
Coarse-Grained MD Simulations of Opioid Interactions with the μ-Opioid Receptor and the Surrounding Lipid Membrane


Abstract
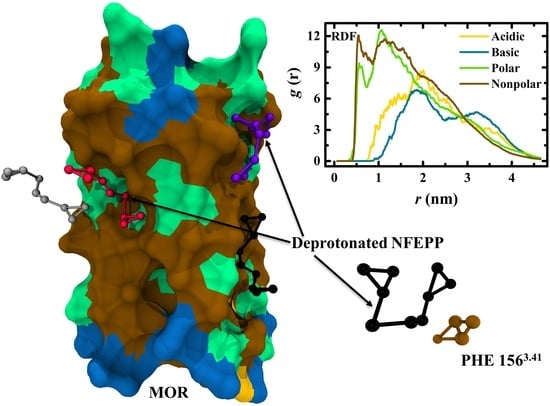
:
1. Introduction
2. Models and Methods
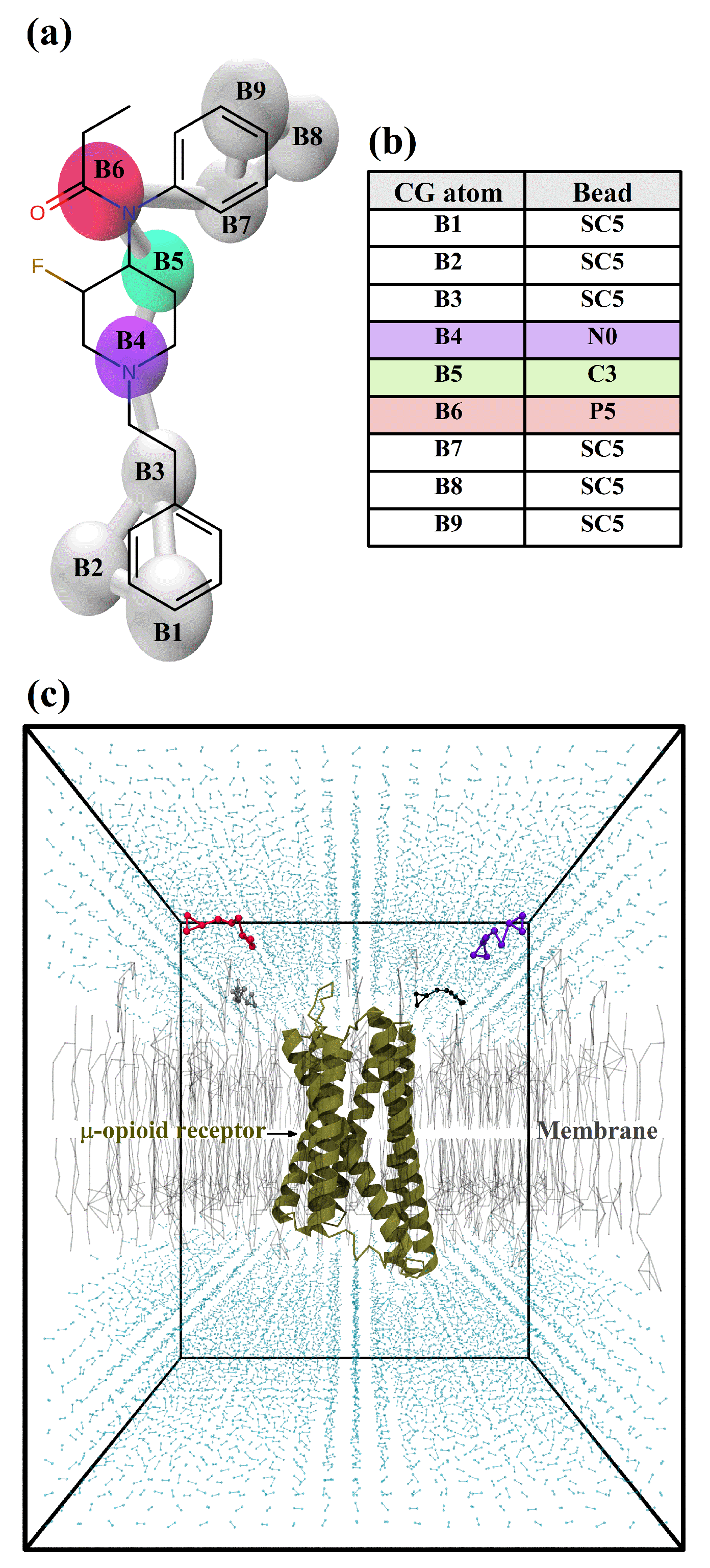
2.1. Fully Atomistic Molecular Dynamics Simulations
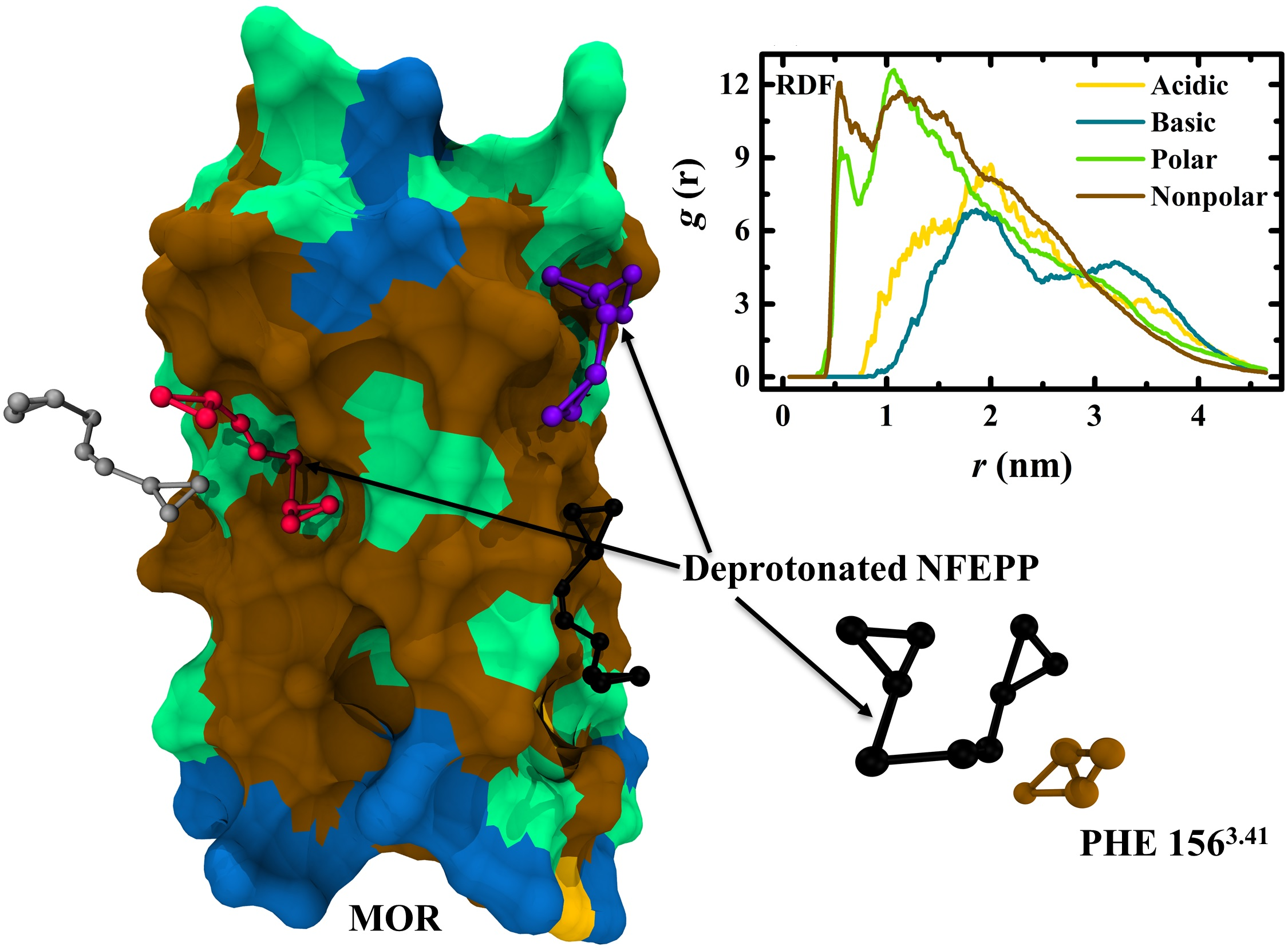
2.2. Coarse-Grained Molecular Dynamics Simulations
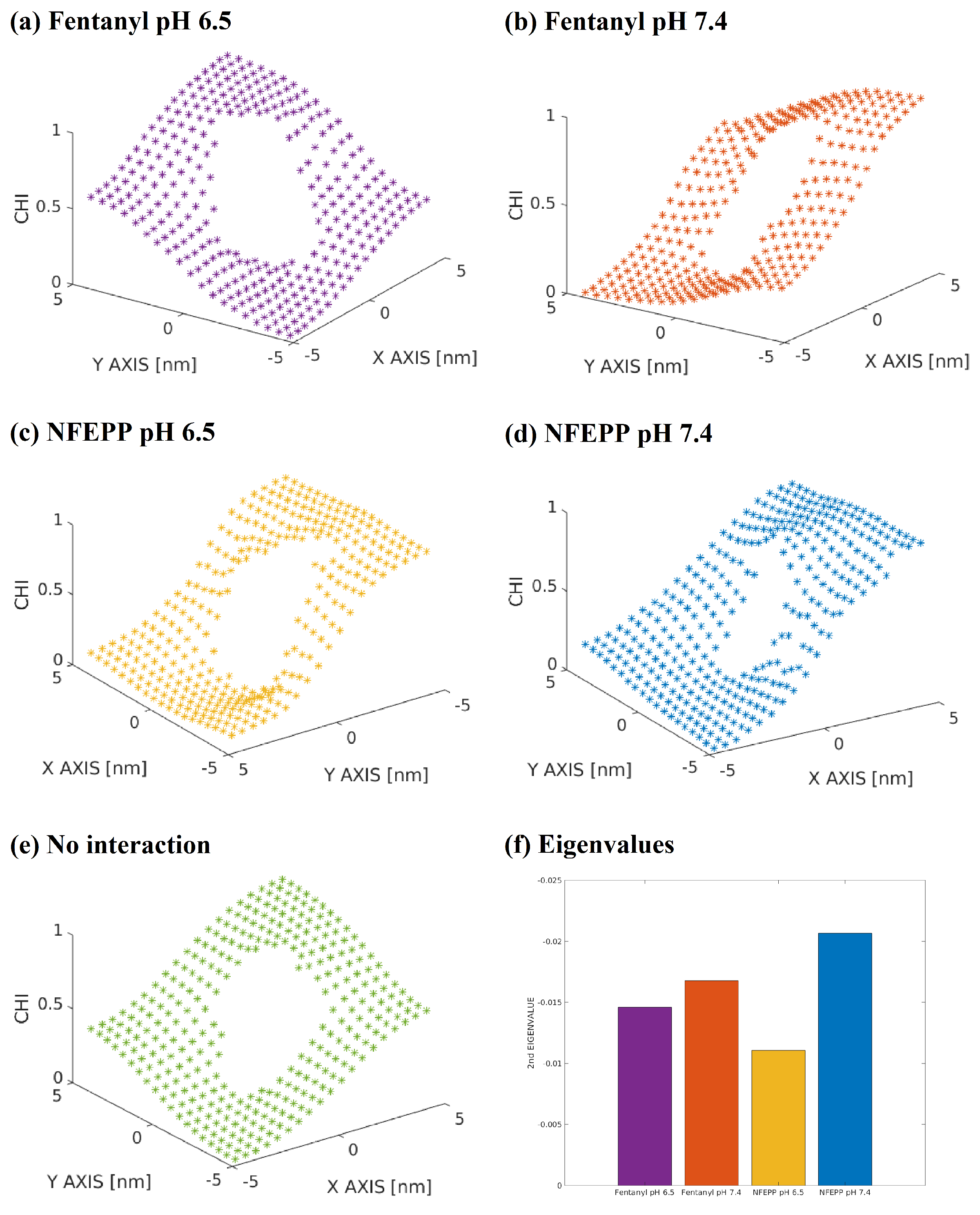
2.3. Square Root Approximation
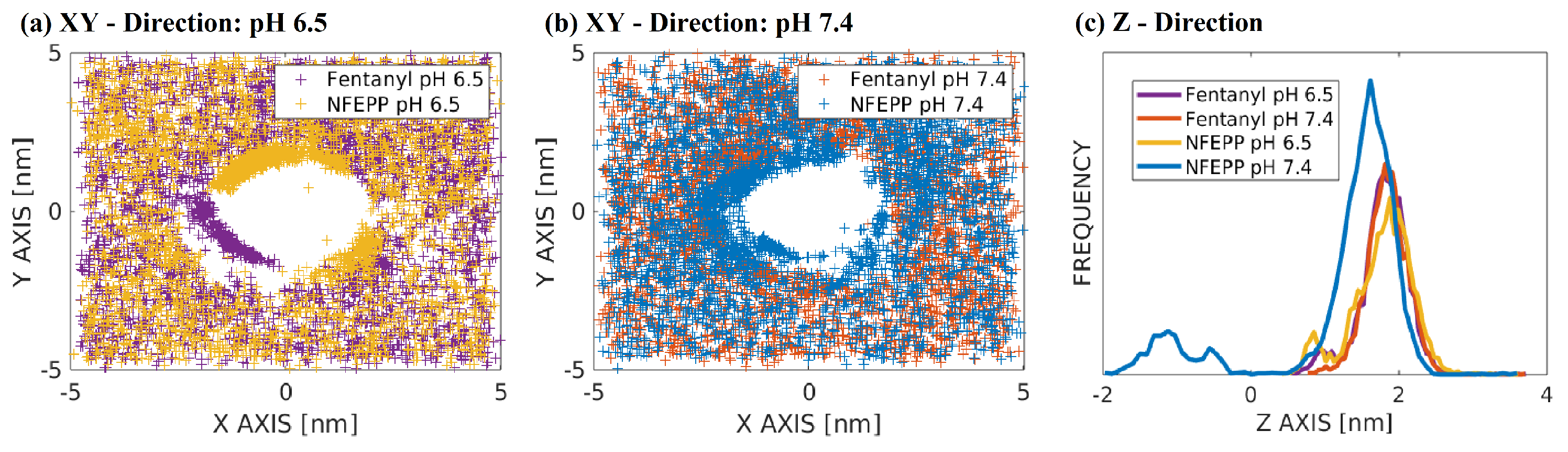
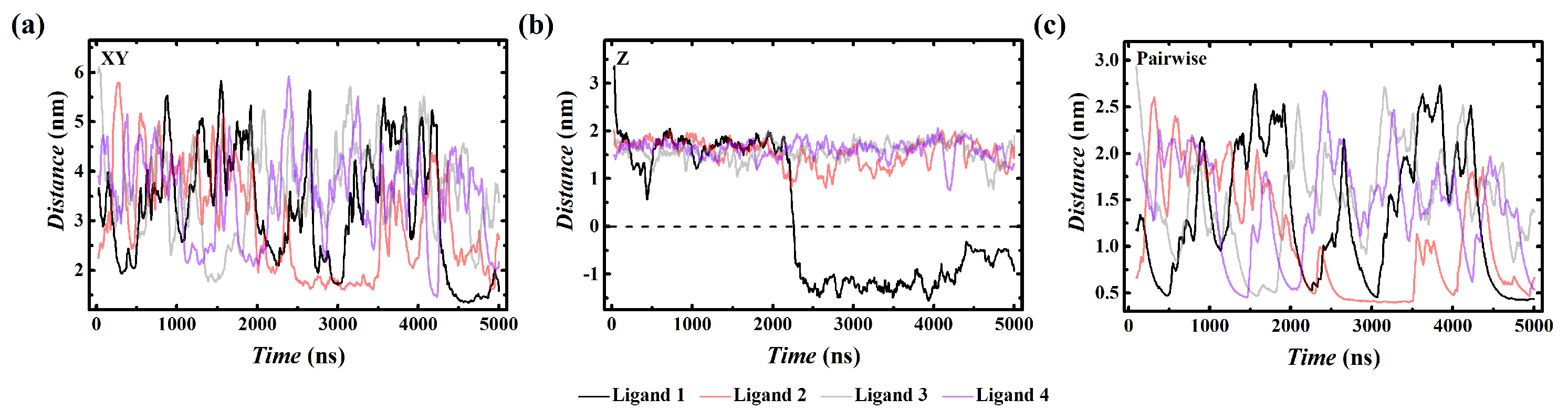
3. Results
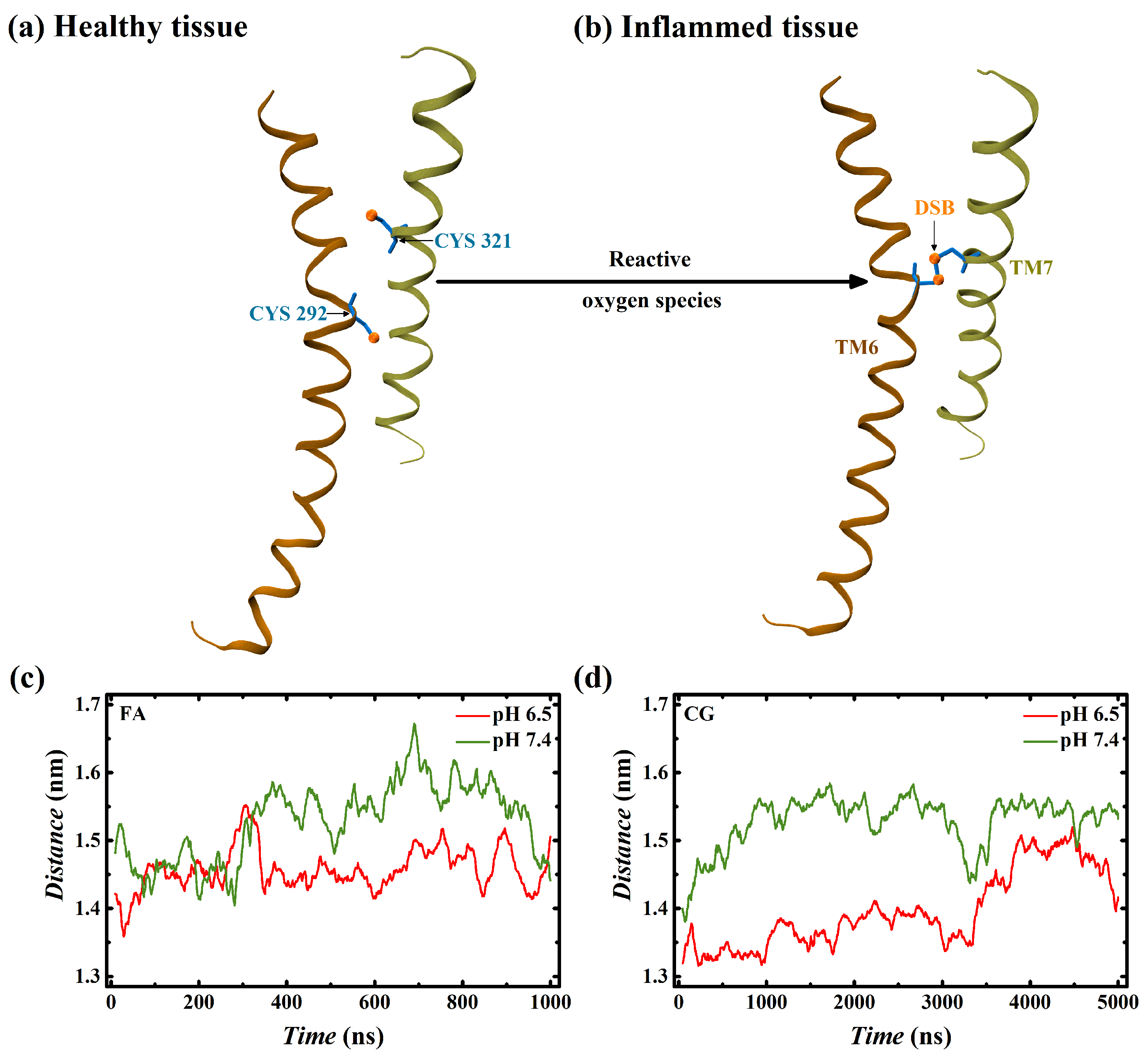
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Spahn, V.; Del-Vecchio, G.; Labuz, D.; Rodriguez-Gaztelumendi, A.; Massaly, N.; Temp, J.; Stein, C. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 2017, 355, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Stein, C. New concepts in opioid analgesia. Expert Opin. Investig. Drugs 2018, 27, 765–775. [Google Scholar] [CrossRef]
- Ray, S.; Sunkara, V.; Schütte, C.; Weber, M. How to calculate pH-dependent binding rates for receptor-ligand systems based on thermodynamic simulations with different binding motifs. Mol. Simul. 2020, 46, 1443–1452. [Google Scholar] [CrossRef]
- Lešnik, S.; Bertalan, E.; Bren, U.; Bondar, A.N. Opioid Receptors and Protonation-Coupled Binding of Opioid Drugs. Int. J. Mol. Sci. 2021, 22, 13353. [Google Scholar] [CrossRef] [PubMed]
- Settimo, L.; Bellman, K.; Knegtel, R.M.A. Comparison of the Accuracy of Experimental and Predicted pKa Values of Basic and Acidic Compounds. Pharm. Res. 2014, 31, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Bowen, W.D.; Pert, C.B. Conformational malleability of opiate receptors: Sulfhydryl modification alters ion-induced μ/δ-ligand selectivity shifts in rat striatal sections. Cell. Mol. Neurobiol. 1982, 2, 115–128. [Google Scholar] [CrossRef]
- Yang, F.; Guo, L.; Li, Y.; Wang, G.; Wang, J.; Zhang, C.; Sun, J.P. Structure, function and pharmacology of human itch receptor complexes. Nature 2021, 600, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Kang, H.J.; Singh, I.; Chen, H.; Zhang, C.; Ye, W.; Roth, B.L. Structure, function and pharmacology of human itch GPCRs. Nature 2021, 600, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.M.; Jakob, U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013, 288, 26489–26496. [Google Scholar] [CrossRef] [Green Version]
- Sutcliffe, K.J.; Corey, R.A.; Alhosan, N.; Cavallo, D.; Groom, S.; Santiago, M.; Kely, E. Interaction With the Lipid Membrane Influences Fentanyl Pharmacology. Adv. Drug Alcohol Res. 2022, 2, 10280. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Thallmair, S.; Conflitti, P.; Ram írez Palacios, C.; Alessandri, R.; Raniolo, S.; Marrink, S.J. Protein—ligand binding with the coarse-grained Martini model. Nat. Commun. 2020, 11, 3714. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- wwPDB Consortium. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [Green Version]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Xu, H.E. Structure of the μ-opioid receptor-G-protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, A.; Dalton, J.A.R.; Giraldo, J. Structural Assessment of Agonist Efficacy in the μ-Opioid Receptor: Morphine and Fentanyl Elicit Different Activation Patterns. J. Chem. Inf. Model. 2021, 61, 1251–1274. [Google Scholar] [CrossRef]
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.-P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers—Aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Patel, D.S.; Ståhle, J.; Park, S.J.; Kern, N.B.; Kim, S.; Lee, J.; Cheng, X.; Valvano, M.A.; Holst, O.; et al. CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J. Chem. Theory Comput. 2019, 15, 775–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, I.S.; Lin, F.Y.; Vanommeslaeghe, K.; Lemkul, J.A.; Armacost, K.A.; Brooks, C.L.; MacKerell, A.D. Parametrization of halogen bonds in the CHARMM general force field: Improved treatment of ligand-protein interactions. Bioorganic Med. Chem. 2016, 24, 4812–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, I.; Charles, L.; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Yesylevskyy, S.O.; Schäfer, L.V.; Sengupta, D.; Marrink, S.J. Polarizable Water Model for the Coarse-Grained MARTINI Force Field. PLoS Comput. Biol. 2010, 6, e810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periole, X.; Cavalli, M.; Marrink, S.J.; Ceruso, M.A. Combining an Elastic Network With a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. [Google Scholar] [CrossRef] [Green Version]
- Ansell, T.B.; Song, W.; Sansom, M.S.P. The Glycosphingolipid GM3 Modulates Conformational Dynamics of the Glucagon Receptor. Biophys. J. 2020, 119, 300–313. [Google Scholar] [CrossRef]
- Barnoud, J. CG Builder. Available online: https://jbarnoud.github.io/cgbuilder/ (accessed on 25 September 2021).
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Ehlert, S.; Stahn, M.; Spicher, S.; Grimme, S. Robust and Efficient Implicit Solvation Model for Fast Semiempirical Methods. J. Chem. Theory Comput. 2021, 17, 4250–4261. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [Green Version]
- Spicher, S.; Grimme, S. Robust Atomistic Modeling of Materials, Organometallic, and Biochemical Systems. Angew. Chem. Int. Ed. 2020, 59, 15665–15673. [Google Scholar] [CrossRef]
- Zhou, J.; Rao, X.; Liu, X.; Li, T.; Zhou, L.; Zheng, Y.; Zhu, Z. Temperature dependent optical and dielectric properties of liquid water studied by terahertz time-domain spectroscopy. AIP Adv. 2019, 9, 035346. [Google Scholar] [CrossRef] [Green Version]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.A.; Ingólfsson, H.I.; Böckmann, R.A.; Tieleman, D.P.; Marrink, S.J. Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, H.I.; Melo, M.N.; van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; de Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid Organization of the Plasma Membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Baoukina, S.; Monticelli, L.; Risselada, H.J.; Marrink, S.J.; Tieleman, D.P. The molecular mechanism of lipid monolayer collapse. Proc. Natl. Acad. Sci. USA 2008, 105, 10803–10808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, C.A.; Sovova, Z.; van Eerden, F.J.; de Vries, A.H.; Marrink, S.J. Martini Force Field Parameters for Glycolipids. J. Chem. Theory Comput. 2013, 9, 1694–1708. [Google Scholar] [CrossRef] [Green Version]
- Gu, R.X.; Ingólfsson, H.I.; deVries, A.H.; Marrink, S.J.; Tieleman, D.P. Ganglioside-Lipid and Ganglioside-Protein Interactions Revealed by Coarse-Grained and Atomistic Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 3262–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrink, S.J.; de Vries, A.H.; Harroun, T.A.; Katsaras, J.; Wassall, S.R. Cholesterol Shows Preference for the Interior of Polyunsaturated Lipid Membranes. J. Am. Chem. Soc. 2008, 130, 10–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, M.N.; Ingólfsson, H.I.; Marrink, S.J. Parameters for Martini sterols and hopanoids based on a virtual-site description. J. Chem. Phys. 2015, 143, 243152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorent, J.H.; Levental, K.R.; Ganesan, L.; Rivera-Longsworth, G.; Sezgin, E.; Doktorova, M.; Levental, I. Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat. Chem. Biol. 2020, 16, 644–652. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Khan, H.M.; Souza, P.C.T.; Thallmair, S.; Barnoud, J.; deVries, A.H.; Marrink, S.J.; Reuter, N. Capturing Choline–Aromatics Cation–π Interactions in the MARTINI Force Field. J. Chem. Theory Comput. 2020, 16, 2550–2560. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- de Jong, D.H.; Baoukina, S.; Ingólfsson, H.I.; Marrink, S.J. Martini straight: Boosting performance using a shorter cutoff and GPUs. Comput. Phys. Commun. 2016, 199, 1–7. [Google Scholar] [CrossRef]
- MATLAB. Version R2019b; The MathWorks Inc.: Natick, MA, USA, 2010. [Google Scholar]
- Lie, H.C.; Fackeldey, K.; Weber, M. A Square Root Approximation of Transition Rates for a Markov State Model. SIAM J. Matrix Anal. Appl. 2013, 34, 738–756. [Google Scholar] [CrossRef] [Green Version]
- Donati, L.; Heida, M.; Keller, B.G.; Weber, M. Estimation of the infinitesimal generator by square-root approximation. J. Phys. Condens. Matter 2018, 30, 425201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, L. Reweighting Methods for Molecular Dynamics. 2019. Available online: http://dx.doi.org/10.17169/refubium-2305 (accessed on 31 August 2022).
- Qiao, B.; Jiménez-Ángeles, F.; Nguyen, T.D.; de la Cruz, M.O. Water follows polar and nonpolar protein surface domains. Proc. Natl. Acad. Sci. USA 2019, 116, 19274–19281. [Google Scholar] [CrossRef] [Green Version]
- Heida, M. Convergences of the square-root approximation scheme to the Fokker-Planck operator. Math. Model. Methods Appl. Sci. 2018, 28, 2599–2635. [Google Scholar] [CrossRef] [Green Version]
- Donati, L.; Weber, M.; Keller, B.G. Markov models from the square root approximation of the Fokker-Planck equation: Calculating the grid-dependent flux. J. Phys. Condens. Matter. 2021, 33, 115902. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, R.; Barnoud, J.; Gertsen, A.S.; Patmanidis, I.; de Vries, A.H.; Souza, P.C.T.; Marrink, S.J. Martini 3 Coarse-Grained Force Field: Small Molecules. Adv. Theory Simul. 2022, 5, 2100391. [Google Scholar] [CrossRef]
- Sampaio, J.L.; Gerl, M.J.; Klose, C.; Ejsing, C.S.; Beug, H.; Simons, K.; Shevchenko, A. Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. USA 2011, 108, 1903–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulkner, C.; Santos-Carballal, D.; Plant, D.F.; de Leeuw, N.H. Atomistic Molecular Dynamics Simulations of Propofol and Fentanyl in Phosphatidylcholine Lipid Bilayers. ACS Omega 2020, 5, 14340–14353. [Google Scholar] [CrossRef]
- Yuan, X.; Raniolo, S.; Limongelli, V.; Xu, Y. The Molecular Mechanism Underlying Ligand Binding to the Membrane-Embedded Site of a G-Protein-Coupled Receptor. J. Chem. Theory Comput. 2018, 14, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Lüllmann, H.; Martins, B.S.; Peters, T. pH-dependent accumulation of fentanyl, lofentanil and alfentanil by beating guineapig atria. BJA Br. J. Anaesth. 1985, 57, 1012–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bower, S. Plasma protein binding of fentanyl. J. Pharm. Pharmacol. 1981, 33, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, G.; Labuz, D.; Temp, J.; Seitz, V.; Kloner, M.; Negrete, R.; Rodriguez-Gaztelumendi, A.; Weber, M.; Machelska, H.; Stein, C. pKa of opioid ligands as a discriminating factor for side effects. Sci. Rep. 2019, 9, 19344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas, R.; Huang, X.P.; Roth, B.L.; Dockendorff, C. β-Fluorofentanyls Are pH-Sensitive Mu Opioid Receptor Agonists. ACS Med. Chem. Lett. 2019, 10, 1353–1356. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upper Leaflet | Percent | Acronym |
|---|---|---|
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine | 20 | POPC |
| 1,2-dioleoyl-sn-glycero-3-phosphocholine | 20 | DOPC |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine | 5 | POPE |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | 5 | DOPE |
| N-stearoyl-D-erythro-sphingosylphosphorylcholine | 15 | DOSM |
| N-stearoyl-D-erythro-monosialodihexosylganglioside | 10 | DPG3 |
| Cholesterol | 25 | Chol |
| Lower leaflet | Percent | Acronym |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine | 5 | POPC |
| 1,2-dioleoyl-sn-glycero-3-phosphocholine | 5 | DOPC |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine | 20 | POPE |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | 20 | DOPE |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine | 8 | POPS |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine | 7 | DOPS |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoinositol-bisphosphate | 10 | POP2 |
| Cholesterol | 25 | Chol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ray, S.; Fackeldey, K.; Stein, C.; Weber, M. Coarse-Grained MD Simulations of Opioid Interactions with the μ-Opioid Receptor and the Surrounding Lipid Membrane. Biophysica 2023, 3, 263-275. https://doi.org/10.3390/biophysica3020017
Ray S, Fackeldey K, Stein C, Weber M. Coarse-Grained MD Simulations of Opioid Interactions with the μ-Opioid Receptor and the Surrounding Lipid Membrane. Biophysica. 2023; 3(2):263-275. https://doi.org/10.3390/biophysica3020017
Chicago/Turabian StyleRay, Sourav, Konstantin Fackeldey, Christoph Stein, and Marcus Weber. 2023. "Coarse-Grained MD Simulations of Opioid Interactions with the μ-Opioid Receptor and the Surrounding Lipid Membrane" Biophysica 3, no. 2: 263-275. https://doi.org/10.3390/biophysica3020017