. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Kannappan, S.; Palanisamy, N.; Anuradha, C.V. Suppression of Hepatic Oxidative Events and Regulation of ENOS Expression in the Liver by Naringenin in Fructose-Administered Rats. Eur. J. Pharmacol. 2010, 645, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Zaki, S.M.; Fattah, S.A.; Hassan, D.S. The Differential Effects of High-Fat and High-Fructose Diets on the Liver of Male Albino Rat and the Proposed Underlying Mechanisms. Folia Morphol. (Warsz) 2019, 78, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beydogan, A.B.; Coskun, Z.M.; Bolkent, S. The Protective Effects of Δ9-Tetrahydrocannabinol against Inflammation and Oxidative Stress in Rat Liver with Fructose-Induced Hyperinsulinemia. J. Pharm. Pharmacol. 2019, 71, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Abdelmegeed, M.A.; Song, B.J. Diet High in Fructose Promotes Liver Steatosis and Hepatocyte Apoptosis in C57BL/6J Female Mice: Role of Disturbed Lipid Homeostasis and Increased Oxidative Stress. Food Chem. Toxicol. 2017, 103, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.M.; Cheng, Y.J.; Wu, L.Y.; Kuo, C.H.; Lee, Y.S.; Wu, M.C.; Huang, C.Y.; Ting, H.; Lee, S.-D. Activated Apoptotic and Anti-Survival Effects on Rat Hearts with Fructose Induced Metabolic Syndrome. Cell Biochem. Funct. 2014, 32, 133–141. [Google Scholar] [CrossRef]
- Park, J.H.; Ku, H.J.; Kim, J.K.; Park, J.-W.; Lee, J.H. Amelioration of High Fructose-Induced Cardiac Hypertrophy by Naringin. Sci. Rep. 2018, 8, 9464. [Google Scholar] [CrossRef] [Green Version]
- Kornfeld, O.S.; Qvit, N.; Haileselassie, B.; Shamloo, M.; Bernardi, P.; Mochly-Rosen, D. Interaction of Mitochondrial Fission Factor with Dynamin Related Protein 1 Governs Physiological Mitochondrial Function In Vivo. Sci. Rep. 2018, 8, 14034. [Google Scholar] [CrossRef] [Green Version]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. Mitochondrial Fission Protein 1: Emerging Roles in Organellar Form and Function in Health and Disease. Front. Endocrinol. (Lausanne) 2021, 12, 274. [Google Scholar] [CrossRef]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial Dynamics and Apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial Fission Mediates High Glucose-Induced Cell Death through Elevated Production of Reactive Oxygen Species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Luo, A.; Liu, X. The Imbalance of Mitochondrial Fusion/Fission Drives High-Glucose-Induced Vascular Injury. Biomolecules 2021, 11, 1779. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Chau, Y.Y.; Ng, H.Y.; Chen, C.H.; Wang, P.W.; Liou, C.W.; Lin, T.K.; Chen, J.B. Empagliflozin Protects HK-2 Cells from High Glucose-Mediated Injuries via a Mitochondrial Mechanism. Cells 2019, 8, 1085. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, S.; Wang, C.Y.; Wang, Y.; Liu, H.X.; Cui, Y.; Zhang, L.-D. Advanced Glycation End Products Induce Oxidative Stress and Mitochondrial Dysfunction in SH-SY5Y Cells. Vitr. Cell. Dev. Biol.-Anim. 2015, 51, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.K.; Woo, J.S.; Kim, Y.H.; Son, D.W.; Lee, S.W.; Song, G.S. Sildenafil Ameliorates Advanced Glycation End Products-Induced Mitochondrial Dysfunction in HT-22 Hippocampal Neuronal Cells. J. Korean Neurosurg. Soc. 2016, 59, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Ottum, M.S.; Mistry, A.M. Advanced Glycation End-Products: Modifiable Environmental Factors Profoundly Mediate Insulin Resistance. J. Clin. Biochem. Nutr. 2015, 57, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced Glycation End-Products: A Review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; del Castillo, M.D.; de la Maza, M.P.; Filip, R.; Gugliucci, A.; Luevano-Contreras, C.; Macías-Cervantes, M.H.; Markowicz Bastos, D.H.; Medrano, A.; Menini, T.; et al. Dietary Advanced Glycation End Products and Their Role in Health and Disease. Adv. Nutr. 2015, 6, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Aragno, M.; Mastrocola, R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 2017, 9, 385. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Matsui, T. Pathologic Role of Dietary Advanced Glycation End Products in Cardiometabolic Disorders, and Therapeutic Intervention. Nutrition 2016, 32, 157–165. [Google Scholar] [CrossRef]
- Ruiz, H.H.; Ramasamy, R.; Schmidt, A.M. Advanced Glycation End Products: Building on the Concept of the “Common Soil” in Metabolic Disease. Endocrinology 2020, 161, bqz006. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Wu, L. Accumulation of Endogenous Methylglyoxal Impaired Insulin Signaling in Adipose Tissue of Fructose-Fed Rats. Mol. Cell. Biochem. 2007, 306, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Mori, T.; Jiang, Y.; Hu, C.; Osaki, Y.; Yoneki, Y.; Sun, Y.; Hosoya, T.; Kawamata, A.; Ogawa, S.; et al. Methylglyoxal Contributes to the Development of Insulin Resistance and Salt Sensitivity in Sprague-Dawley Rats. J. Hypertens. 2009, 27, 1664–1671. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jing, J.; Yu, S.; Song, M.; Tan, H.; Cui, B.; Huang, L. Advanced Glycation Endproducts Induce Apoptosis of Endothelial Progenitor Cells by Activating Receptor RAGE and NADPH Oxidase/JNK Signaling Axis. Am. J. Transl. Res. 2016, 8, 2169–2178. [Google Scholar]
- Akhter, F.; Chen, D.; Akhter, A.; Sosunov, A.A.; Chen, A.; McKhann, G.M.; Yan, S.F.; Yan, S.S. High Dietary Advanced Glycation End Products Impair Mitochondrial and Cognitive Function. J. Alzheimer’s Dis. 2020, 76, 165–178. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, X.; Yuan, Y. Linagliptin Protects Human Chondrogenic ATDC5 Cells against Advanced Glycation End Products (AGEs)-Induced Apoptosis via a Mitochondria-Dependent Pathway. Chem. Biol. Interact. 2020, 315, 108901. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Yap, F.Y.T.; Tong, D.C.K.; Andrikopoulos, S.; Gasser, A.; Thallas-Bonke, V.; Webster, D.E.; Miyazaki, J.I.; Kay, T.W.; Slattery, R.M.; et al. Advanced Glycation End Products Are Direct Modulators of β-Cell Function. Diabetes 2011, 60, 2523–2532. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, J.; Heslehurst, N.; Hall, J.; Schoenaker, D.A.J.M.; Hutchinson, J.; Cade, J.E.; Poston, L.; Barrett, G.; Crozier, S.R.; Barker, M.; et al. Before the Beginning: Nutrition and Lifestyle in the Preconception Period and Its Importance for Future Health. Lancet 2018, 391, 1830–1841. [Google Scholar] [CrossRef]
- Musial, B.; Vaughan, O.R.; Fernandez-Twinn, D.S.; Voshol, P.; Ozanne, S.E.; Fowden, A.L.; Sferruzzi-Perri, A.N. A Western-Style Obesogenic Diet Alters Maternal Metabolic Physiology with Consequences for Fetal Nutrient Acquisition in Mice. J. Physiol. 2017, 595, 4875–4892. [Google Scholar] [CrossRef] [Green Version]
- Kereliuk, S.M.; Brawerman, G.M.; Dolinsky, V.W. Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring. Int. J. Mol. Sci. 2017, 18, 1451. [Google Scholar] [CrossRef]
- Grilo, L.F.; Diniz, M.S.; Tocantins, C.; Areia, A.L.; Pereira, S.P. The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming? Obesities 2022, 2, 236–255. [Google Scholar] [CrossRef]
- Cordero, P.; Vinciguerra, M.; Oben, J.A. Maternal Perinatal Nutrition and Offspring Programming; Elsevier Inc.: Amsterdam, The Netherlands, 2020; ISBN 9780128045725. [Google Scholar]
- Barker, D.J.P. The Developmental Origins of Adult Disease. J. Am. Coll. Nutr. 2004, 23, 588S–595S. [Google Scholar] [CrossRef] [PubMed]
- Lopes, G.A.D.; Ribeiro, V.L.B.; Barbisan, L.F.; Marchesan Rodrigues, M.A. Fetal Developmental Programing: Insights from Human Studies and Experimental Models. J. Matern.-Fetal Neonatal Med. 2017, 30, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, M.; Raji, L.; Prabhu, D.; Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. High-Fructose Diet Is as Detrimental as High-Fat Diet in the Induction of Insulin Resistance and Diabetes Mediated by Hepatic/Pancreatic Endoplasmic Reticulum (ER) Stress. Mol. Cell. Biochem. 2016, 423, 93–104. [Google Scholar] [CrossRef]
- Saad, A.F.; Dickerson, J.; Kechichian, T.B.; Yin, H.; Gamble, P.; Salazar, A.; Patrikeev, I.; Motamedi, M.; Saade, G.R.; Costantine, M.M. High-Fructose Diet in Pregnancy Leads to Fetal Programming of Hypertension, Insulin Resistance, and Obesity in Adult Offspring. Am. J. Obstet. Gynecol. 2016, 215, 378.e1–378.e6. [Google Scholar] [CrossRef]
- Toop, C.R.; Muhlhausler, B.S.; O’Dea, K.; Gentili, S. Impact of Perinatal Exposure to Sucrose or High Fructose Corn Syrup (HFCS-55) on Adiposity and Hepatic Lipid Composition in Rat Offspring. J. Physiol. 2017, 595, 4379–4398. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.D.; Debosch, B.J. Maternal Fructose Diet-Induced Developmental Programming. Nutrients 2021, 13, 3278. [Google Scholar] [CrossRef]
- Koo, S.; Kim, M.; Cho, H.M.; Kim, I. Maternal High-Fructose Intake during Pregnancy and Lactation Induces Metabolic Syndrome in Adult Offspring. Nutr. Res. Pract. 2021, 15, 160–172. [Google Scholar] [CrossRef]
- Rodríguez, L.; Otero, P.; Panadero, M.I.; Rodrigo, S.; Álvarez-Millán, J.J.; Bocos, C. Maternal Fructose Intake Induces Insulin Resistance and Oxidative Stress in Male, but Not Female, Offspring. J. Nutr. Metab. 2015, 2015, 158091. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, L.; Panadero, M.I.; Roglans, N.; Otero, P.; Rodrigo, S.; Álvarez-Millán, J.J.; Laguna, J.C.; Bocos, C. Fructose Only in Pregnancy Provokes Hyperinsulinemia, Hypoadiponectinemia, and Impaired Insulin Signaling in Adult Male, but Not Female, Progeny. Eur. J. Nutr. 2016, 55, 665–674. [Google Scholar] [CrossRef]
- Clayton, Z.E.; Vickers, M.H.; Bernal, A.; Yap, C.; Sloboda, D.M. Early Life Exposure to Fructose Alters Maternal, Fetal and Neonatal Hepatic Gene Expression and Leads to Sex-Dependent Changes in Lipid Metabolism in Rat Offspring. PLoS ONE 2015, 10, e0141962. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Ozaki, H.; Serita, Y.; Sato, S. Maternal Fructose Intake during Pregnancy Modulates Hepatic and Hypothalamic AMP-Activated Protein Kinase Signalling in a Sex-Specific Manner in Offspring. Clin. Exp. Pharmacol. Physiol. 2014, 41, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Chan, J.Y.H.; Hsu, C.N. Maternal Fructose Intake Affects Transcriptome Changes and Programmed Hypertension in Offspring in Later Life. Nutrients 2016, 8, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, F.; Coleman, R.A. Fuel Availability and Fate in Cardiac Metabolism: A Tale of Two Substrates. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1425–1433. [Google Scholar] [CrossRef] [Green Version]
- Leu, S.; Wu, K.L.H.; Lee, W.C.; Tain, Y.L.; Chan, J.Y.H. Maternal Fructose Intake Exacerbates Cardiac Remodeling in Offspring with Ventricular Pressure Overload. Nutrients 2021, 13, 3267. [Google Scholar] [CrossRef]
- Torrealba, N.; Aranguiz, P.; Alonso, C.; Rothermel, B.A.; Lavandero, S. Mitochondria in Structural and Functional Cardiac Remodeling. Adv. Exp. Med. Biol. 2017, 982, 277–306. [Google Scholar] [CrossRef]
- Mericq, V.; Piccardo, C.; Cai, W.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H.; Uribarri, J. Maternally Transmitted and Food-Derived Glycotoxins: A Factor Preconditioning the Young to Diabetes? Diabetes Care 2010, 33, 2232–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Uribarri, J. Dietary Advanced Glycation End Products and Their Potential Role in Cardiometabolic Disease in Children. Horm. Res. Paediatr. 2016, 85, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csongová, M.; Gurecká, R.; Koborová, I.; Celec, P.; Domonkos, E.; Uličná, O.; Somoza, V.; Šebeková, K. The Effects of a Maternal Advanced Glycation End Product-Rich Diet on Somatic Features, Reflex Ontogeny and Metabolic Parameters of Offspring Mice. Food Funct. 2018, 9, 3432–3446. [Google Scholar] [CrossRef]
- Francisco, F.A.; Barella, L.F.; Silveira, S.D.S.; Saavedra, L.P.J.; Prates, K.V.; Alves, V.S.; Franco, C.C.D.S.; Miranda, R.A.; Ribeiro, T.A.; Tófolo, L.P.; et al. Methylglyoxal Treatment in Lactating Mothers Leads to Type 2 Diabetes Phenotype in Male Rat Offspring at Adulthood. Eur. J. Nutr. 2018, 57, 477–486. [Google Scholar] [CrossRef]
- Mortensen, O.H.; Larsen, L.H.; Ørstrup, L.K.H.; Hansen, L.H.L.; Grunnet, N.; Quistorff, B. Developmental Programming by High Fructose Decreases Phosphorylation Efficiency in Aging Offspring Brain Mitochondria, Correlating with Enhanced UCP5 Expression. J. Cereb. Blood Flow Metab. 2014, 34, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Munetsuna, E.; Yamazaki, M.; Mizuno, G.; Sadamoto, N.; Ando, Y.; Fujii, R.; Shiogama, K.; Ishikawa, H.; Suzuki, K.; et al. Maternal Fructose–Induced Oxidative Stress Occurs via Tfam and Ucp5 Epigenetic Regulation in Offspring Hippocampi. FASEB J. 2019, 33, 11431–11442. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of Metformin and Other Biguanides on Oxidative Phosphorylation in Mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Qi, X.; Fu, Y.; Chen, Q.; Cheng, P.; Yu, X.; Sun, X.; Wu, J.; Li, W.; Zhang, Q.; et al. Flavonoids Extracted from Mulberry (Morus alba L.) Leaf Improve Skeletal Muscle Mitochondrial Function by Activating AMPK in Type 2 Diabetes. J. Ethnopharmacol. 2020, 248, 112326. [Google Scholar] [CrossRef]
- Gutierrez, J.A.; Liu, W.; Perez, S.; Xing, G.; Sonnenberg, G.; Kou, K.; Blatnik, M.; Allen, R.; Weng, Y.; Vera, N.B.; et al. Pharmacologic Inhibition of Ketohexokinase Prevents Fructose-Induced Metabolic Dysfunction. Mol. Metab. 2021, 48, 101196. [Google Scholar] [CrossRef]
- van Poelje, P.D.; Potter, S.C.; Erion, M.D. Fructose-1, 6-Bisphosphatase Inhibitors for Reducing Excessive Endogenous Glucose Production in Type 2 Diabetes. Handb. Exp. Pharmacol. 2011, 203, 279–301. [Google Scholar] [CrossRef]
- Mazzoli, A.; Spagnuolo, M.S.; Nazzaro, M.; Gatto, C.; Iossa, S.; Cigliano, L. Fructose Removal from the Diet Reverses Inflammation, Mitochondrial Dysfunction, and Oxidative Stress in Hippocampus. Antioxidants 2021, 10, 487. [Google Scholar] [CrossRef]
- Huang, H.M.; Wu, C.W.; Chen, I.C.; Lee, Y.C.; Huang, Y.S.; Hung, C.Y.; Wu, K.L.H. Maternal High-Fructose Diet Induced Early-Onset Retinopathy via the Suppression of Synaptic Plasticity Mediated by Mitochondrial Dysfunction. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E1173–E1182. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diniz, M.S.; Tocantins, C.; Grilo, L.F.; Pereira, S.P. The Bitter Side of Sugar Consumption: A Mitochondrial Perspective on Diabetes Development. Diabetology 2022, 3, 583-595. https://doi.org/10.3390/diabetology3040044
Diniz MS, Tocantins C, Grilo LF, Pereira SP. The Bitter Side of Sugar Consumption: A Mitochondrial Perspective on Diabetes Development. Diabetology. 2022; 3(4):583-595. https://doi.org/10.3390/diabetology3040044
Chicago/Turabian StyleDiniz, Mariana S., Carolina Tocantins, Luís F. Grilo, and Susana P. Pereira. 2022. "The Bitter Side of Sugar Consumption: A Mitochondrial Perspective on Diabetes Development" Diabetology 3, no. 4: 583-595. https://doi.org/10.3390/diabetology3040044