1. Introduction
The decomposition of small molecules into fragments is a frequent task in drug design. One of the areas where this task is ubiquitously used is Fragment-Based Drug Discovery (FBDD), which has gained significant momentum over the last decades [
1,
2,
3,
4].
A similar request arose when using our recently proposed reverse fragment-based drug discovery (R-FBDD) [
5,
6]. R-FBDD proposes a simple and still useful way to infer the contributions of specific fragments to the interaction energy of the entire ligand with its target using scoring functions. It should be noted that the approach itself is not limited to a specific scheme of ligand decomposition, and the utility of the approach was shown using manual ligand breakdowns. However, to streamline the chemoinformatics applications of the R-FBDD approach, it would be more convenient to couple the analysis provided by R-FBDD with the automatic procedure to break ligands into fragments. We initially decided to adopt one of the existing methods to decompose organic molecules into fragments. For that purpose, several requirements were put forward for the breakdown scheme sought. First, fragments should not overlap since it is important to more or less unambiguously assign the contribution to the binding energy of each of them. In the case of overlapping fragments, it would require additional efforts, which should help reveal what contribution each part of the ligand makes. The second requirement is the ability to naturally use the resulting fragments in medicinal chemists’ practice by appealing to partially intuitive perception, which is however readily shared and persistent among the medicinal chemistry community. This requirement is not easily captured by strict definitions, but it is definitely related to the interpretation of the results obtained in terms of actionable insights. In short, a medicinal chemist should generally agree with the decomposition in terms of the meaningfulness and novelty (related to the intellectual property) of the resulting fragments. It is common wisdom that the contributions of the ligand fragments differ in their significance, and this difference is, in particular, laid in the foundation of the FBDD approach itself, in which the initial hits are required to form (despite low absolute magnitude values of the interaction energy) energetically dense interactions, which are characterized by different ligand efficiency metrics [
7,
8,
9,
10]. Efficient interactions are only possible when most parts of a ligand form complementary interactions with a receptor. Different fragments are responsible for different types of intermolecular interactions, which have been both found useful in numerous drug discovery practices and received a theoretically sound physicochemical interpretation [
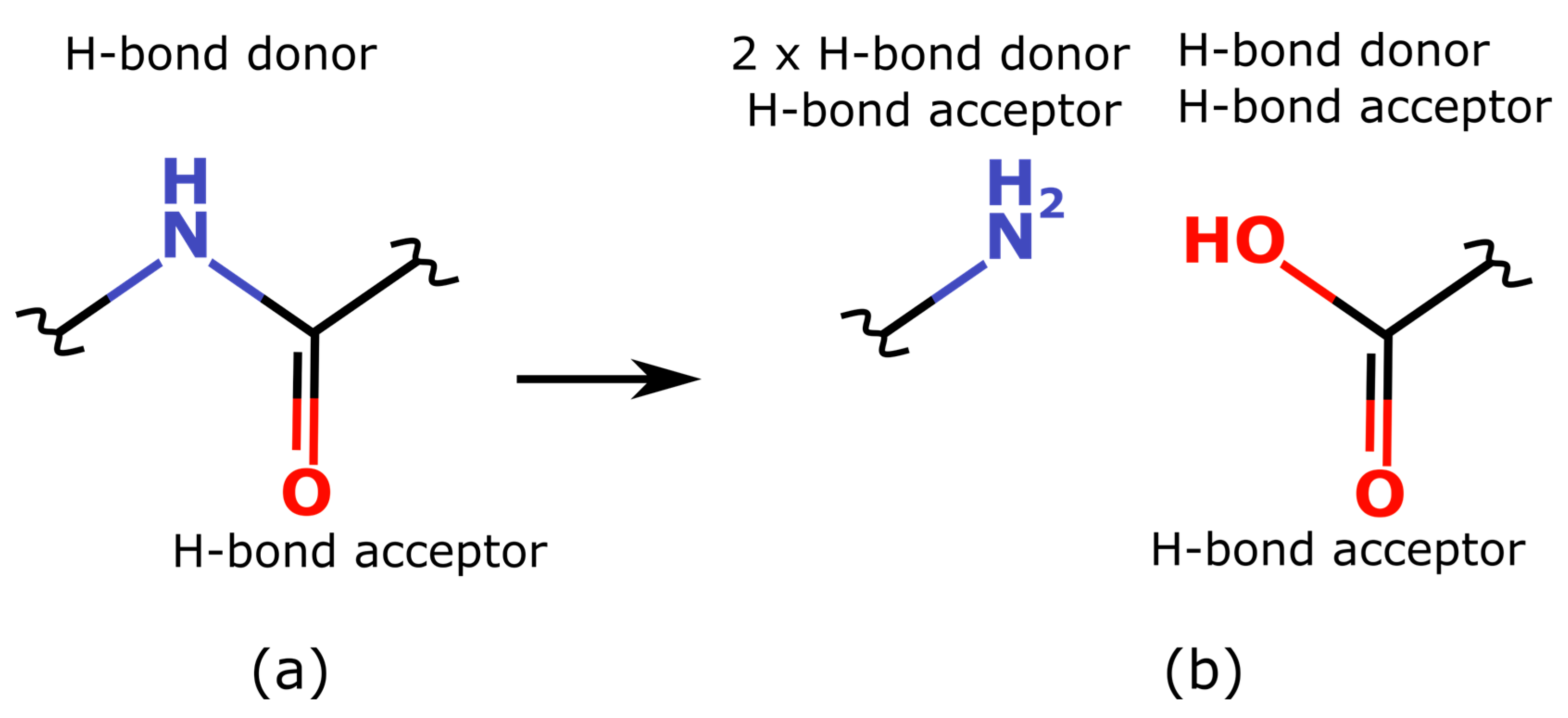
11]. For example, a moiety with an amide bond can act as a hydrogen bond donor, a hydrogen bond acceptor, and a planar conjugated system capable of forming interactions with aromatic systems, known as “amide stacking” [
12,
13,
14,
15]. The phenyl moiety, in turn, is responsible for both hydrophobic interactions and π–π stacking. In drug design, the concept of a pharmacophore is used to build spatial models in which the mutual arrangement of the features responsible for certain preferred interactions is directly related to ligand activity. From the point of view of the pharmacophore, the nitrogen atoms of the amine, aromatic amine, and nitrogen from the amide fragment are different atoms. Therefore, at a practical level, one should obtain a set of fragments with functional groups, reflecting the most significant contribution from the neighboring atoms (such as conjugation) in order for the resulting fragments to adequately capture the main types of intermolecular interactions that are formed between each fragment of the entire ligand and the receptor. We believe the fragments possessing the above properties will be meaningful from the point of view of medicinal chemistry. At the next step we decided to check how the existing methods and programs fulfill the formulated requirements for the resulting fragments.
There exist a number of approaches and methods to break molecules into fragments, such as RECAP, [
16] BRICS, [
17] Murcko scaffolds, [
18] MolBLOCKS, [
19] DAIM, [
20] eMolFrags, and [
21] rdScaffoldNetwork [
22]. However, the main disadvantage of those approaches for our requirements is the lack of medicinal chemical interpretation of the resulting molecular fragments. Each method was considered as a candidate to fulfill the requirements above, aimed at obtaining meaningful fragments to capture different types of interactions between a ligand and a receptor.
Retrosynthetic Combinatorial Analysis Procedure (RECAP) [
16] is perhaps historically the first method that describes the decomposition of molecules into fragments by retrosynthetically breaking along 11 types of bonds that can be formed by common chemical reactions. Mainly, RECAP is aimed to identify “building blocks”, such as monomers or scaffolds, for a set of ligands. Such building blocks could be subsequently used to produce virtual libraries covering the intended chemical space. A similar approach, called Breaking of Retrosynthetically Interesting Chemical Substructures (BRICS) [
17], in which a larger set of 16 rules is used to decompose ligands into fragments, is in fact an extension of RECAP. Both RECAP and BRICS are designed to avoid overlapping between the resulting fragments. However, in both RECAP and BRICS, a fragment meaningful for interaction analysis (e.g., the amide fragment) could be broken into chemical reagents, leading to a missed interpretation of the fragment’s interactions with a target. Bemis and Murcko decomposition is used for the determination of the frameworks (ring systems and linkers) and side-chain fragments [
18]. This algorithm finds numerical chemoinformatics applications to the ligand framework clustering and enumeration in different libraries. This method aims to provide a single framework for a molecule, defined as a combination of rings and inter-ring linkers, excluding all terminal substituents. Thus, the Murcko scaffold provides a very general (not specific) form of the scaffold, which for most practically interesting ligands is larger than the specific pockets in a target that impose specific physicochemical-based interactions. An additional source of violating our requirements is that the Murcko method generally ignores the conjugation within rings and linkers, as well as the exo-conjugated groups, whose presence can significantly alter the conjugation of the system and hence the imposed interaction patterns. Certain terminal substituents, which are not entering the Murcko scaffold, might also be significant in terms of the intermolecular interactions of a ligand with a receptor. The program MolBLOCKS [
19] uses the rules of RECAP, BRICS, and CCQ (cleaving a bond between two carbon atoms of which at least one is connected to a heteroatom) [
23], which also eventually leads to the decomposition of fragments in terms of synthesis. The main additional benefit of MolBLOCKS is the capability to perform statistical analysis on top of the retrosynthetic fragmentation. Thus, in regard to our requirements, MolBLOCKS is equivalent to RECAP and BRICS. Decomposition and Identification of Molecules (DAIM) is a program for decomposing ligands into fragments for fragment-based docking [
20]. The main purpose of DAIM decomposition is to select the most “anchoring” fragments of a ligand to use in a subsequent elaborate fragment-based docking algorithm. In particular, DAIM tries not to split the conjugated systems of a molecule but shreds all rotatable bonds, that leads to excessive fragmentation of aliphatic linkers, which is not suitable for our purposes. The decomposition in the eMolFrags program is based on the BRICS rules, [
21] and aims at extracting “building blocks” for subsequent use in combinatorial synthesis software in order to expand the chemical space defined by possible ligand analogs. In eMolFrags, linkers are defined as the fragments obtained by subtracting the well-defined “bricks” (conjugated and ring blocks) from the ligand; they are rather reasonable from a medicinal chemist’s point of view. However, the reliance on retrosynthetic decomposition still leads to the breaking of certain conjugated fragments. The RDKit module rdScaffoldNetwork [
22] is a versatile and flexible tool to build a nested hierarchy of fragments. rdScaffoldNetwork tries to preserve the conjugation between the fragments, but it generates a set of overlapping fragments. The latter complicates the decomposition of the whole ligand properties into fragment contributions.
All the above-described methods perform the decomposition either in terms of synthetic rules (labile bonds, for example) or in terms of ring systems, linkers, and substituents. Our goal is to provide a qualitatively different decomposition and to find a set of fragments that can be readily interpreted from the point of view of binding and physicochemical properties. In other words, it should be a set of fragments meaningful to medicinal chemists. Unlike the existing decomposition methods, it should allow one to interpret the intermolecular interaction in terms of the complete and non-overlapped sets of fragments, which can help guide a rational design in the right direction. This is achieved by understanding and exploiting the interaction of fragments with pockets of the binding site. Meaningful fragments with the correct types of atoms and conjugated bonds have certain functional groups that are responsible for various intermolecular interactions. Such a set should definitely be important in the analysis of drugs since fragments, which were optimized in drugs, should already possess appropriate ADMET properties [
24].
Alternatively, some of the observed decomposition methods and programs are not suitable for our purposes since they offer sets of overlapping fragments. The presence of overlapping fragments can greatly complicate the analysis and lead to incorrect conclusions since a part of the contribution to the binding energy can be erroneously attributed to the presence of an overlapping fragment. Of course, there are cases where a certain fragment seems to be conjugated to several neighboring fragments, so any bond breaking would lead to the loss of such conjugation. Such cumbersome cases are described below, and we generally believe that the automatic fragmentation procedure should produce maximally unambiguous results of decomposition.
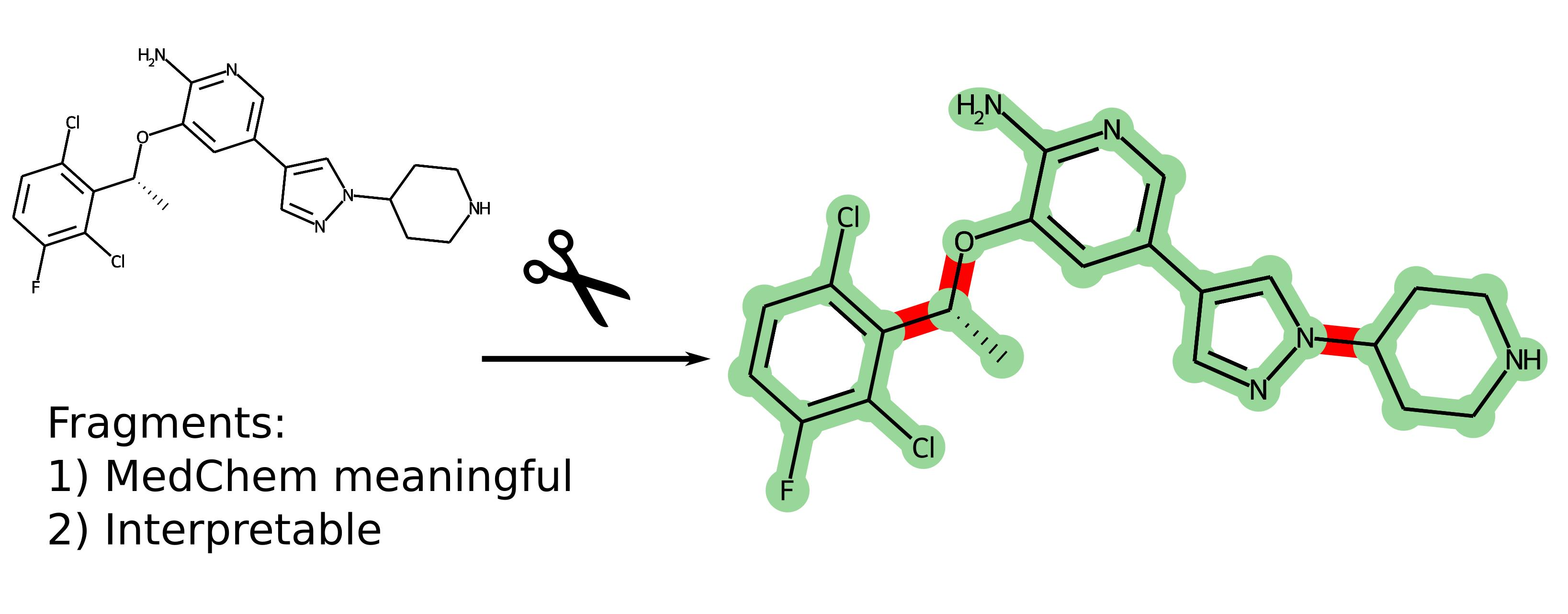
In what follows, we propose a simple means to formulate the requirements for the fragments to be meaningful for medicinal chemistry, the main principle being that strongly conjugated systems should not be broken upon decomposition. The above thinking is both close in spirit and complementary to the tools of the FBDD approach. One of the planned consumers of this decomposition is the Reverse Fragment-Based Drug Discovery (R-FBDD) approach [
5,
6], in which the contributions of individual non-overlapping fragments to the binding energy of the whole ligand are estimated based on the complex geometry by using, in particular, scoring functions.
It is evident that the existing methods of splitting molecules into fragments were coiled using a different set of requirements, aiming at different goals. Thus, in this work, we offer new requirements and a new method, MedChemFrag, with a defined set of rules for the decomposition of molecules.
3. Results and Discussion
Our main goal is to split ligands into fragments that are close to the expectation of the FBDD approach and that retain the character of interaction fingerprints as they occur in the whole ligand. The latter property ensures a relevant interpretation of the intermolecular interactions of the fragments with the binding site.
We are interested in the simplest breakdown that satisfies the requirements, which we believe reflects the medicinal chemist’s experience the most. We propose to use two such requirements: (1) the conjugated fragments should not be broken, and (2) the linkers should be as integral as possible (not excessively broken into pieces).
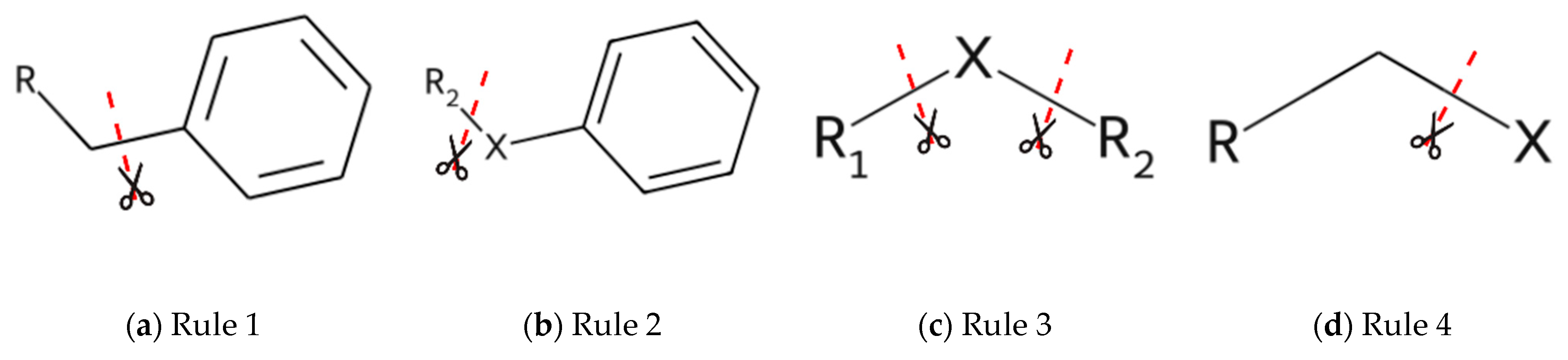
The new decomposition method is based on the following principles: (1) the conjugated bonds are not broken; (2) the ring bonds are preserved; and (3) one-atom (non-hydrogen) fragments are not allowed. Based on these principles, we derive a set of decomposition rules (
Figure 1):
To break a bond between any ring and an sp3-carbon of a fragment (excluding a –CH3 fragment);
To break a bond in fragment R1-X-R2 between X and R2, where R1—aromatic ring, R2 —any fragment with a carbon atom that is connected to X and X = –O–, –S–, >NR3, >NH, >C=O, >C=NR3, >C=CH2, >C=S, –C(=O)O–, –N=N–, –NHC(=O)–, –C(=O)NH–, –S(O2)NH–, or –NHS(O2)–; R3—any fragment;
To break two bonds in fragment R1-X-R2 between X, R1 and R2, where R1 and R2—any fragment with a non-ring atom that is connected with X and X = –O–, –S–, –NH–, >C=O, >C=NR3, >C=CH2, >C=S, –C(=O)O–, –N=N–, –NHC(=O)–, –C(=O)NH–, –S(O2)NH–, –NHS(O2)NH–; R3—any fragment;
To break a bond between an sp3-carbon of a fragment (excluding the –CH3 fragment) and X, where X = –C(=O)OR3, –OC(O)R3, –N(R3)C(=O)R3, –C(=O)N(R3), –NHR3, –N(R3)2, –OR3, –SR3, –S(O2)NHR3, –NHS(O2)R3; R3—any fragment.
This set of principles allows one to meaningfully extract heterocyclic fragments with exo-conjugated bonds, as well as the largest saturated linkers, without breaking all rotatable bonds inside the linker. The main targets for fragmentation are non-conjugated bonds between rings and linkers. It should be noted that the algorithm decomposes molecules into non-overlapping fragments.
The method was applied to a set of molecules from the DrugBank database [
26], downloaded from the drugbank.ca website (version 5.1.8, released 3 January 2021), containing 14,470 drugs. The drugs were filtered by their approval status and the presence of the PDB code. Only molecules found in the scPDB database were used [
27]. As a result, 302 drugs were selected. NADH (DB00157), Sapropterin (DB00360), Flavin mononucleotide (DB03247), and Flavin adenine dinucleotide (DB03147) were removed from the set as cofactors of many complexes. As a result, a set of 298 drugs was obtained. Drugs were broken down into fragments using the SMARTS queries corresponding to the breakdown rules described above (
Table S1), applying the specially developed Python script using the RDKit library.
After defragmentation, we examined the resulting fragments (
Table S2). In cases where the drugs were not divided into fragments (
Table S3), such molecules were removed from further analysis.
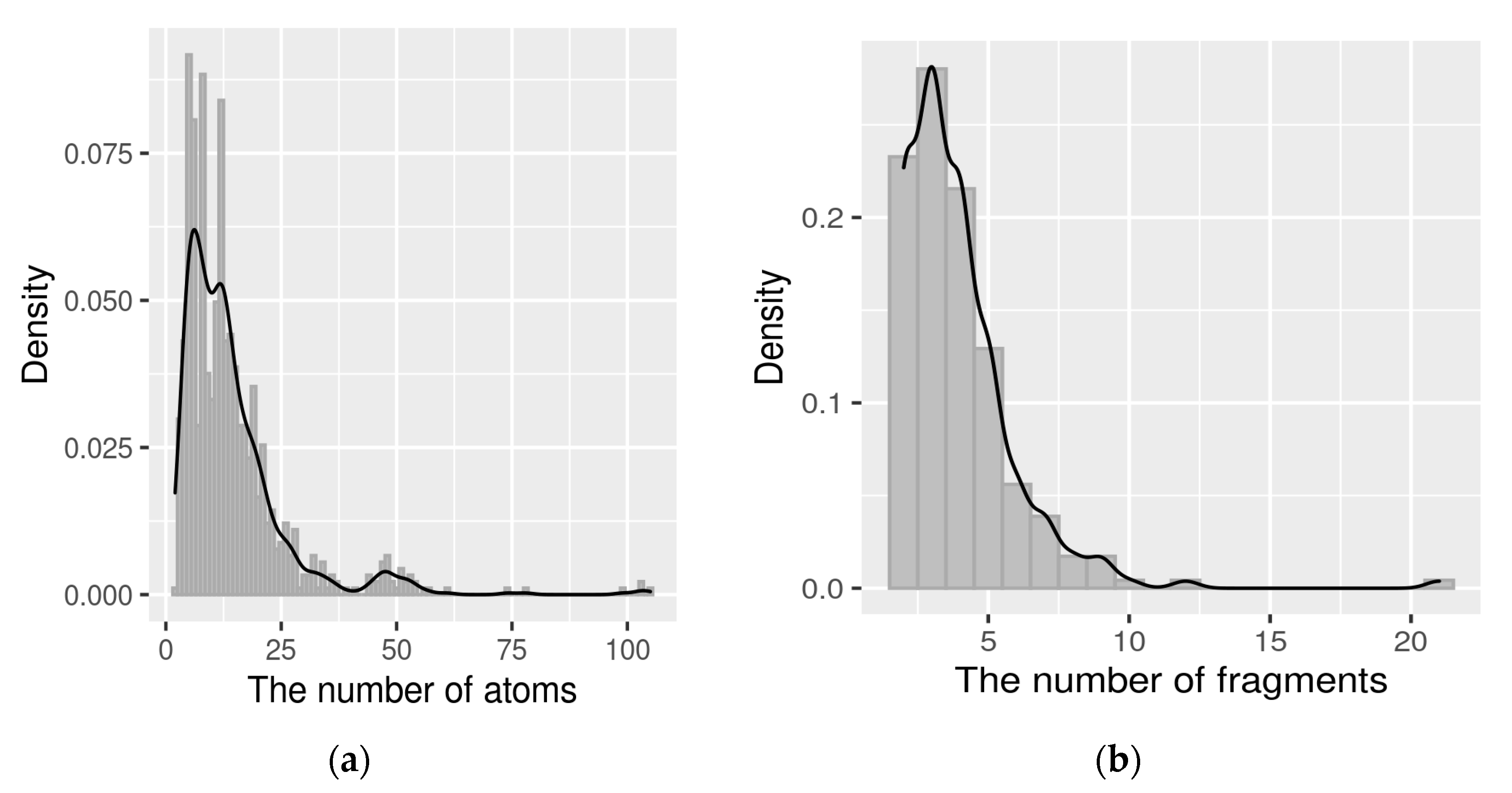
To assess the quality of the breakdown, the distribution of the number of heavy atoms in a fragment (
Figure 2a) and the number of fragments in a molecule were considered (
Figure 2b).
Most of the fragments contain up to 15 atoms, which is consistent with the concept of fragment-like compounds since their molecular weight is less than 300 Da [
28]. Most of the molecules also broke up into a reasonable number of fragments, up to five fragments. This number could be rationalized by considering drug discovery practices. First, it is hard and impractical to work with a large number of fragments. Second, in the fragment approach, the structure is sequentially grown into a drug in several iterations by adding new fragments, and, as a rule, the number of iterations rarely exceeds five. Third, we have an expectation of how many fragments we should receive, namely 2–3 medium and large fragments (medicinal and chemically significant) with a mass of up to 300 Da and 2–3 small fragments, including linkers. In sum, the molecular weight of these fragments should give a value that meets modern requirements for drugs [
29]. Therefore, the number of explicit fragments in the structure, up to five, seems reasonable.
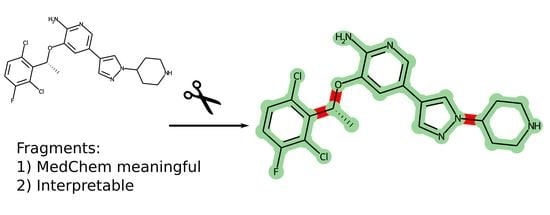
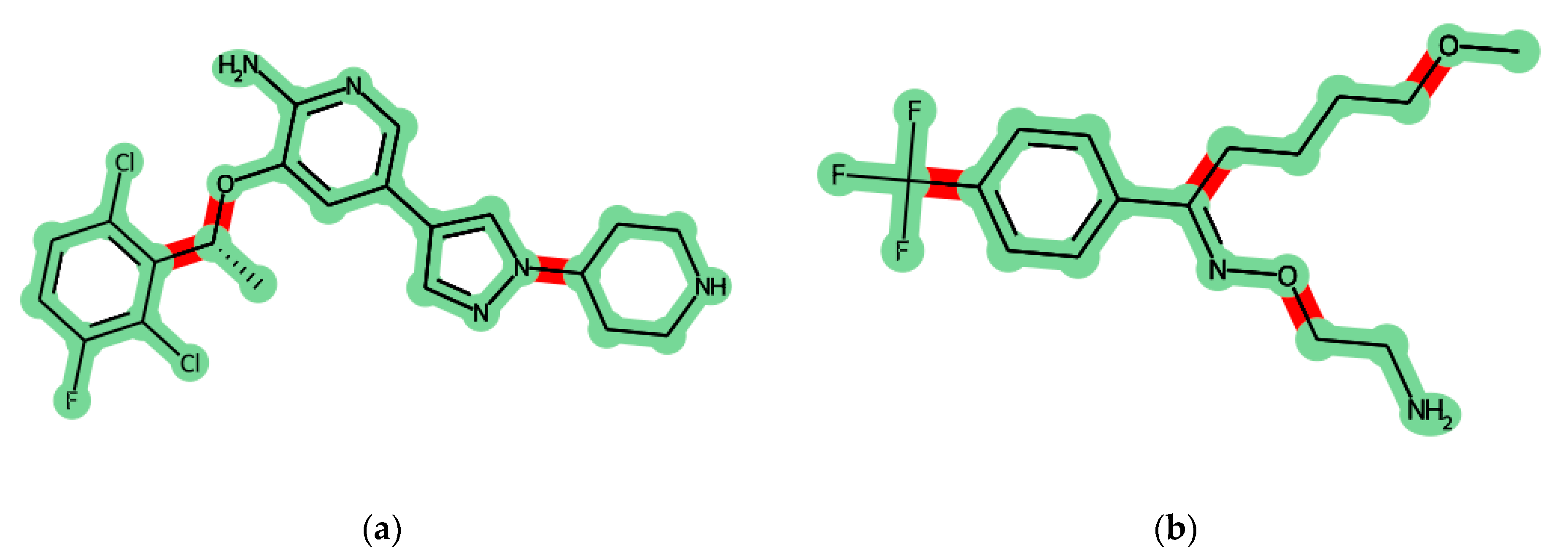
Figure 3 shows an example of some molecules decomposed into fragments. Fragments are mostly meaningful in terms of medicinal chemistry, e.g., the central large conjugated two-ring system in
Figure 3a and the conjugated system including the benzene ring and the oxime fragment in
Figure 3b. The steric and electronic features of the fragments are retained. Also, one should pay attention to the oxime fragment in
Figure 3b. The labile bonds in the saturated linkers are not broken, and their interaction features are also retained.
It should be noted that in certain cases, the application of the third rule may violate the main principles. When applying the breakdown rules, structures were found for which the application of the rules is ambiguous (
Figure 4a,b). According to the rule, single-atom fragments or several fragments with broken conjugated bonds could be obtained.
Figure 4 shows examples of such violations when small linkers, such as ether oxygen (single-atom fragment) and diazo fragments, are located between two aromatic ring systems. Since it is impossible to unambiguously determine with which ring to leave the fragment, it was decided to cleave both bonds (
Figure 4b,c,e,f). In accordance with the breakdown requirements, the resulting fragments must be meaningful and not overlap. In view of the fact that it is practically impossible to define in advance the rules for the relative preference for such mutually conjugated systems, it was decided to deviate from the principles and divide the molecule along all these bonds in such cases. This decision helps to completely avoid such uncertainties in the breakdown of molecules. In this case, to remove the ambiguity, conjugated bonds are broken, and the decision of which fragment to end up with remains with the medicinal chemist. Since there can be several such fragments in the structure, the number of possible variants of fragment sets grows. Thus, breaking conjugacy in such cases is offset by the increased convenience and flexibility of the fragment decompositions, which become more general.
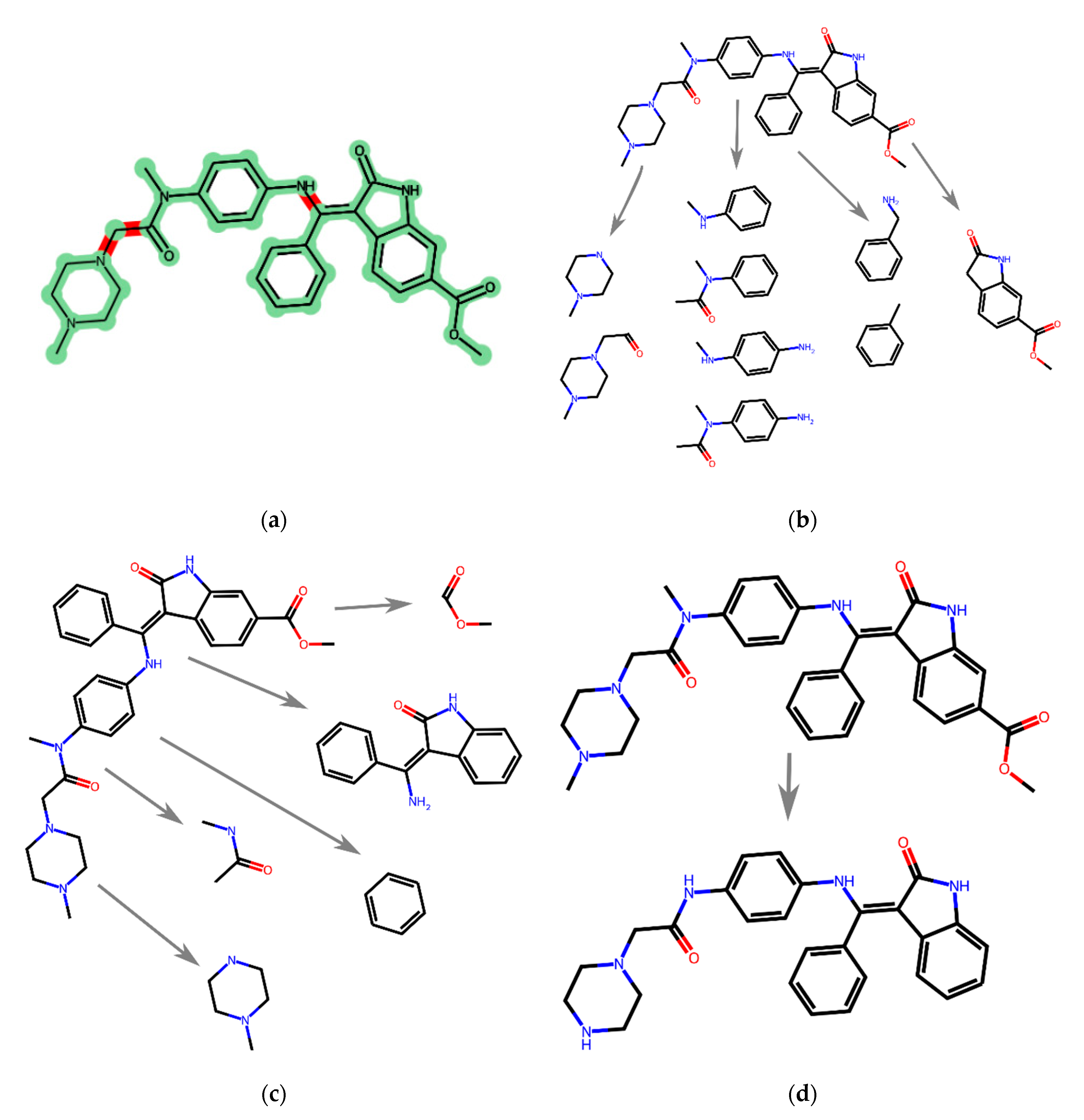
For comparison purposes, several representatives of the existing fragment decomposition approaches are analyzed below using the same example molecule.
Figure 5b shows the breakdown using MolBLOCKS according to the RECAP rules;
Figure 5c shows the breakdown using eMolFrag, which performs the primary breakdown according to the BRICS rules.
Figure 5d shows the Murcko breakdown. The structure proposed for comparison is shown in
Figure 5a. Our algorithm divides this structure into several fragments (
Figure 5a), among which there are (1) a large fragment consisting of the phenyl fragment and the 2-oxoindole fragment connected through a double conjugated bond; (2) an aromatic fragment conjugated with an amide bond through a nitrogen atom, as well as with an amine functional group at the para-position. These fragments are meaningful from the point of view of medicinal chemistry since they retain their steric and electronic features after breaking the molecule into such fragments. The main intermolecular interactions are preserved after breakdown (see
Figure S1). Similar fragments are partially observed in
Figure 5b,c, but they lack some conjugated bonds. In this particular example in
Figure 5b (MolBLOCKS program), overlapping fragments are observed. For the eMolFrag program, this is also possible. After splitting according to Murcko, one large fragment without substituents was obtained, which does not meet the requirements presented by us above.
Figure 6 shows an example of the difference between the proposed approach and retrosynthetic ones. The proposed method leads to fragments that do not introduce new interactions into the environment but, if possible, preserve the existing ones. Thus, they try to preserve the interaction pattern of the entire group, which could be otherwise significantly changed upon retrosynthetic breakdown.
Our article presents a decomposition method based on a new paradigm and its pilot implementation. Metrics for assessing the quality and completeness of the method are still under development. Even in its current implementation, this method gives meaningful results (see
Table S2) and can be used in various computer-aided drug discovery (CADD) tasks, in particular in the construction of new molecules with appropriate tools (for example, eSynth [
30]) or in the correlation of the specific fragment in ligands with activity against a particular receptor.
Thus, we obtained a simple and robust method for the decomposition of molecules into fragments. The decomposition results in reasonably small and meaningful fragments, with the electronic properties of the fragments closely related to their properties within a ligand. The meaningfulness of fragments helps to keep the fragment’s immediate chemical environment, which may be important in the analysis of binding.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





