
Recent Advances in Application of Alkoxy Radical in Organic Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
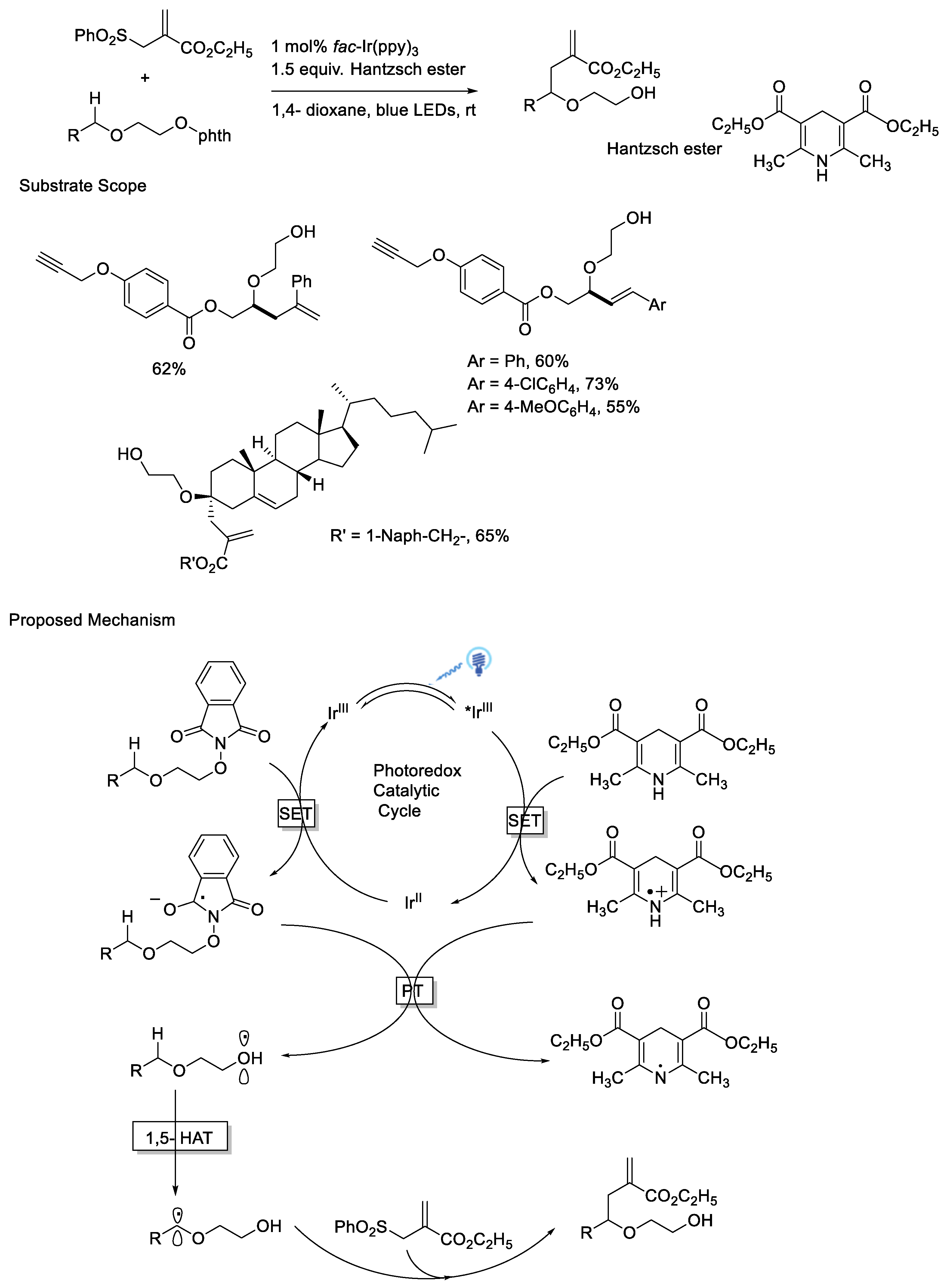{kind=link}
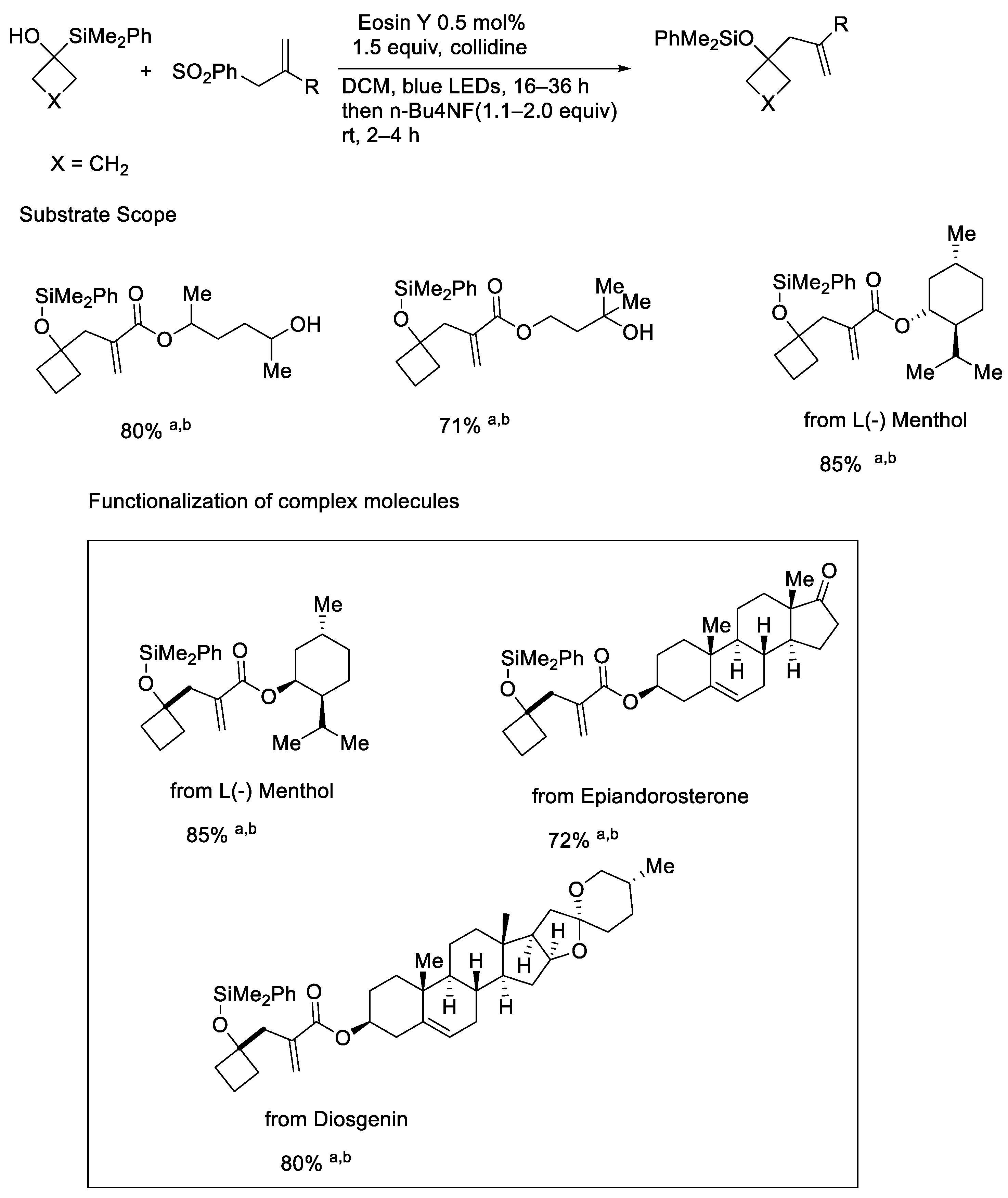{kind=link}
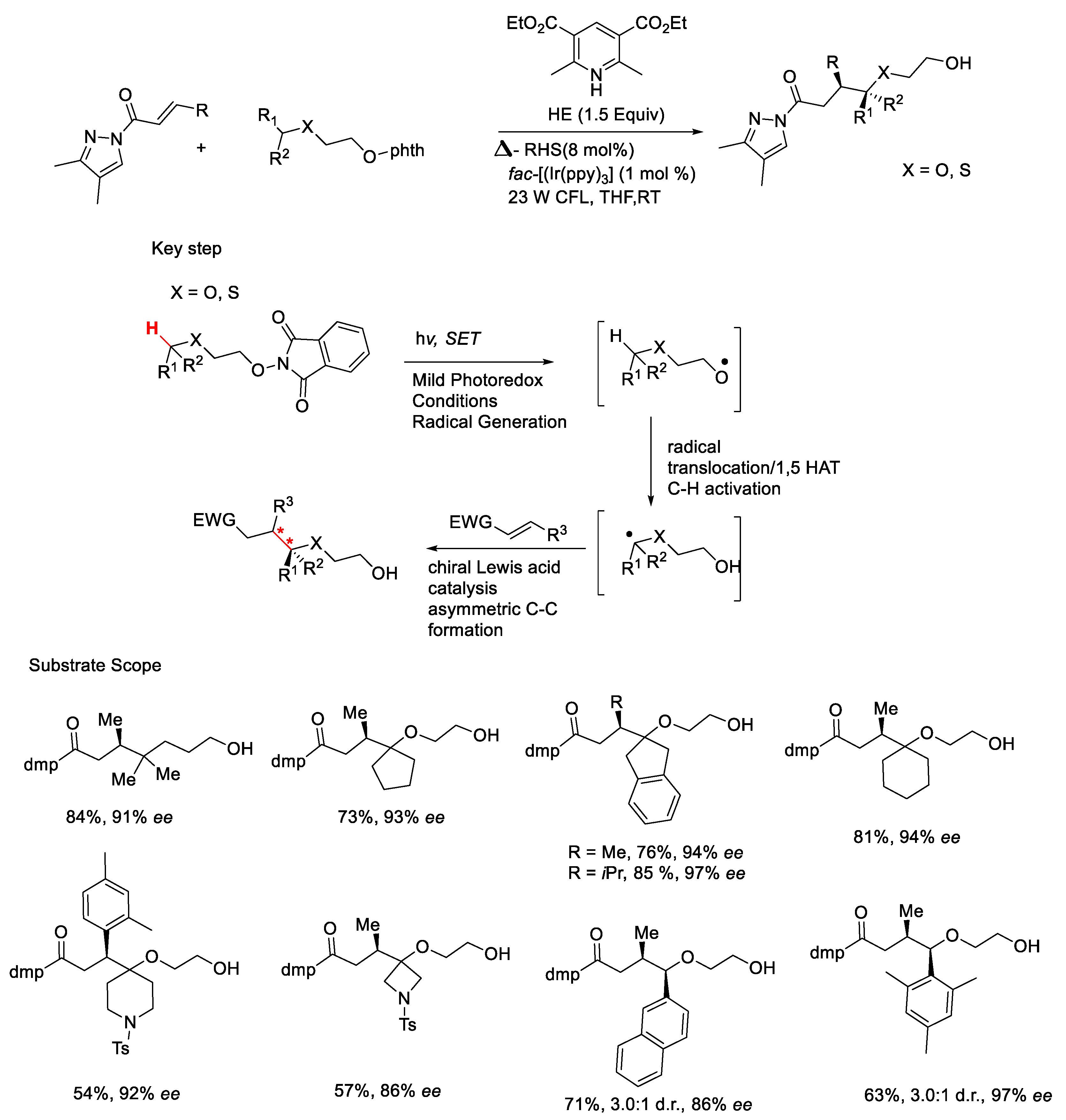{kind=link}
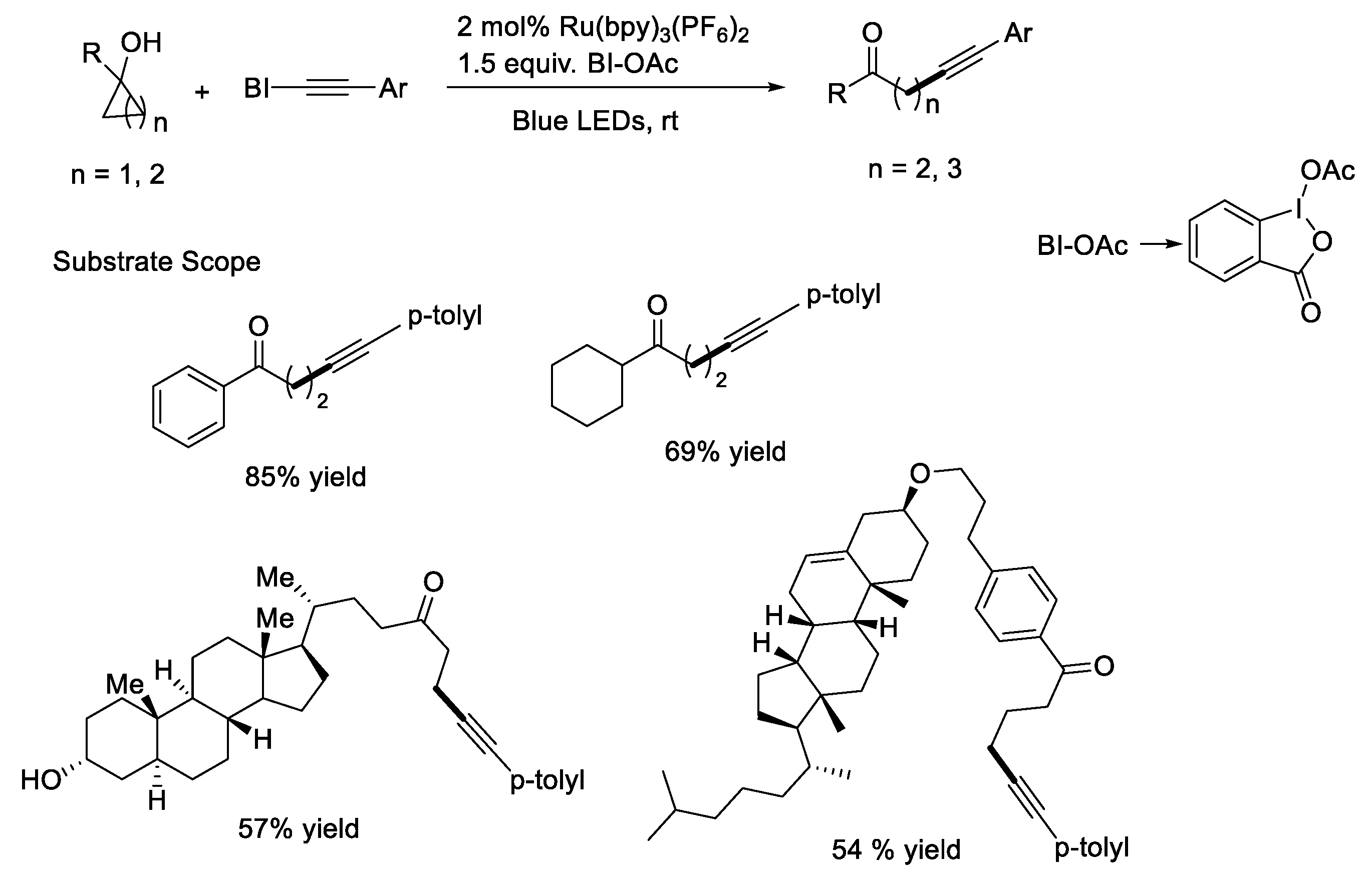{kind=link}
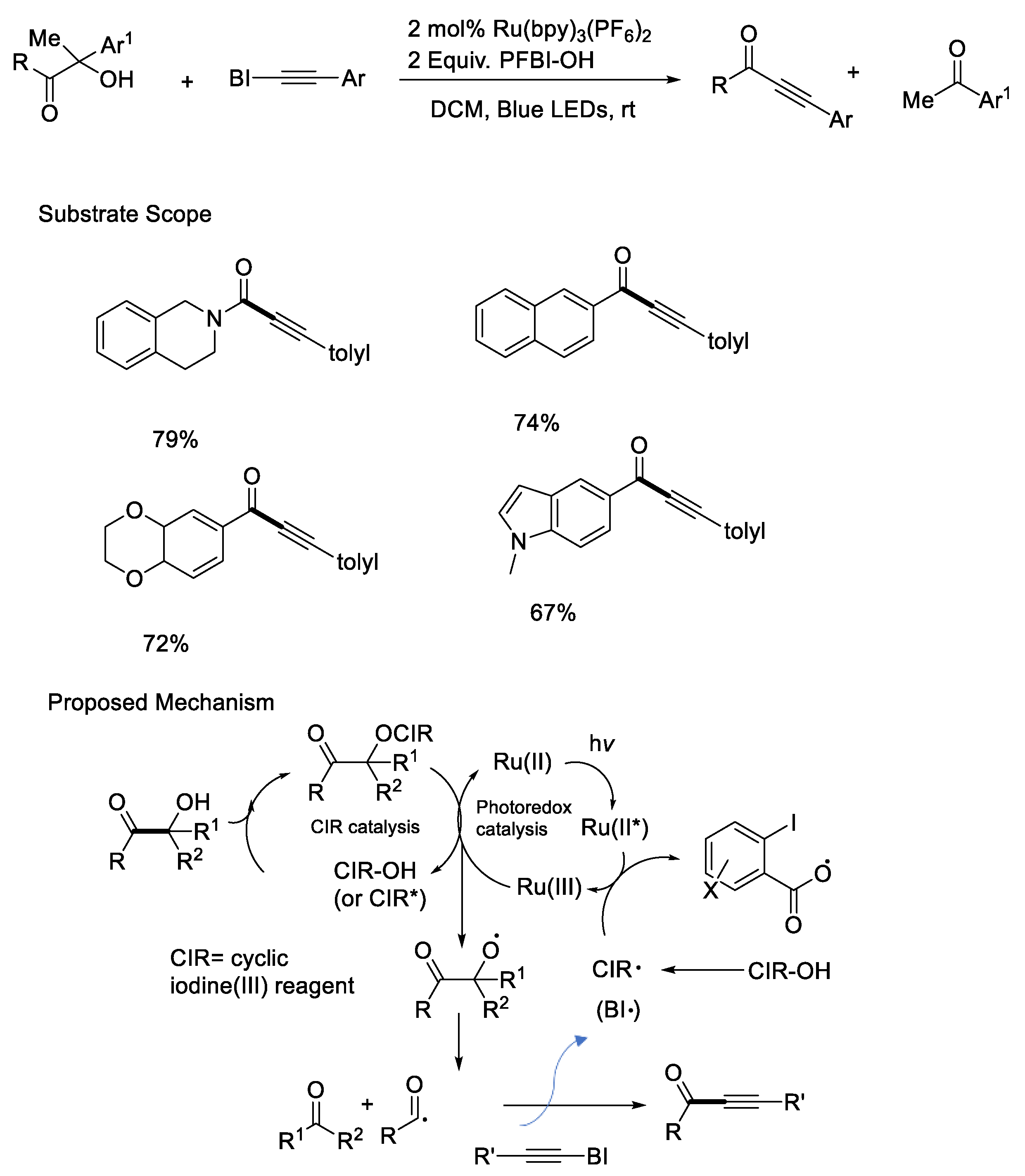{kind=link}
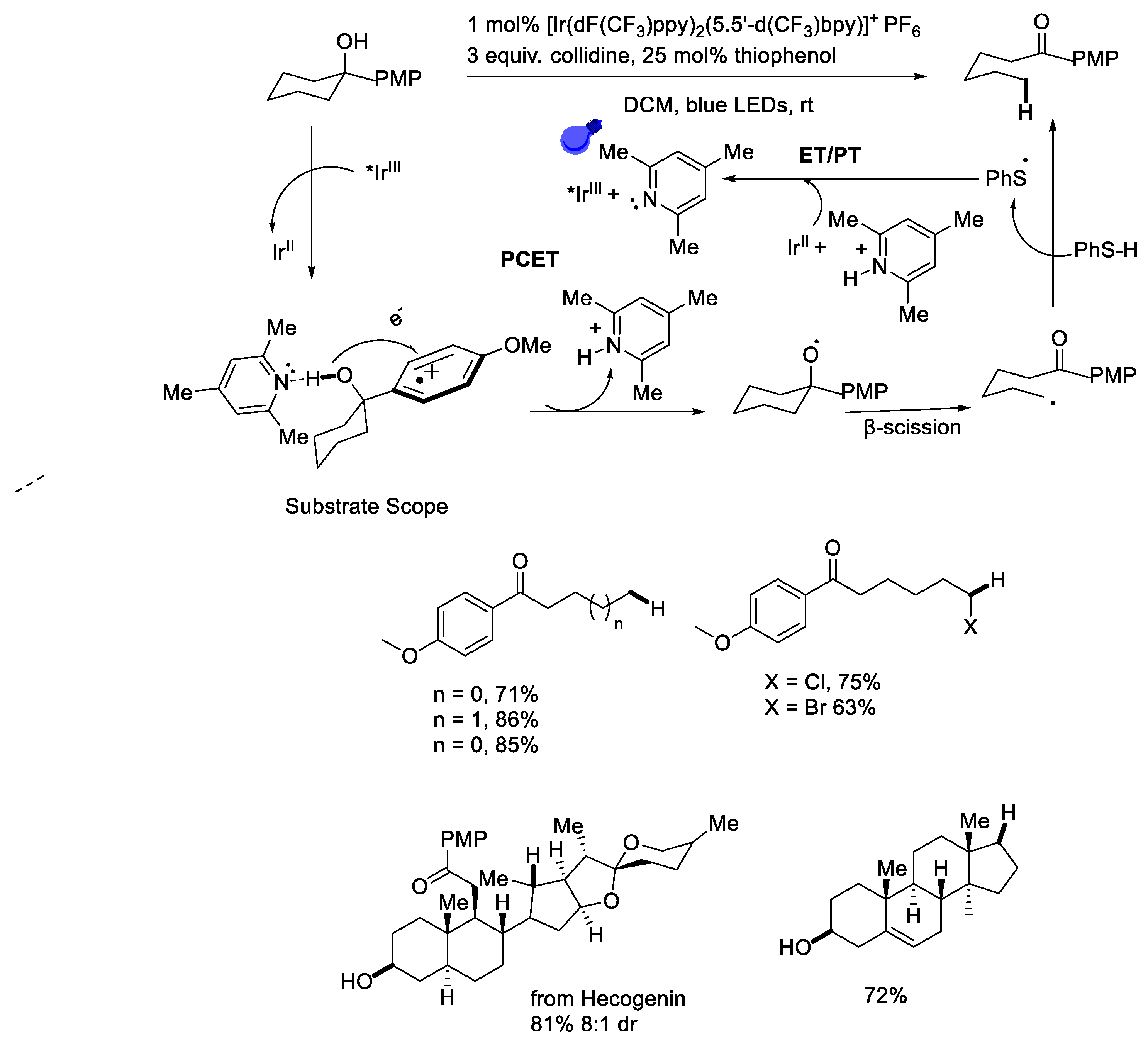{kind=link}
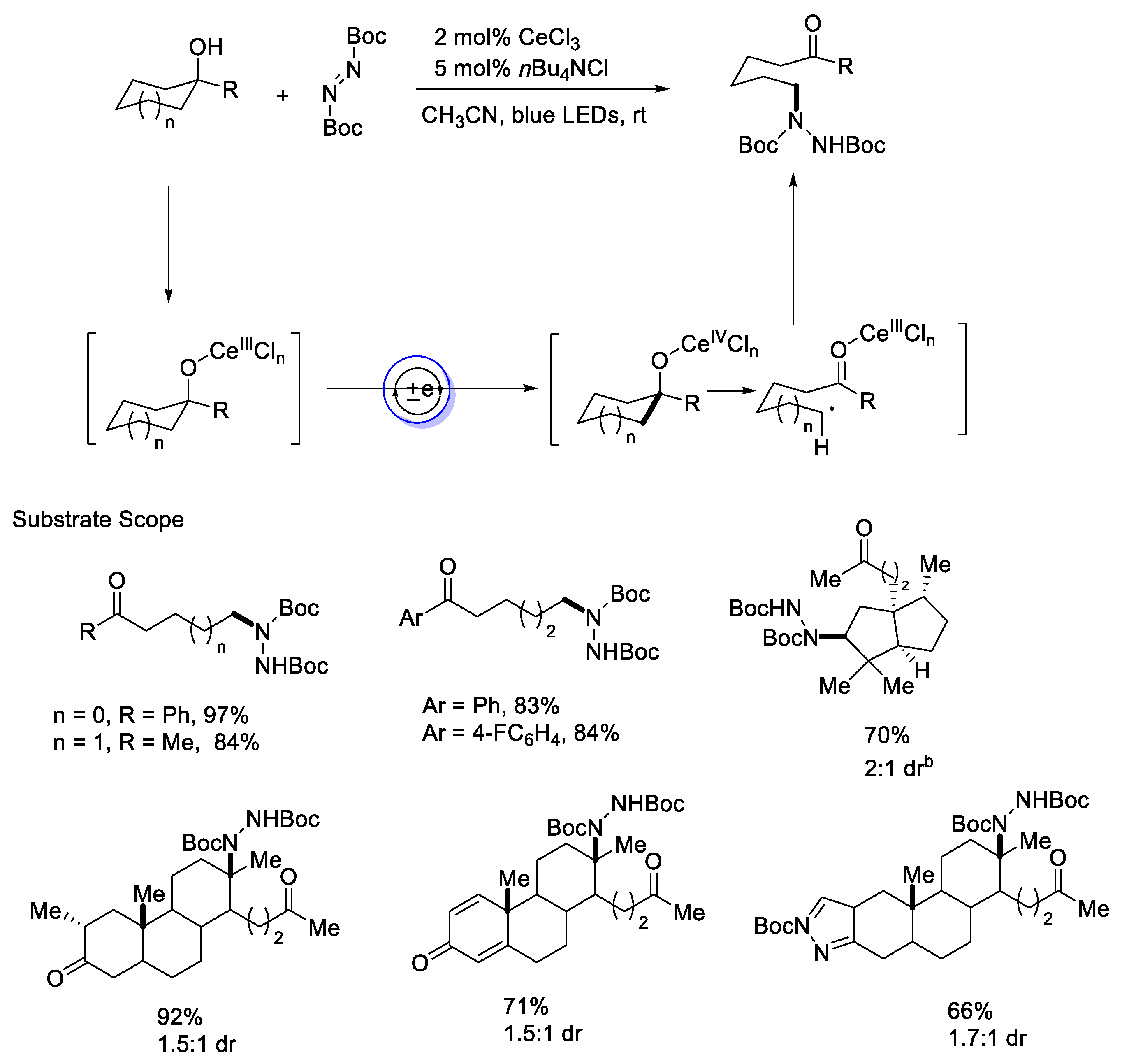{kind=link}
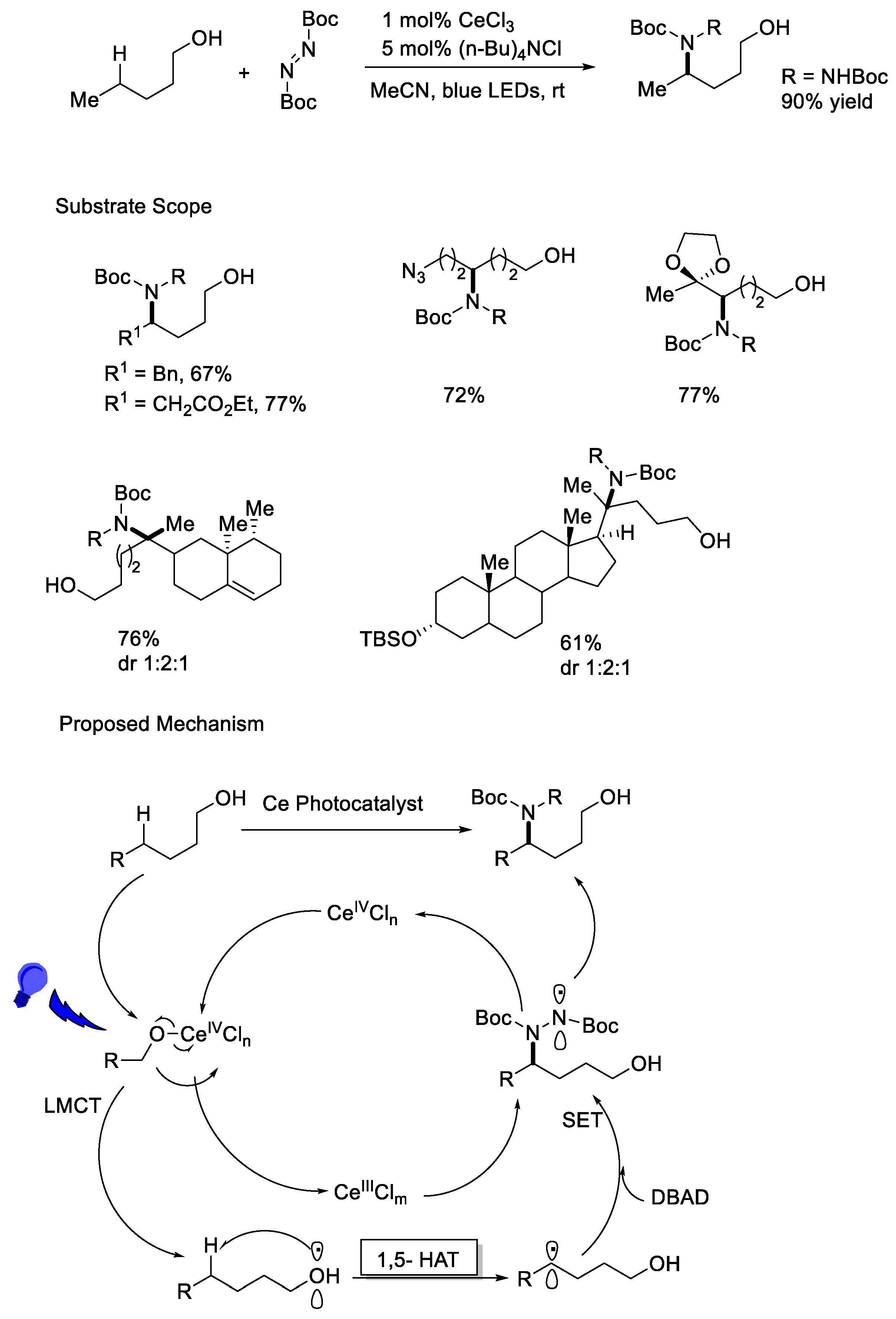{kind=link}
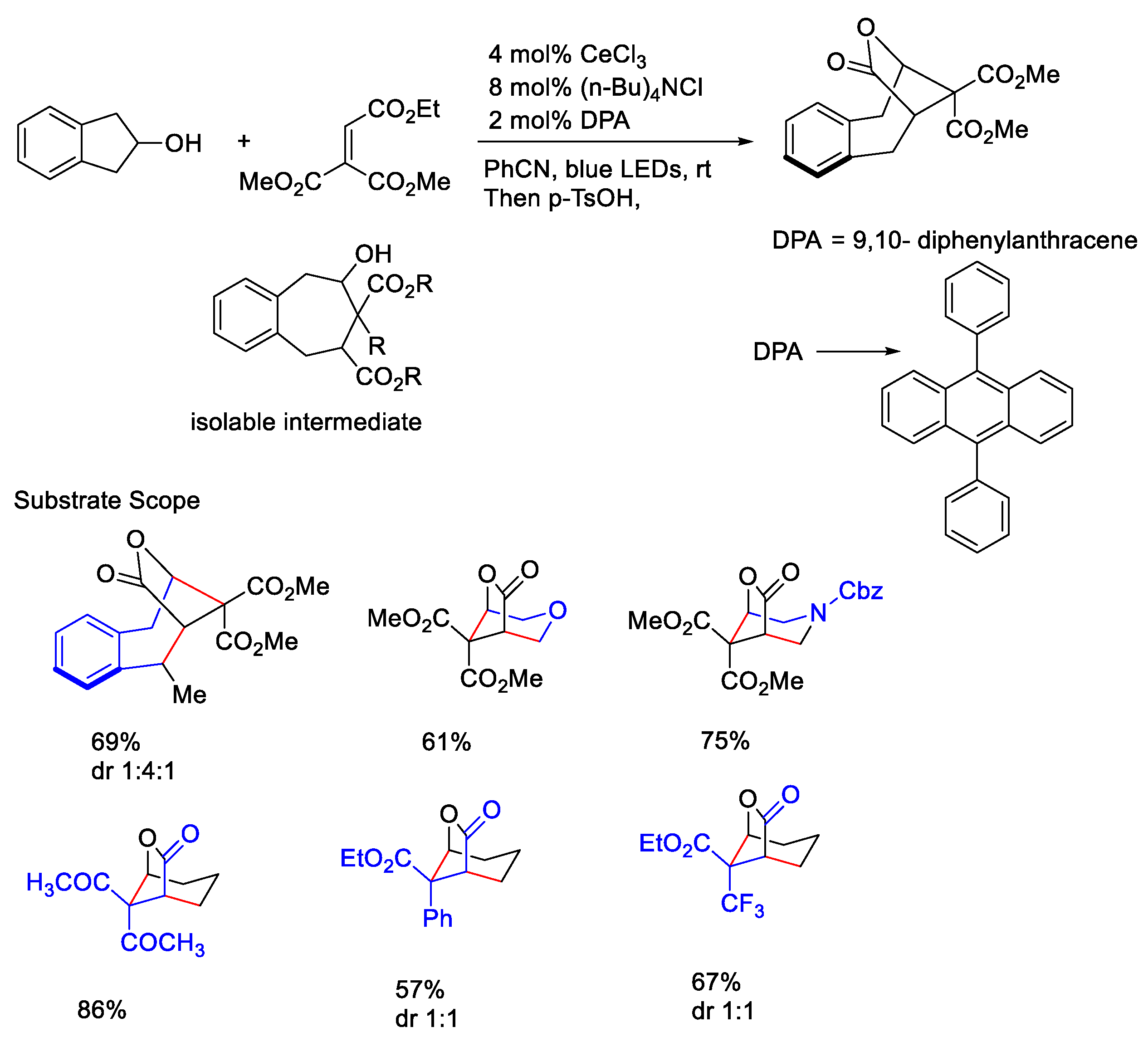{kind=link}
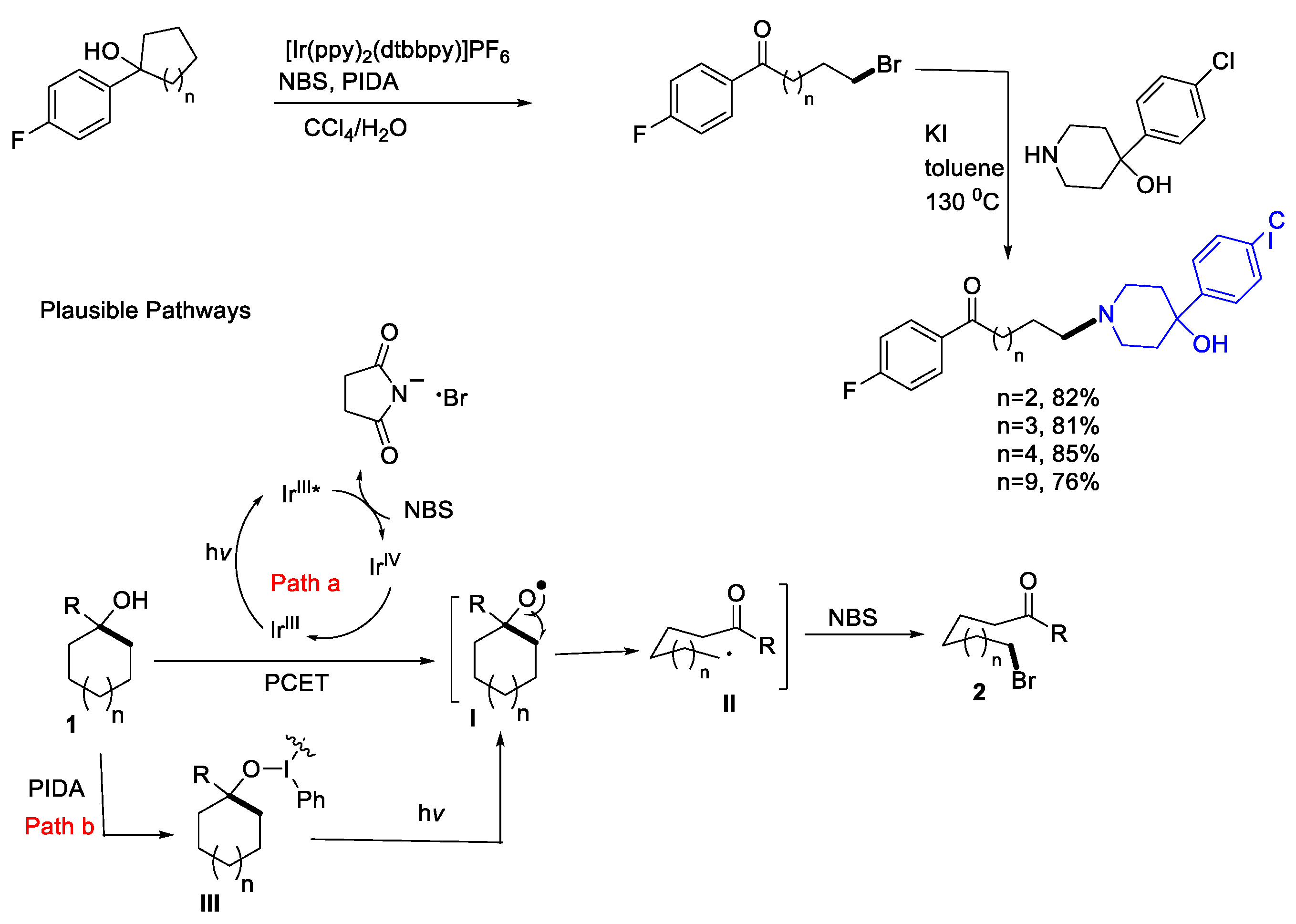{kind=link}
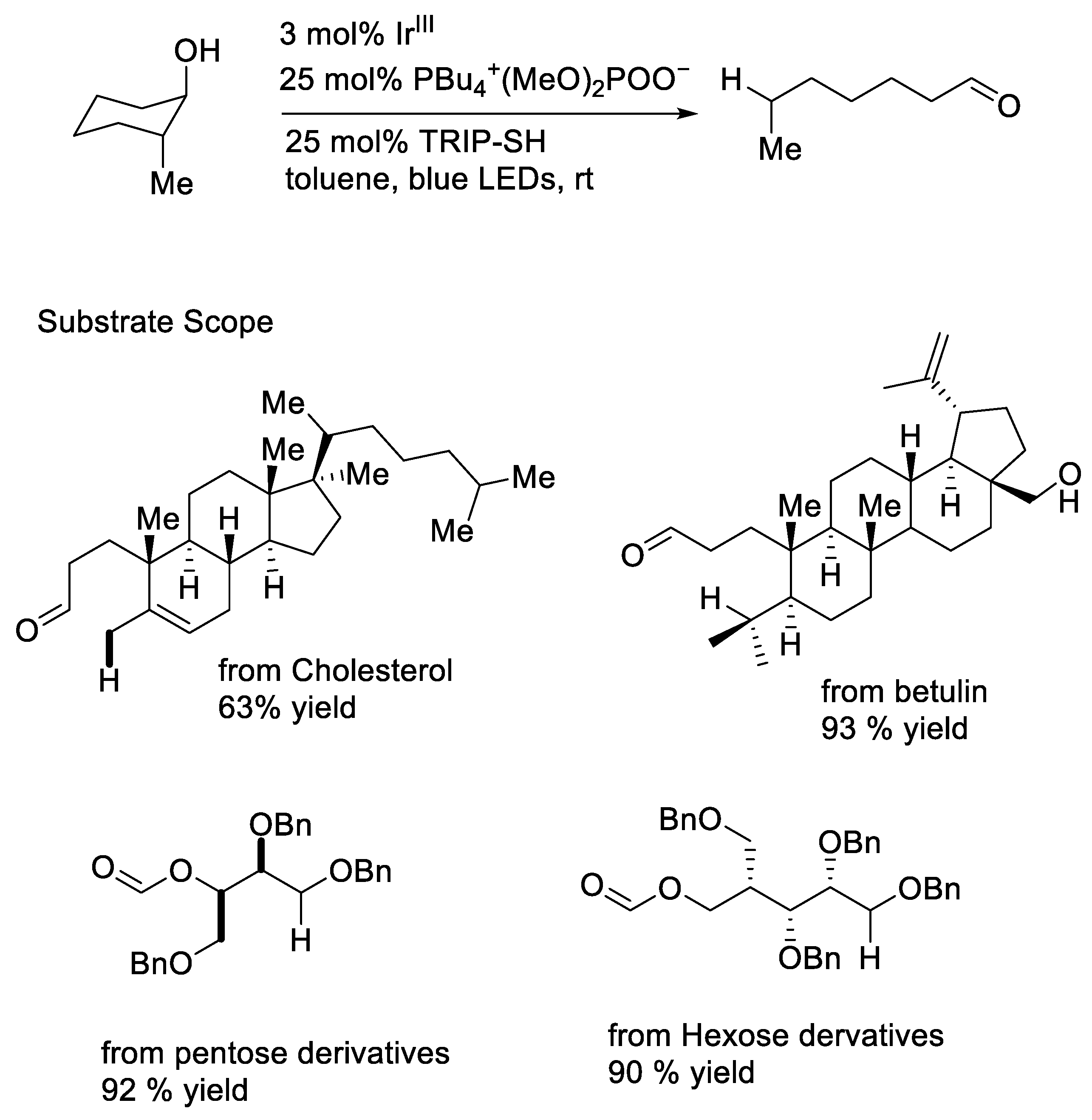{kind=link}
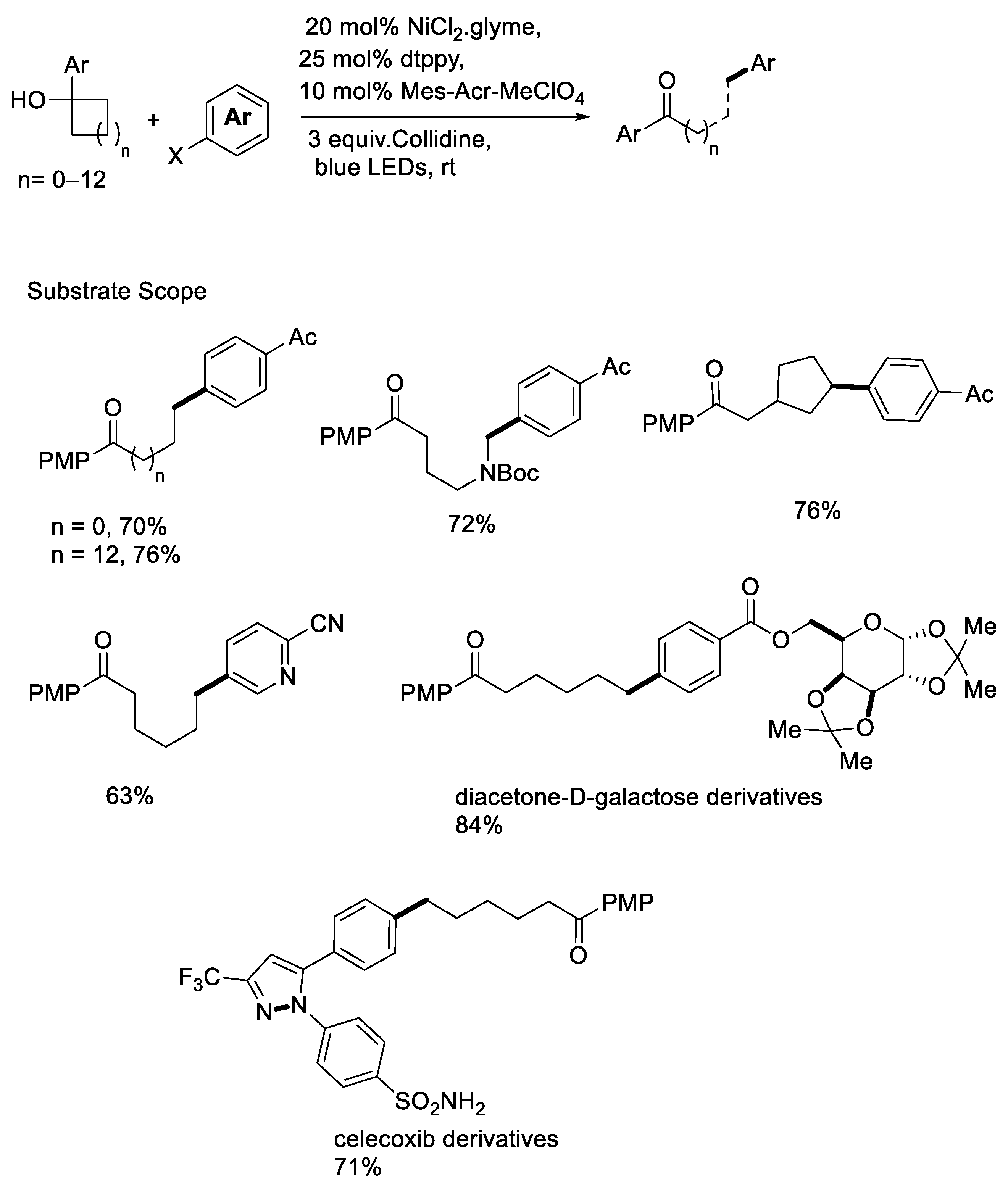{kind=link}
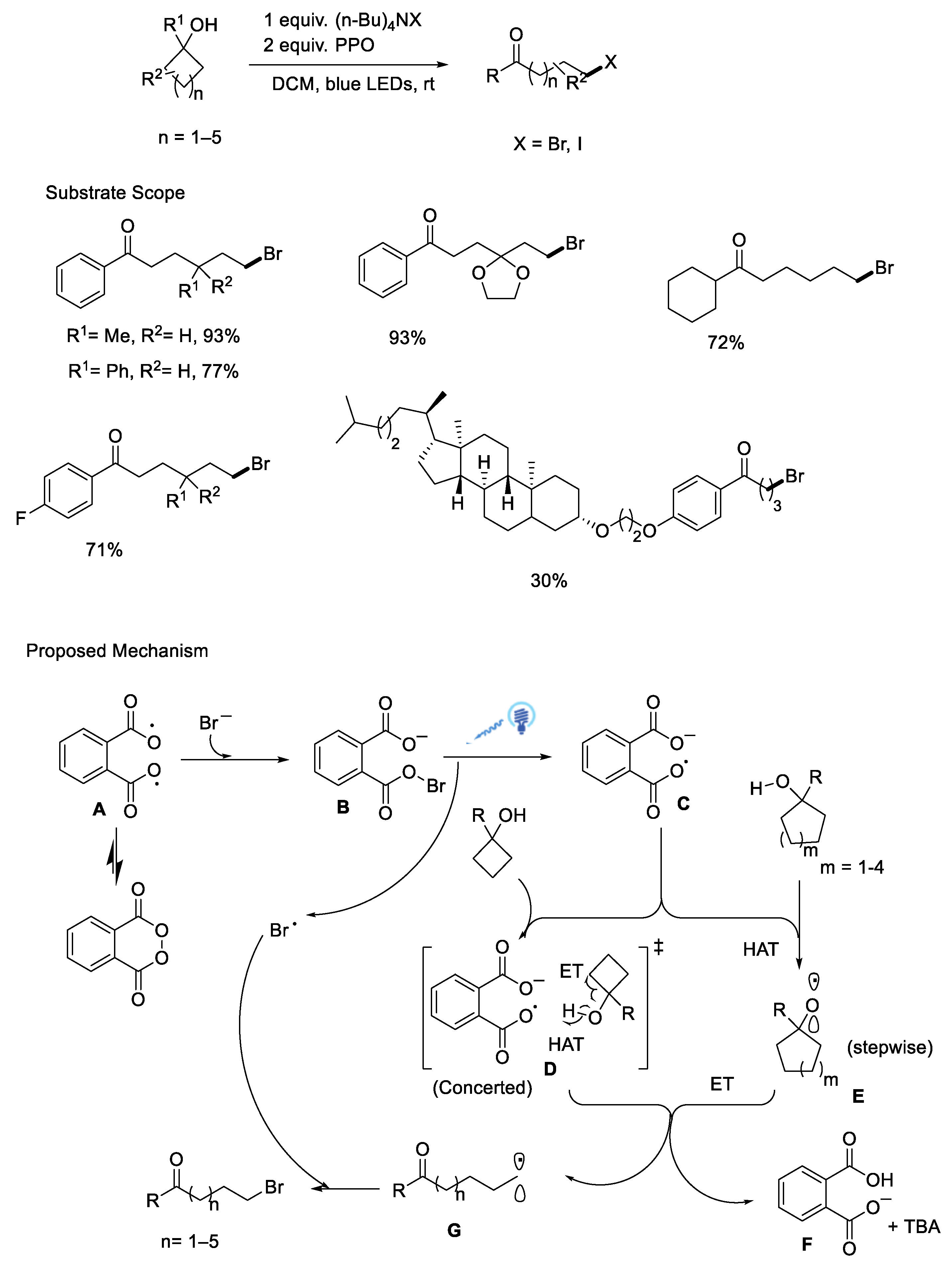{kind=link}
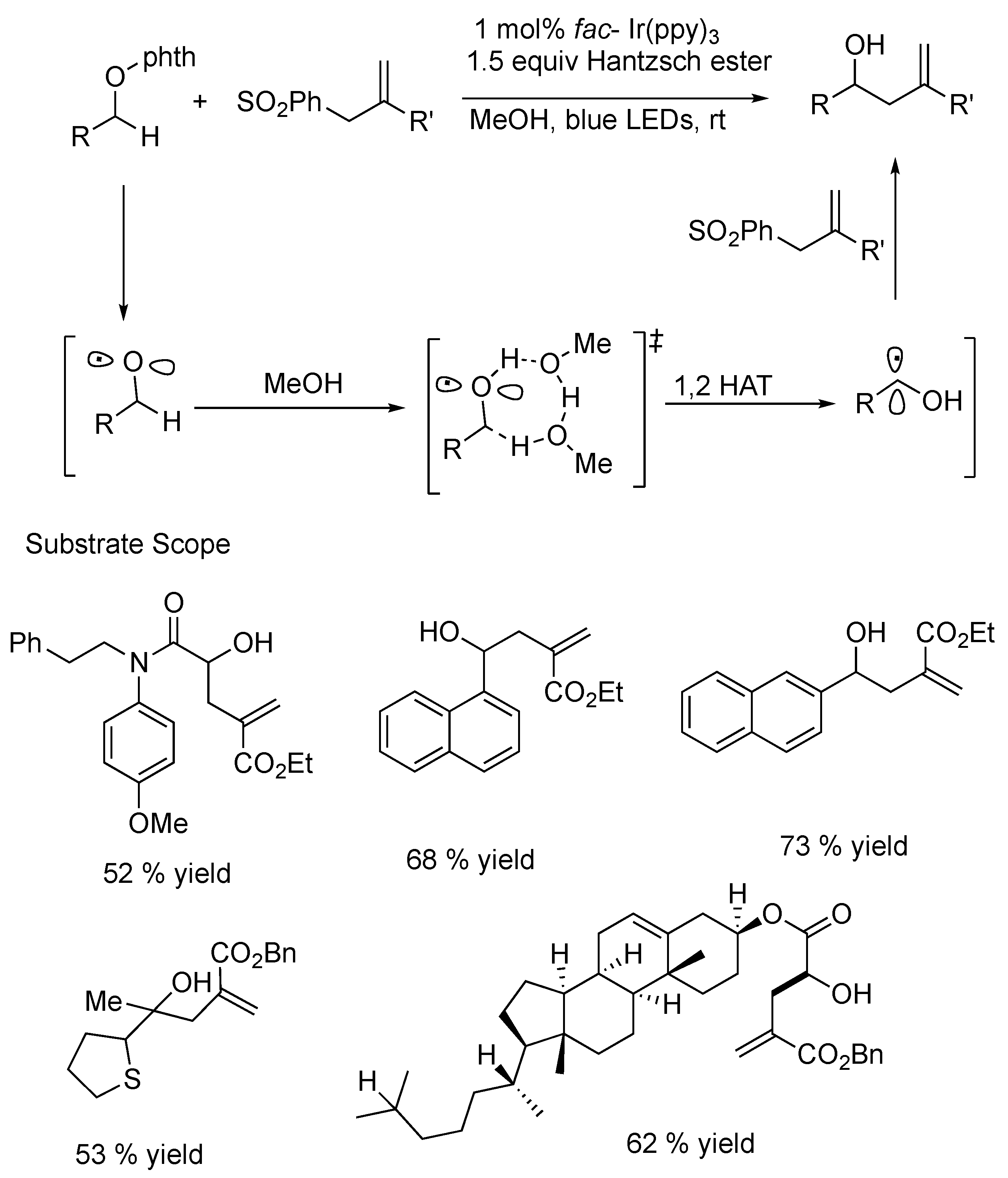{kind=link}
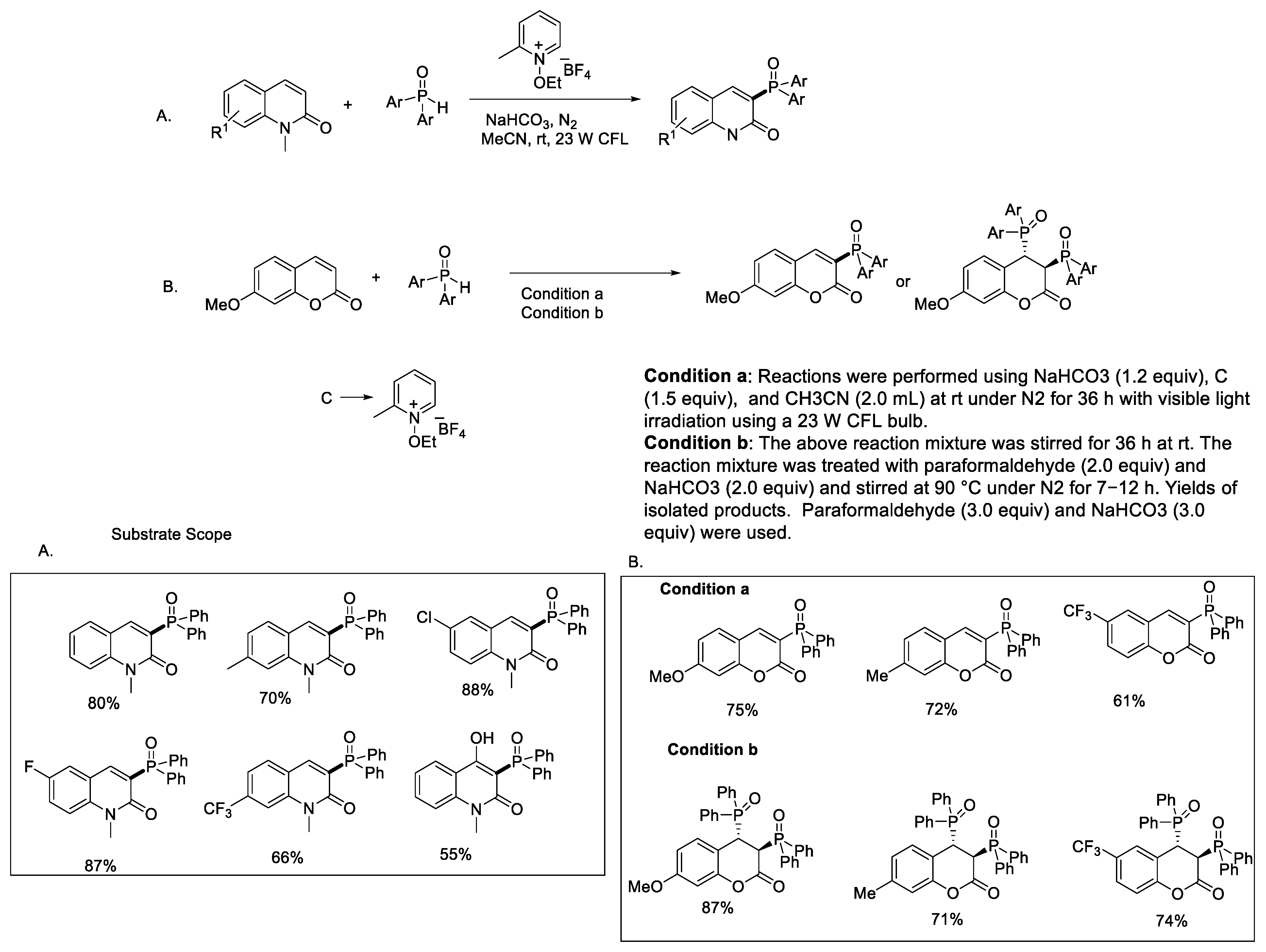{kind=link}
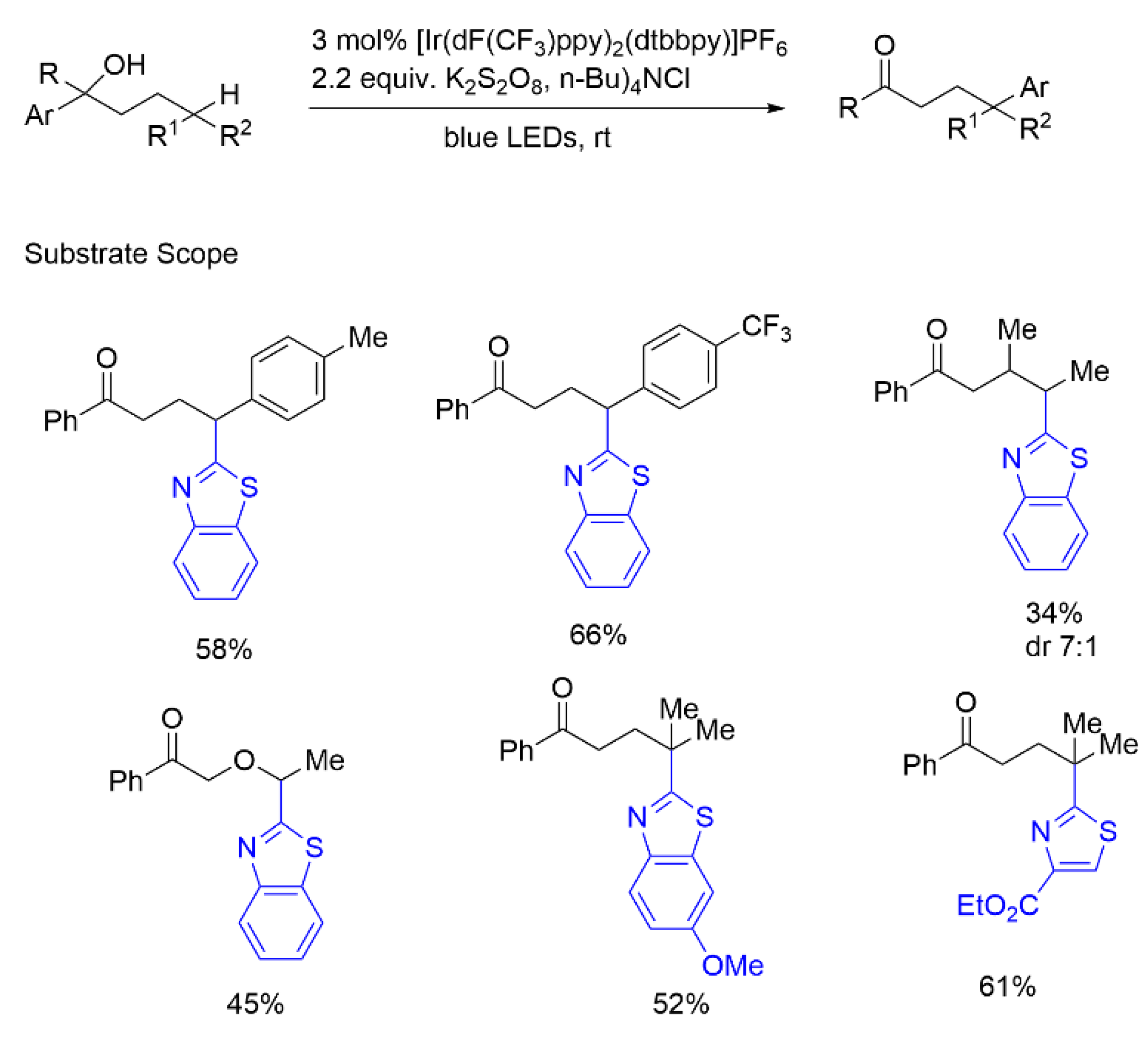{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
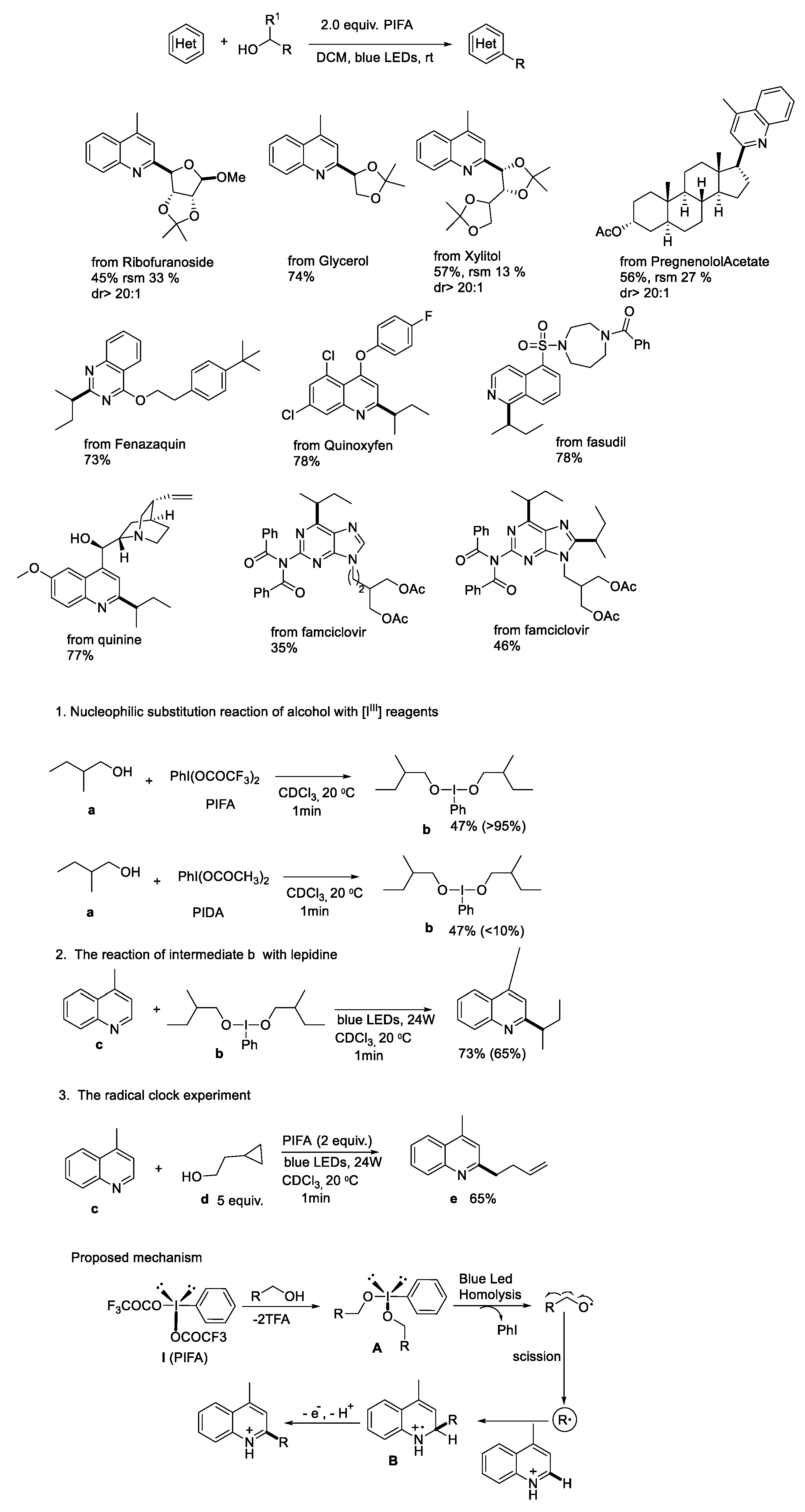{kind=link}
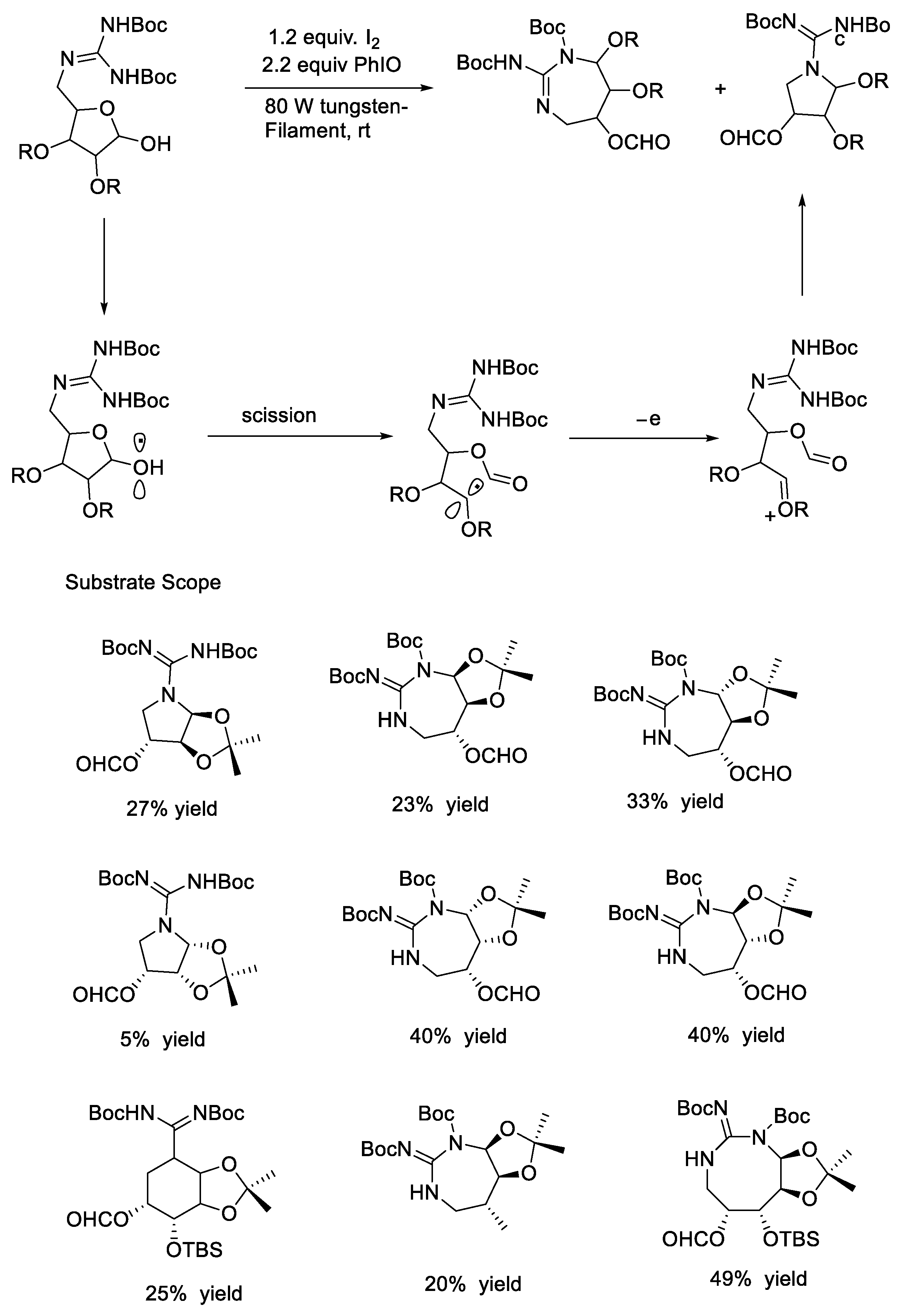{kind=link}
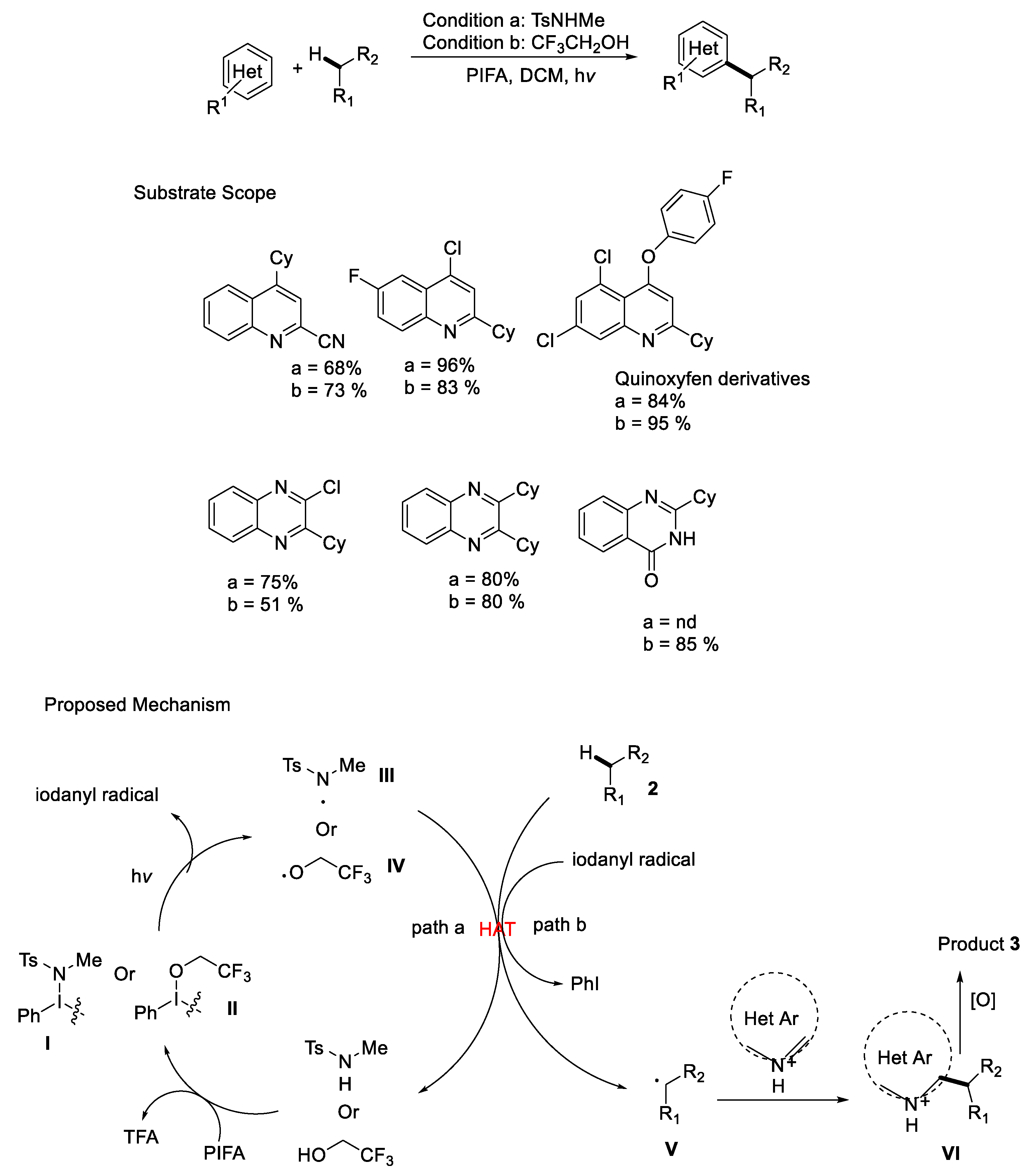{kind=link}
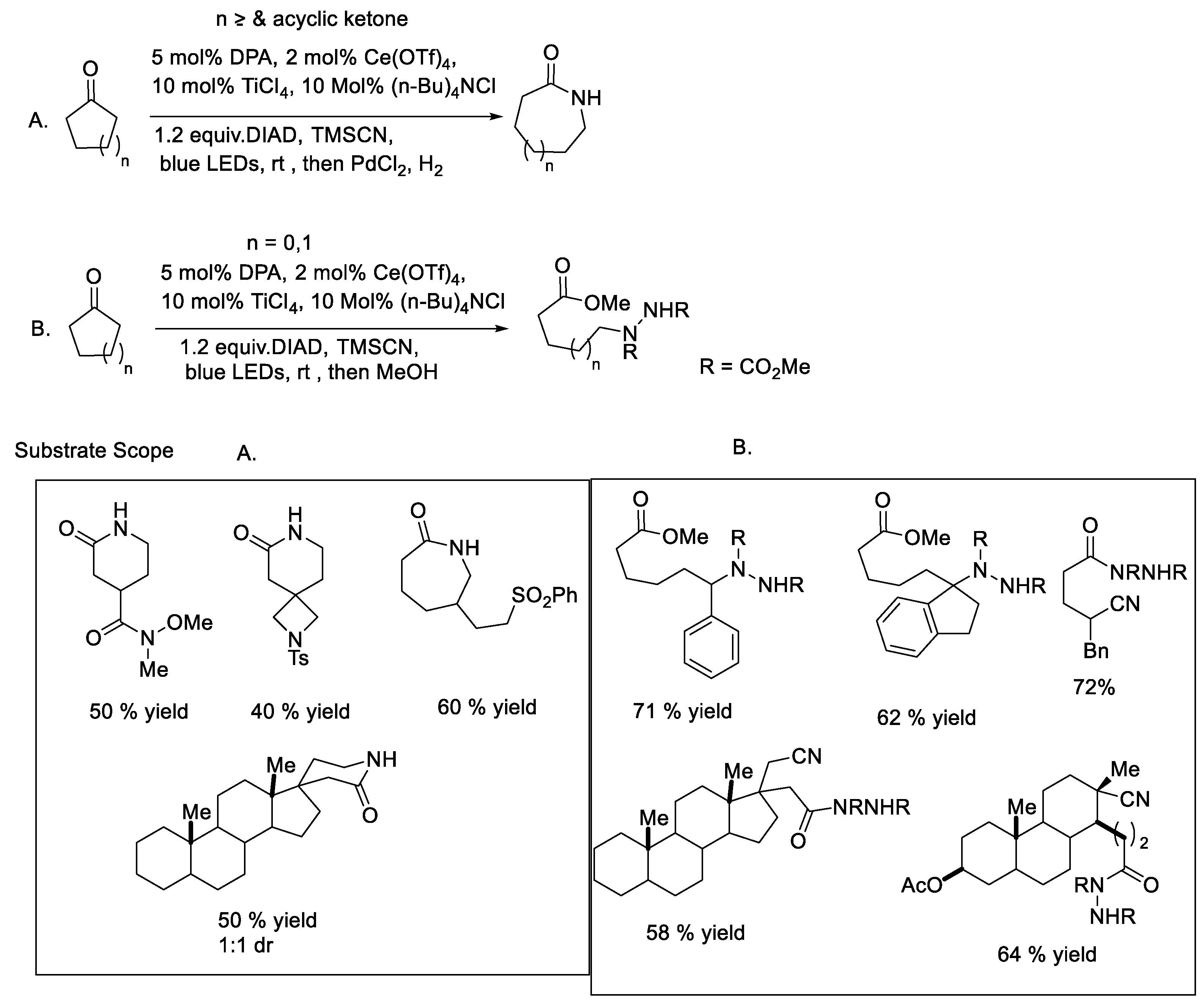{kind=link}
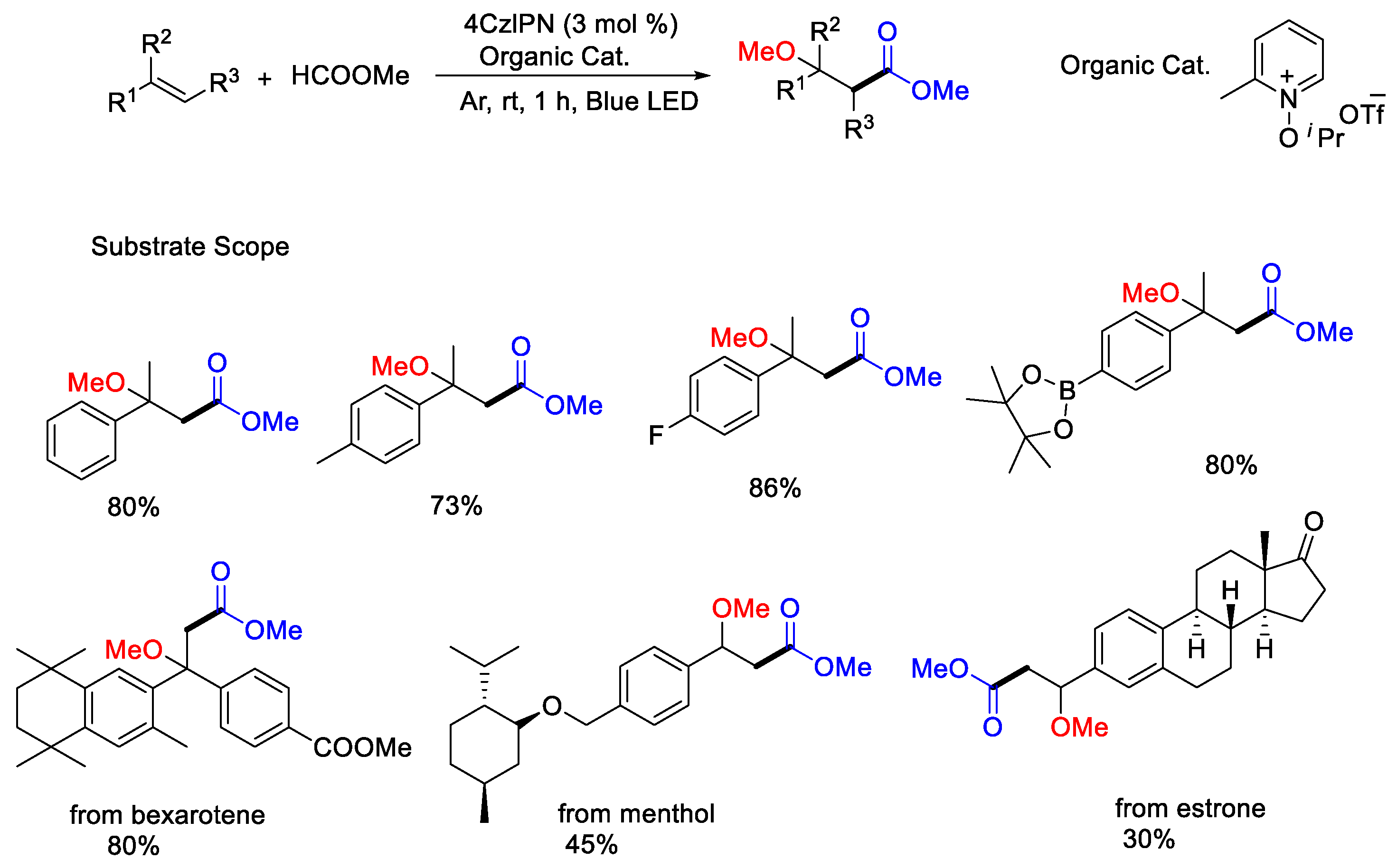{kind=link}
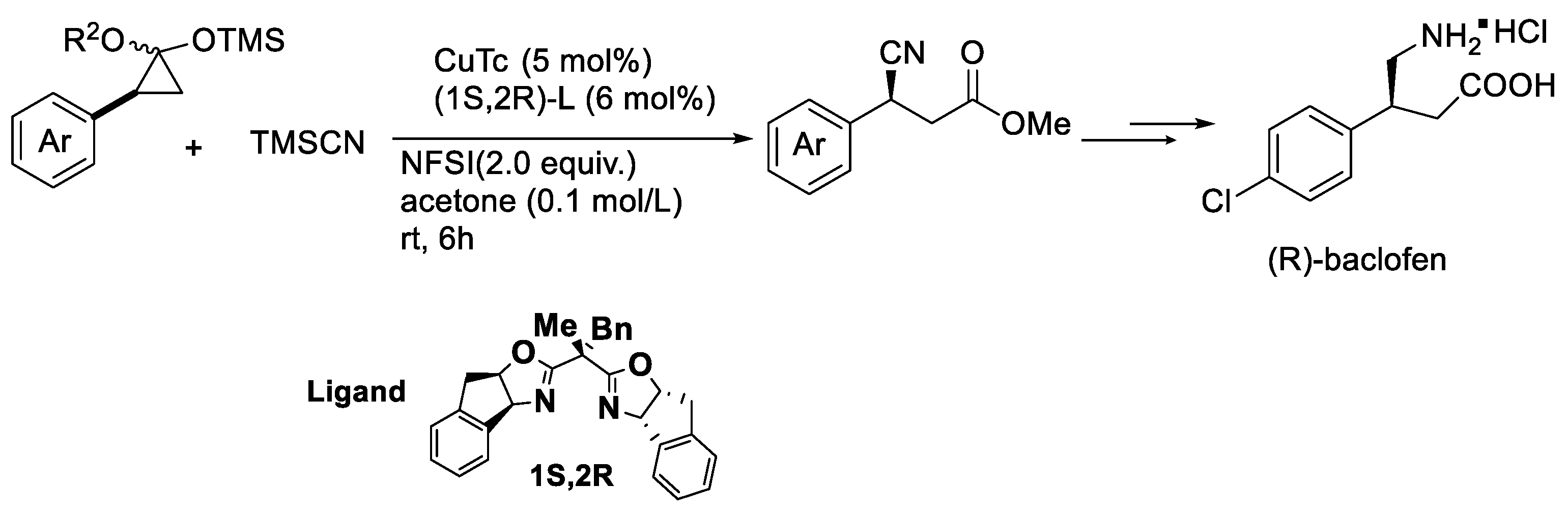{kind=link}
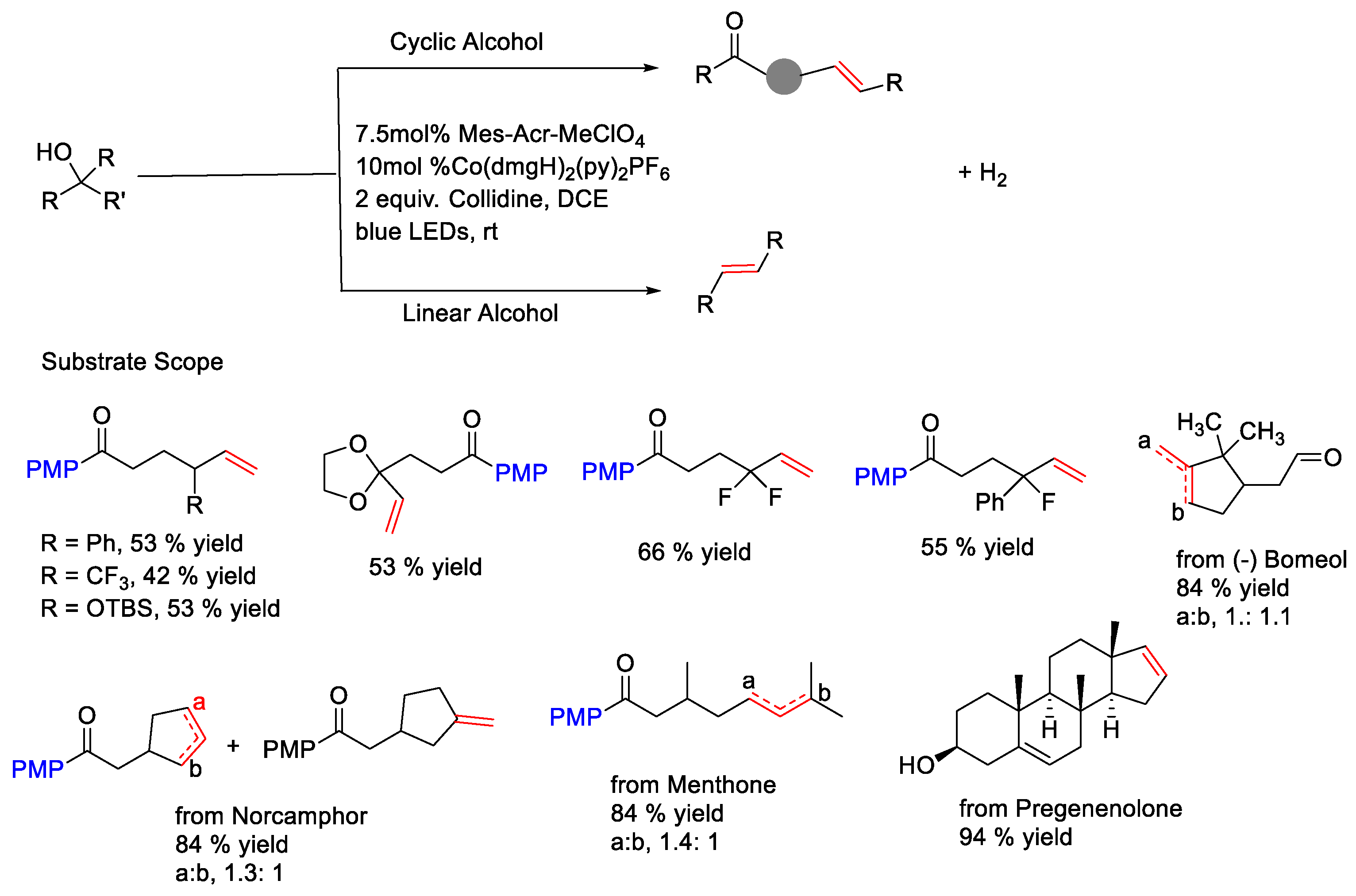{kind=link}
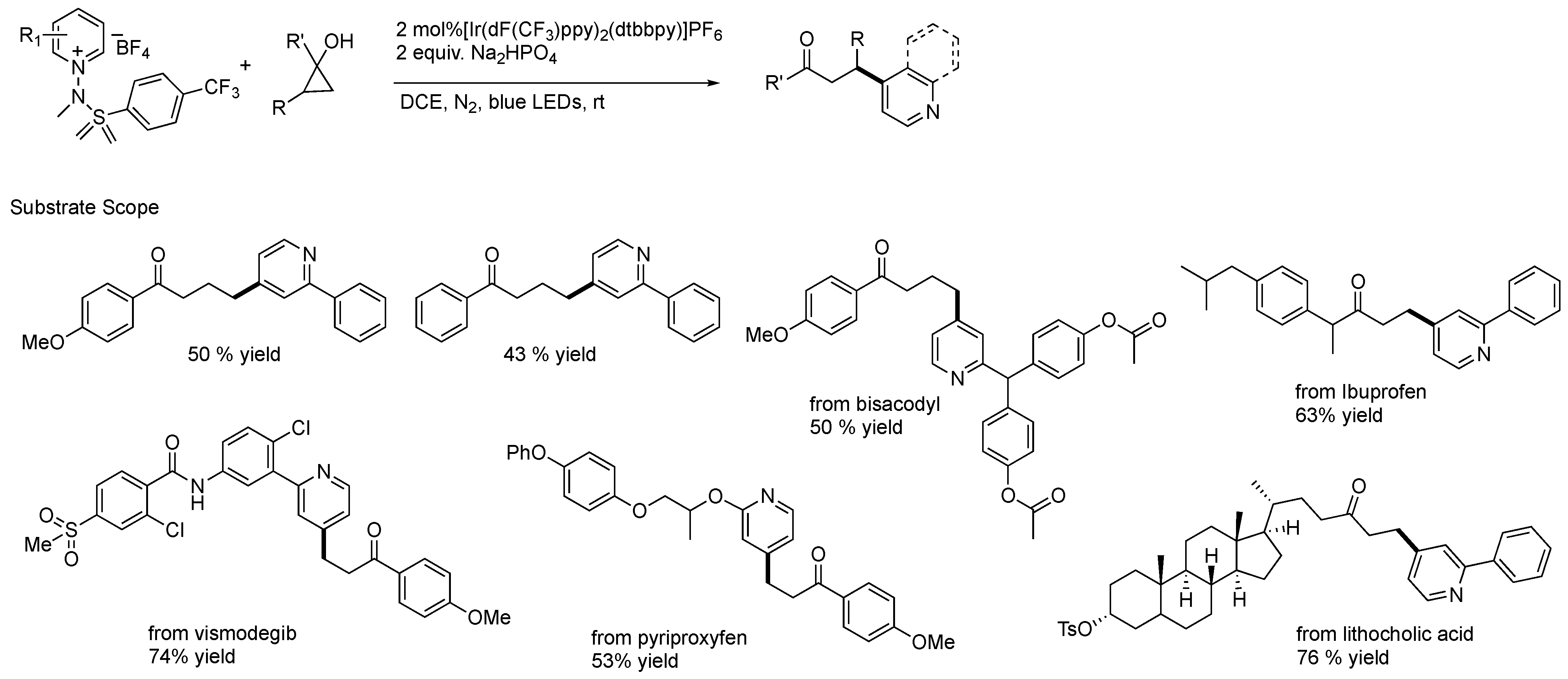{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
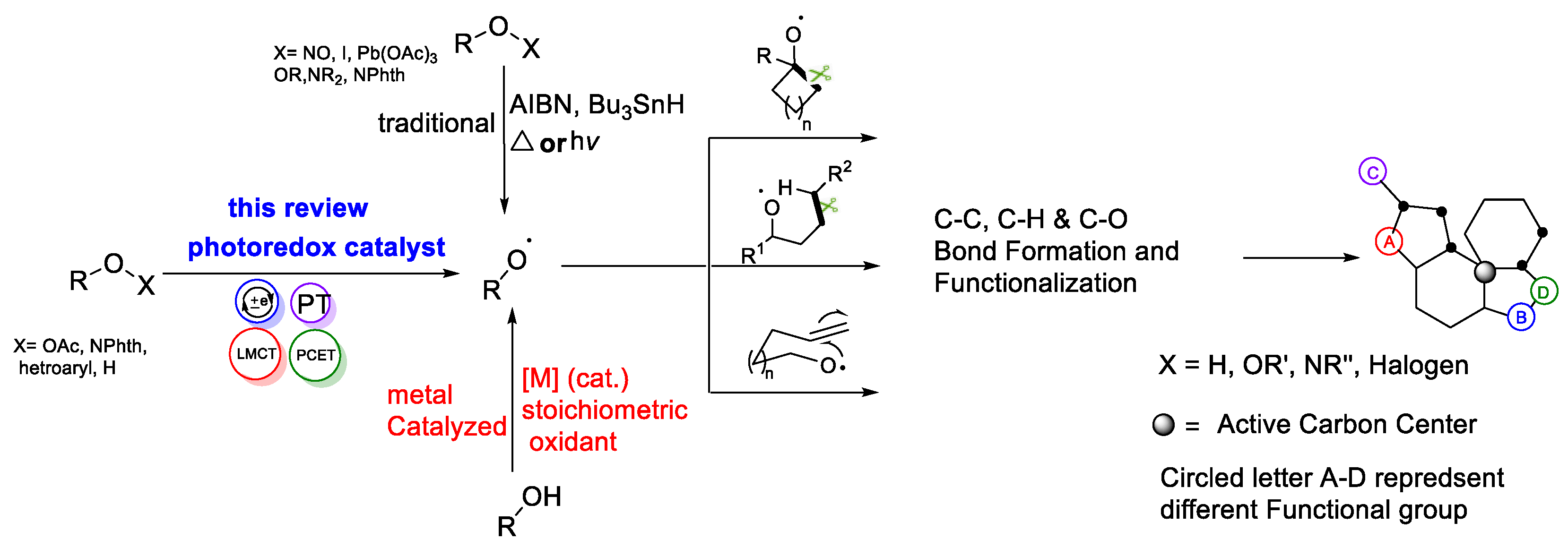
Abstract
:1. Introduction
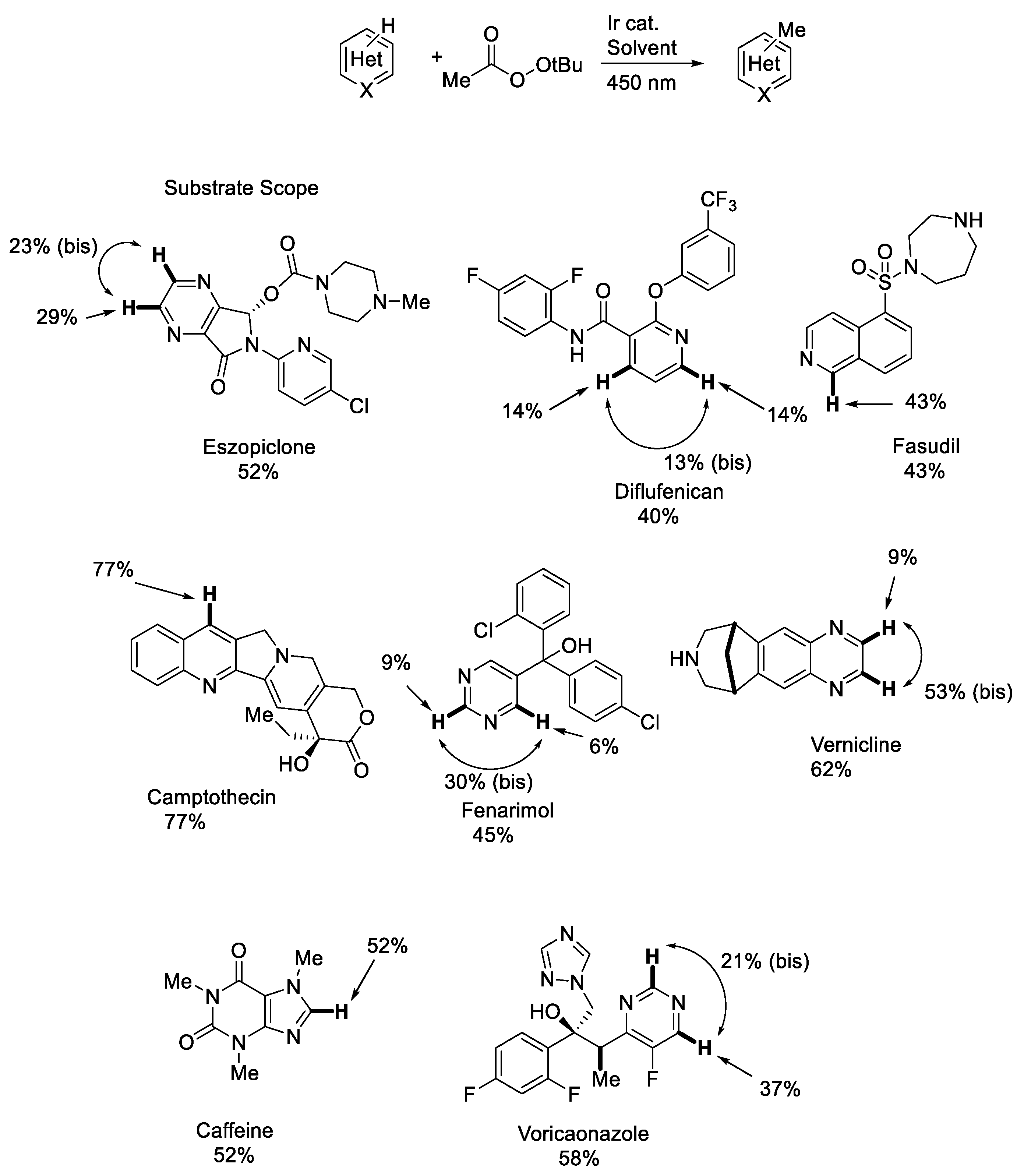
2. Photocatalytic Generation of Alkoxy Radicals
3. Application of Alkoxy Radicals in Natural Product Synthesis
4. Conclusions and Outlook
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BDE | Bond dissociation energy |
| BI | Benziodoxole |
| CIR | Cyclic iodine(III) reagents |
| DBAD | Di-tert-butyl azodicarboxylate |
| DPA | (9,10-diphenylanthracene) |
| EPR | Electron paramagnetic resonance |
| HAT | Hydrogen atom transfer |
| HPLC | High-performance liquid chromatography |
| HE | Hantzsch ester |
| LMCT | Ligand-to-metal charge transfer |
| LED | Light-emitting diode |
| MLCT | Metal-to-ligand charge transfer |
| PCET | Proton-coupled electron transfer |
| PMP | p-methoxyphenyl |
| PIDA | Iodobenzene diacetate |
| PIFA | [bis(trifluoroacetoxy)iodo]benzene |
| PPO | Phthaloyl peroxide |
| PT | Proton Transfer |
| TBAB | Tetrabutylammonium bromide |
| 4CzIPN | 2,4,5,6-tetra(carbazol-9-yl)benzene-1,3-dicarbonitrile |
References
- Tsui, E.; Wang, H.; Knowles, R.R. Catalytic Generation of Alkoxy Radicals from Unfunctionalized Alcohols. Chem. Sci. 2020, 11, 11124–11141. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; An, Q.; Duan, L.; Feng, K.; Zuo, Z. Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis. Chem. Rev. 2022, 122, 2429–2486. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.H.R.; Beaton, J.M. A Synthesis of Aldosterone Acetate. J. Am. Chem. Soc. 2002, 82, 2641. [Google Scholar] [CrossRef]
- Barton, D.R.; Beaton, J.M.; Geller, L.E.; Pechet, M.M. A New Photochemical Reaction1. J. Am. Chem. Soc. 2002, 83, 4076–4083. [Google Scholar] [CrossRef]
- Jia, K.; Chen, Y. Visible-Light-Induced Alkoxyl Radical Generation for Inert Chemical Bond Cleavage/Functionalization. Chem. Commun. 2018, 54, 6105–6112. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhu, C. Recent Advances in Alkoxy Radical-Promoted C–C and C–H Bond Functionalization Starting from Free Alcohols. Chem. Commun. 2019, 55, 9747–9756. [Google Scholar] [CrossRef]
- Bietti, M.; Lanzalunga, O.; Salamone, M. Structural Effects on the β-Scission Reaction of Alkoxyl Radicals. Direct Measurement of the Absolute Rate Constants for Ring Opening of Benzocycloalken-1-Oxyl Radicals. J. Org. Chem. 2005, 70, 1417–1422. [Google Scholar] [CrossRef]
- Morcillo, S.P. Radical-Promoted C−C Bond Cleavage: A Deconstructive Approach for Selective Functionalization. Angew. Chem. Int. Ed. 2019, 58, 14044–14054. [Google Scholar] [CrossRef]
- Yu, X.Y.; Chen, J.R.; Xiao, W.J. Visible Light-Driven Radical-Mediated C-C Bond Cleavage/Functionalization in Organic Synthesis. Chem. Rev. 2021, 121, 506–561. [Google Scholar] [CrossRef]
- Hartung, J. Stereoselective Construction of the Tetrahydrofuran Nucleus by Alkoxyl Radical Cyclizations. Eur. J. Org. Chem. 2001, 2001, 619–632. [Google Scholar] [CrossRef]
- de Armas, P.; Francisco, C.G.; Suárez, E. Fragmentation of Carbohydrate Anomeric Alkoxy Radicals. Tandem β-Fragmentation-Cyclization of Alcohols. J. Am. Chem. Soc. 1993, 115, 8865–8866. [Google Scholar] [CrossRef]
- Martin, A.; Salazar, J.A.; Suárez, E. Synthesis of Chiral Spiroacetals from Carbohydrates. J. Org. Chem. 1996, 61, 3999–4006. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wickenden, J.G.; Campbell, N.E.; Leung, J.C.T.; Johnson, K.M.; Sammis, G.M. Construction of Carbo- and Heterocycles Using Radical Relay Cyclizations Initiated by Alkoxy Radicals. Org. Lett. 2009, 11, 2019–2022. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Naruko, A.; Nakazawa, Y.; Zhao, L.; Hayashi, Y.; Hirama, M. Total Synthesis of Limonin. Angew. Chem. Int. Ed. 2015, 54, 8538–8541. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.S.; Ji, P.; Zhou, B.; Cheng, J.P. The Essential Role of Bond Energetics in C–H Activation/Functionalization. Chem. Rev. 2017, 117, 8622–8648. [Google Scholar] [CrossRef] [PubMed]
- Pirts, J.N.; Tolberg, R.S.; Martin, T.W.; Thompson, D.D.; Woolfolk, R.W. Intramolecular Free-Radical Reactions. Angew. Chem. Int. Ed. English 1964, 3, 525–538. [Google Scholar]
- Abarca, R.M. Global manual on surveillance of adverse events following immunizaion. WHO Nuevos Sist. Comun. E Inf. 2021, 4, 2013–2015. [Google Scholar]
- Kundu, R.; Ball, Z.T. Copper-Catalyzed Remote Sp3 C-H Chlorination of Alkyl Hydroperoxides. Org. Lett. 2010, 12, 2460–2463. [Google Scholar] [CrossRef]
- Petrović, G.; Čeković, Ž. Alkylation of Remote Non-Activated δ-Carbon Atoms: Addition of δ-Carbon Radicals, Generated by 1,5-Hydrogen Transfer in Alkoxy Radical Intermediates, to Activated Olefins. Tetrahedron 1999, 55, 1377–1390. [Google Scholar] [CrossRef]
- Petrović, G.; Saičić, R.N.; Čeković, Ž. Synthesis of Acetyl Scopine. Intramolecular Reactions of N-Carbethoxy Nortropine-3α-Benzene-Sulfenate. Synlett 1999, 1999, 635–637. [Google Scholar] [CrossRef]
- Martín, A.; Pérez-Martín, I.; Suárez, E. Intramolecular Hydrogen Abstraction Promoted by Amidyl Radicals. Evidence for Electronic Factors in the Nucleophilic Cyclization of Ambident Amides to Oxocarbenium Ions. Org. Lett. 2005, 7, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.; Gallon, F. Ring Closure Reactions of Substituted 4-Pentenyl-l-Oxy Radicals. J. Org. Chem 1995, 60, 6706–6716. [Google Scholar] [CrossRef]
- Kim, S.; Lee, T.A.; Song, Y. Facile Generation of Alkoxy Radicals from N-Alkoxyphthalimides. Synlett 1998, 5, 471–472. [Google Scholar] [CrossRef]
- Zlotorzynska, M.; Sammis, G.M. Photoinduced Electron-Transfer-Promoted Redox Fragmentation of N-Alkoxyphthalimides. Org. Lett. 2011, 13, 6264–6267. [Google Scholar] [CrossRef]
- Hoffmann, N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar] [CrossRef]
- Souillart, L.; Cramer, N. Catalytic C–C Bond Activations via Oxidative Addition to Transition Metals. Chem. Rev. 2015, 115, 9410–9464. [Google Scholar] [CrossRef]
- Xuan, J.; Zhang, Z.G.; Xiao, W.J. Visible-Light-Induced Decarboxylative Functionalization of Carboxylic Acids and Their Derivatives. Angew. Chem. Int. Ed. 2015, 54, 15632–15641. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef]
- R Barton, D.H.; Gellbr Cambridge, L.E.; Pechet, M.; Simpson, S.A.; Tait, T.F.; Wettstein, A.; Neher, R.; Euw, J.V.; Schindler, O.; Barton, H.R.; et al. A New Photochemical Reaction. J. Am. Chem. Soc. 2002, 82, 2640–2641. [Google Scholar] [CrossRef]
- Rivero, A.R.; Fodran, P.; Ondrejková, A.; Wallentin, C.J. Alcohol Etherification via Alkoxy Radicals Generated by Visible-Light Photoredox Catalysis. Org. Lett. 2020, 22, 8436–8440. [Google Scholar] [CrossRef]
- Pitre, S.P.; Overman, L.E. Strategic Use of Visible-Light Photoredox Catalysis in Natural Product Synthesis. Chem. Rev. 2021, 122, 1717–1751. [Google Scholar] [CrossRef] [PubMed]
- Genzink, M.J.; Kidd, J.B.; Swords, W.B.; Yoon, T.P. Chiral Photocatalyst Structures in Asymmetric Photochemical Synthesis. Chem. Rev. 2022, 122, 1654–1716. [Google Scholar] [CrossRef] [PubMed]
- Lechner, V.M.; Nappi, M.; Deneny, P.J.; Folliet, S.; Chu, J.C.K.; Gaunt, M.J. Visible-Light-Mediated Modification and Manipulation of Biomacromolecules. Chem. Rev. 2022, 122, 1752–1829. [Google Scholar] [CrossRef] [PubMed]
- Rossi-Ashton, J.A.; Clarke, A.K.; Unsworth, W.P.; Taylor, R.J.K. Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis. ACS Catal. 2020, 10, 7250–7261. [Google Scholar] [CrossRef]
- Stache, E.E.; Ertel, A.B.; Rovis, T.; Doyle, A.G. Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage-Independent Activation of Strong C-O Bonds. ACS Catal. 2018, 8, 11134–11139. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.; Zhang, Q. New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev. 2016, 45, 3069–3087. [Google Scholar] [CrossRef]
- Yu, X.Y.; Zhao, Q.Q.; Chen, J.; Xiao, W.J.; Chen, J.R. When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [Google Scholar] [CrossRef]
- Bloom, S.; Zhou, L.; Lu, B.; Xiao, W.-J.; Chen, J.-R. Recent Advances in Visible-Light-Mediated Amide Synthesis. Molecules 2022, 27, 517. [Google Scholar]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Twilton, J.; Le, C.C.; Zhang, P.; Shaw, M.H.; Evans, R.W.; MacMillan, D.W.C. The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem. 2017, 1, 0052. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Speckmeier, E.; Fischer, T.G.; Zeitler, K. A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [Google Scholar] [CrossRef] [PubMed]
- Crisenza, G.E.M.; Mazzarella, D.; Melchiorre, P. Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. [Google Scholar] [CrossRef] [PubMed]
- Gentry, E.C.; Knowles, R.R. Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res. 2016, 49, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ishida, N. β-Scission of Alkoxy Radicals in Synthetic Transformations. Chem. Lett. 2017, 46, 1692–1700. [Google Scholar] [CrossRef]
- Gratzel, M. Artificial Photosynthesis: Water Cleavage into Hydrogen and Oxygen by Visible Light. Acc. Chem. Res. 2002, 14, 376–384. [Google Scholar] [CrossRef]
- Meyer, T.J. Chemical Approaches to Artificial Photosynthesis. Acc. Chem. Res. 2002, 22, 163–170. [Google Scholar] [CrossRef]
- Takeda, H.; Ishitani, O. Development of Efficient Photocatalytic Systems for CO2 Reduction Using Mononuclear and Multinuclear Metal Complexes Based on Mechanistic Studies. Coord. Chem. Rev. 2010, 254, 346–354. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Takeda, H.; Ishitani, O. Photocatalytic Reduction of CO2 Using Metal Complexes. J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 106–137. [Google Scholar] [CrossRef]
- DiRocco, D.A.; Dykstra, K.; Krska, S.; Vachal, P.; Conway, D.V.; Tudge, M. Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem. Int. Ed. 2014, 53, 4802–4806. [Google Scholar] [CrossRef]
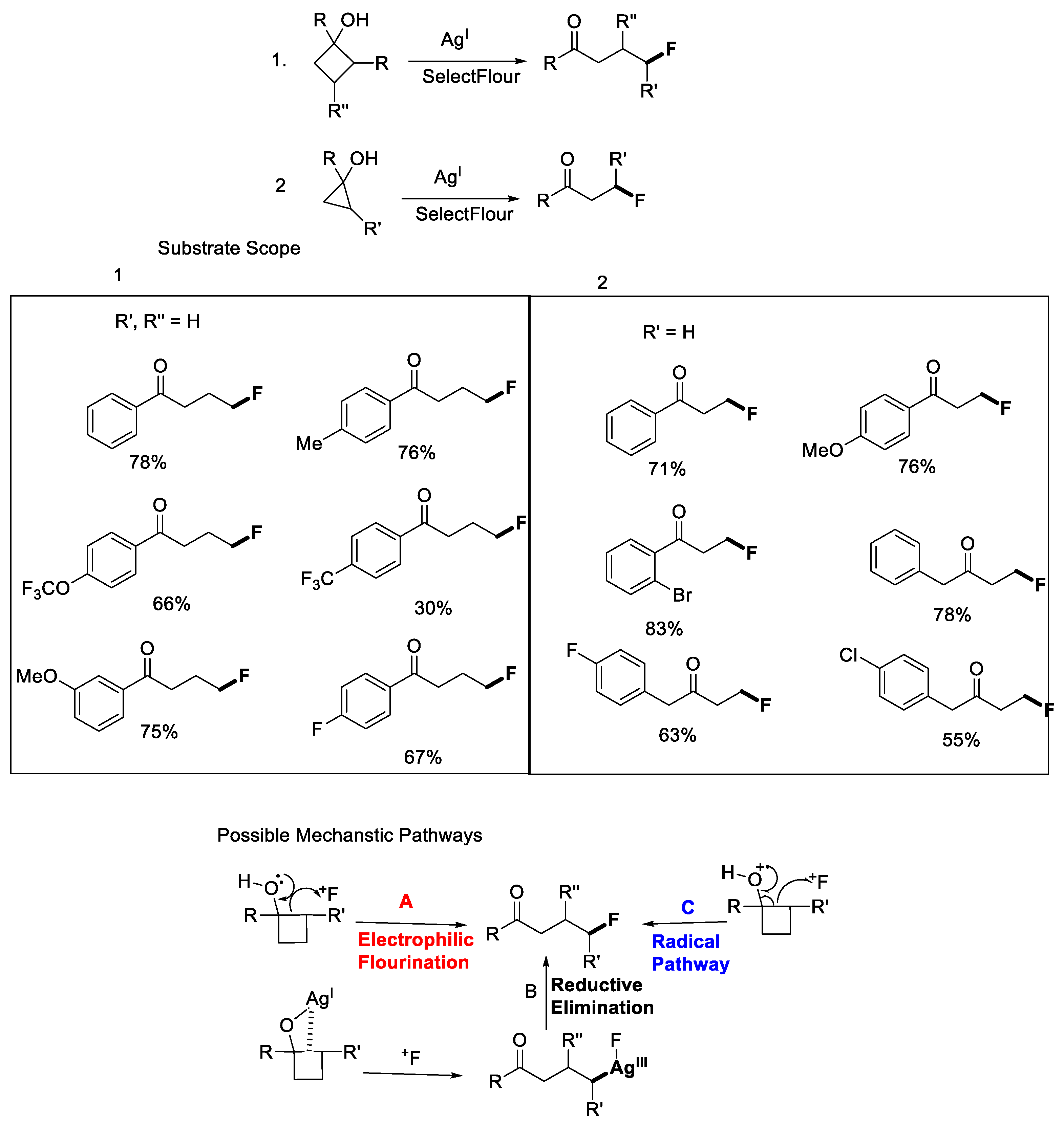
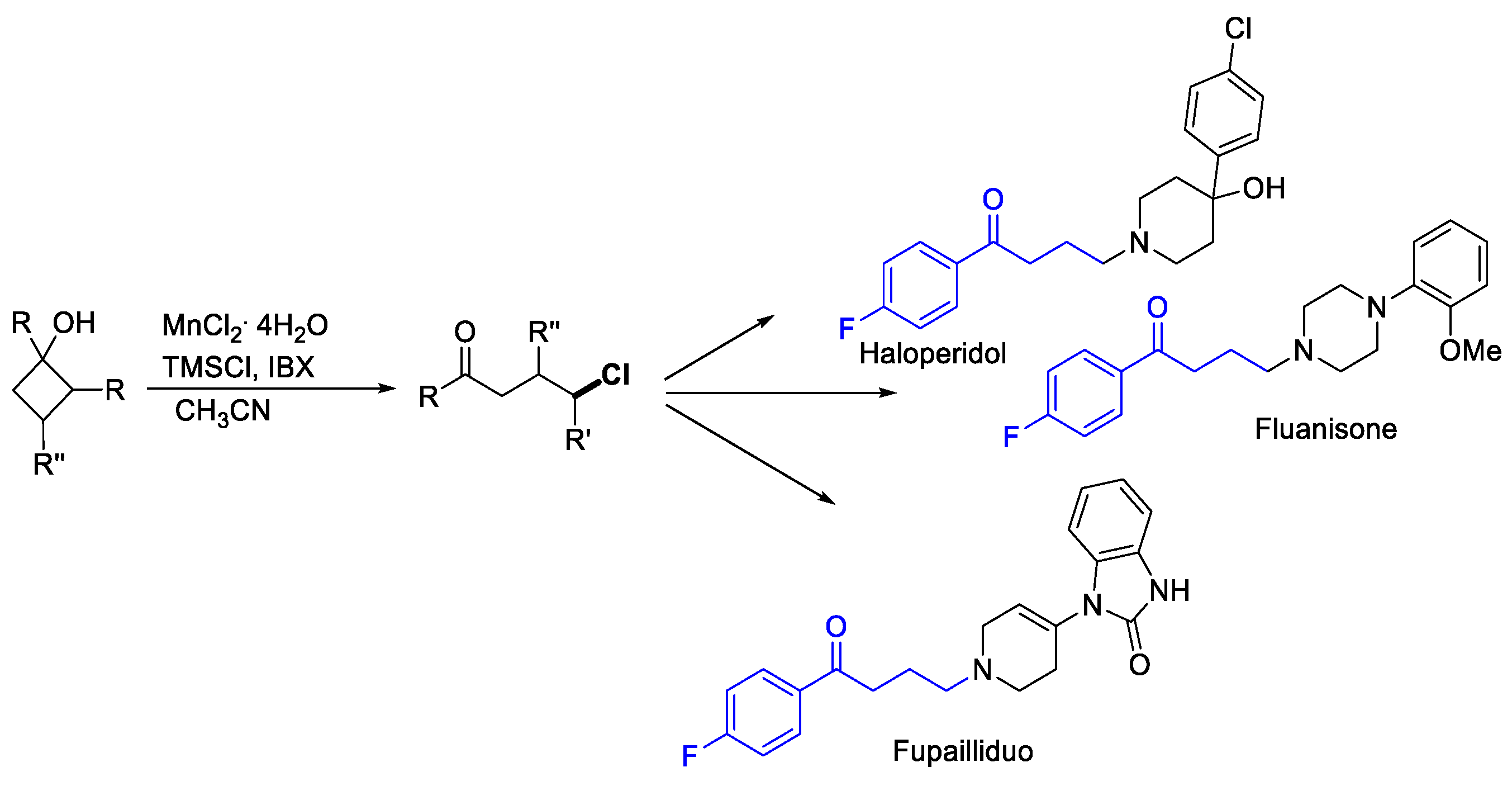
- Zhao, H.; Fan, X.; Yu, J.; Zhu, C. Silver-Catalyzed Ring-Opening Strategy for the Synthesis of β- and γ-Fluorinated Ketones. J. Am. Chem. Soc. 2015, 137, 3490–3493. [Google Scholar] [CrossRef] [PubMed]
- Huan, L.; Zhu, C. Manganese-Catalyzed Ring-Opening Chlorination of Cyclobutanols: Regiospecific Synthesis of γ-Chloroketones †. Org. Chem. Front. 2016, 3, 1467. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Zhang, F.; Hu, C.; Chen, Y. Generation of Alkoxyl Radicals by Photoredox Catalysis Enables Selective C(Sp3)−H Functionalization under Mild Reaction Conditions. Angew. Chem. Int. Ed. 2016, 55, 1872–1875. [Google Scholar] [CrossRef] [PubMed]
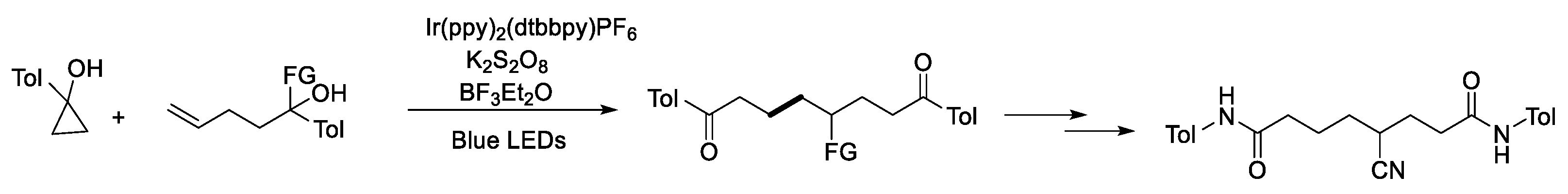
- Zhang, Y.; Zhang, Y.; Shen, X. Alkoxyl-Radical-Mediated Synthesis of Functionalized Allyl Tert-(Hetero)Cyclobutanols and Their Ring-Opening and Ring-Expansion Functionalizations. Chem. Catal. 2021, 1, 423–436. [Google Scholar] [CrossRef]
- Wang, C.; Harms, K.; Meggers, E. Catalytic Asymmetric C−H Functionalization under Photoredox Conditions by Radical Translocation and Stereocontrolled Alkene Addition. Angew. Chem. Int. Ed. 2016, 55, 13495–13498. [Google Scholar] [CrossRef]
- Jia, K.; Zhang, F.; Huang, H.; Chen, Y. Visible-Light-Induced Alkoxyl Radical Generation Enables Selective C(Sp3)-C(Sp3) Bond Cleavage and Functionalizations. J. Am. Chem. Soc. 2016, 138, 1514–1517. [Google Scholar] [CrossRef]
- Jia, K.; Pan, Y.; Chen, Y. Selective Carbonyl-C(Sp 3 ) Bond Cleavage To Construct Ynamides, Ynoates, and Ynones by Photoredox Catalysis. Angew. Chem. Int. Ed. 2017, 56, 2478–2481. [Google Scholar] [CrossRef]
- Yayla, H.G.; Wang, H.; Tarantino, K.T.; Orbe, H.S.; Knowles, R.R. Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O-H Bonds. J. Am. Chem. Soc. 2016, 138, 10794–10797. [Google Scholar] [CrossRef]
- Guo, J.-J.; Nhua Hu, A.; Chen, I.; Sun, J.; Tang, H.; Zuo, Z.; Guo, J.; Hu, A.; Chen, Y.; Sun, J.; et al. Photocatalytic C−C Bond Cleavage and Amination of Cycloalkanols by Cerium(III) Chloride Complex. Angew. Chem. Int. Ed. 2016, 55, 15319–15322. [Google Scholar] [CrossRef]
- Hu, A.; Guo, J.J.; Pan, H.; Tang, H.; Gao, Z.; Zuo, Z. δ-Selective Functionalization of Alkanols Enabled by Visible-Light-Induced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc. 2018, 140, 1612–1616. [Google Scholar] [CrossRef]
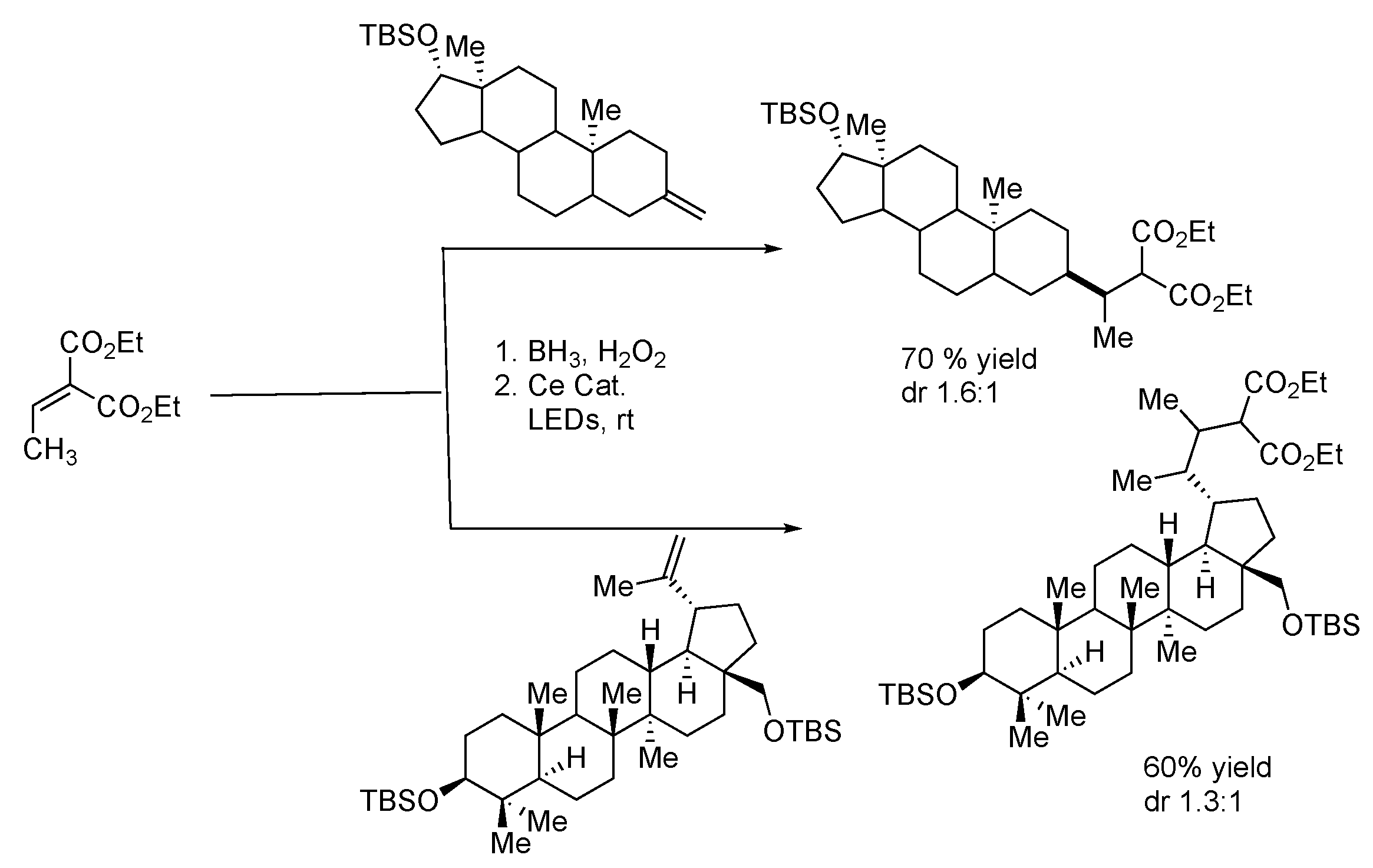
- Hu, A.; Chen, Y.; Guo, J.J.; Yu, N.; An, Q.; Zuo, Z. Cerium-Catalyzed Formal Cycloaddition of Cycloalkanols with Alkenes through Dual Photoexcitation. J. Am. Chem. Soc. 2018, 140, 13580–13585. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mao, J.; Zhu, C. Visible Light-Promoted Ring-Opening Functionalization of Unstrained Cycloalkanols via Inert C–C Bond Scission. Chem. Sci. 2018, 9, 5805–5809. [Google Scholar] [CrossRef] [PubMed]
- Ota, E.; Wang, H.; Frye, N.L.; Knowles, R.R. A Redox Strategy for Light-Driven, Out-of-Equilibrium Isomerizations and Application to Catalytic C-C Bond Cleavage Reactions. J. Am. Chem. Soc. 2019, 141, 1457–1462. [Google Scholar] [CrossRef]
- Huang, L.; Ji, T.; Rueping, M. Remote Nickel-Catalyzed Cross-Coupling Arylation via Proton-Coupled Electron Transfer-Enabled C-C Bond Cleavage. J. Am. Chem. Soc. 2020, 142, 3532–3539. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Yao, Y.; Zhu, D.; Chang, D.; Liu, Y.; Shi, L. Visible-Light-Enhanced Ring Opening of Cycloalkanols Enabled by Brønsted Base-Tethered Acyloxy Radical Induced Hydrogen Atom Transfer-Electron Transfer. Org. Lett. 2018, 20, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, D.; Liu, S.; Ge, Y.; Lan, Y.; Chen, Y. Visible-Light-Induced Alkoxyl Radicals Enable α-C(Sp3)-H Bond Allylation. Iscience 2020, 23, 100755. [Google Scholar]
- Kim, I.; Min, M.; Kang, D.; Kim, K.; Hong, S. Direct Phosphonation of Quinolinones and Coumarins Driven by the Photochemical Activity of Substrates and Products. Org. Lett. 2017, 19, 1394–1397. [Google Scholar] [CrossRef]
- Wu, X.; Wang, M.; Huan, L.; Wang, D.; Wang, J.; Zhu, C. Tertiary-Alcohol-Directed Functionalization of Remote C(Sp3)−H Bonds by Sequential Hydrogen Atom and Heteroaryl Migrations. Angew. Chem. Int. Ed. 2018, 57, 1640–1644. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Tang, N.; Wu, Z.; Wang, D.; Ji, M.; Xu, Y.; Wang, M.; Zhu, C. Metal-Free Alcohol-Directed Regioselective Heteroarylation of Remote Unactivated C(Sp3)–H Bonds. Nat. Commun. 2018, 9, 3343. [Google Scholar] [CrossRef]
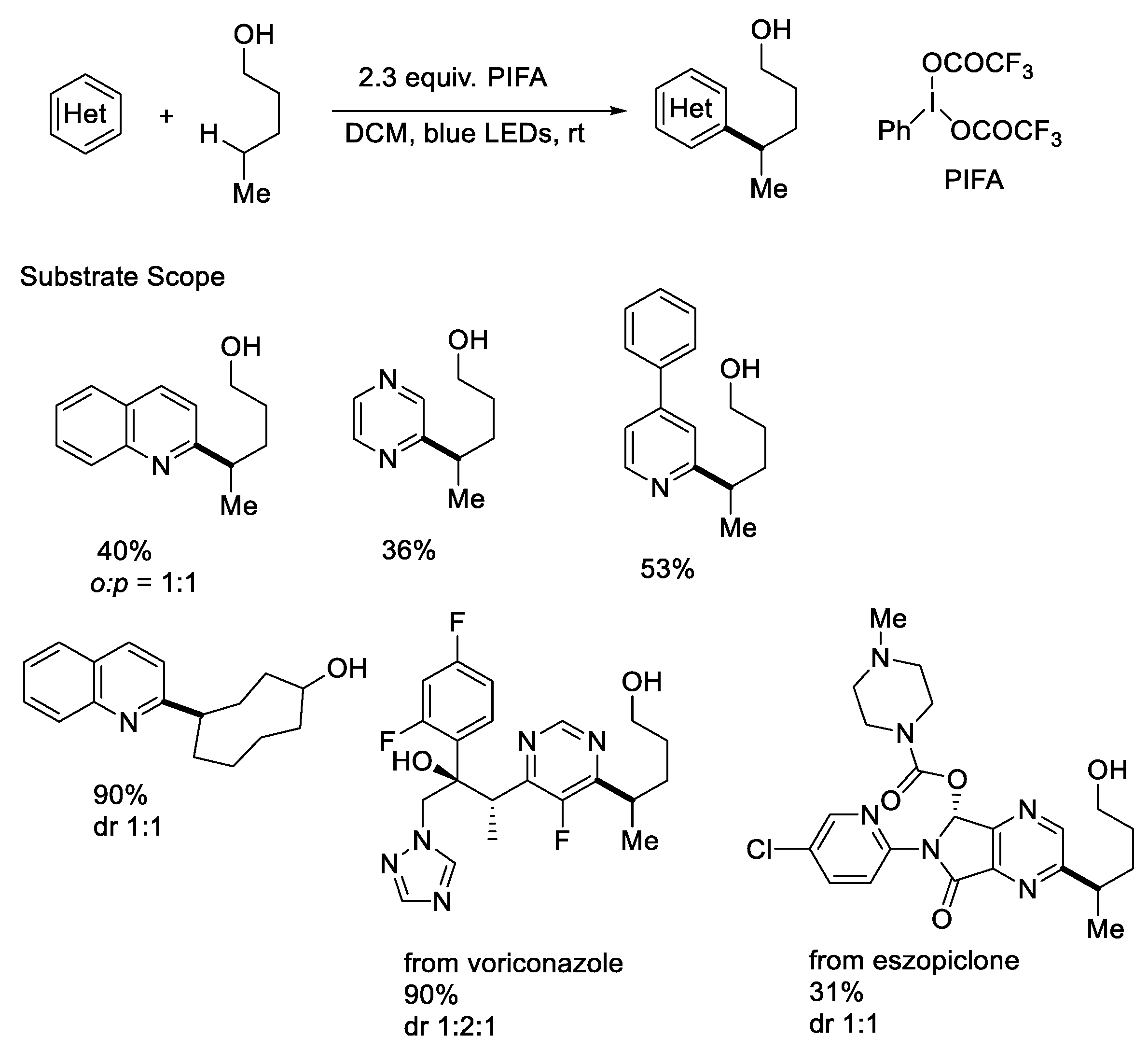
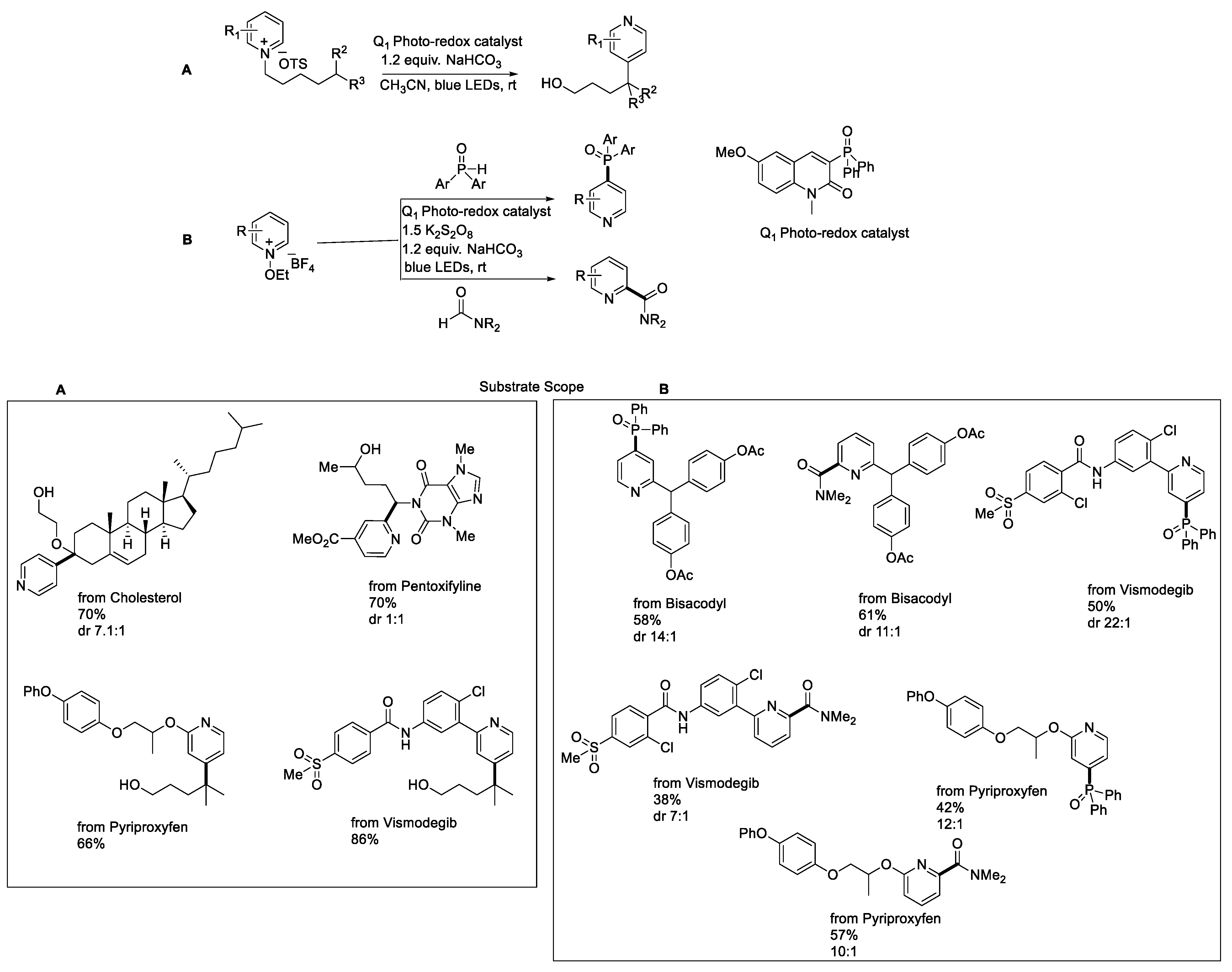
- Kim, I.; Park, B.; Kang, G.; Kim, J.; Jung, H.; Lee, H.; Baik, M.H.; Hong, S. Visible-Light-Induced Pyridylation of Remote C(Sp3)−H Bonds by Radical Translocation of N-Alkoxypyridinium Salts. Angew. Chem. Int. Ed. 2018, 57, 15517–15522. [Google Scholar] [CrossRef]
- Kim, I.; Kang, G.; Lee, K.; Park, B.; Kang, D.; Jung, H.; He, Y.T.; Baik, M.H.; Hong, S. Site-Selective Functionalization of Pyridinium Derivatives via Visible-Light-Driven Photocatalysis with Quinolinone. J. Am. Chem. Soc. 2019, 141, 9239–9248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chang, L.; An, Q.; Wang, X.; Zuo, Z. Dehydroxymethylation of Alcohols Enabled by Cerium Photocatalysis. J. Am. Chem. Soc. 2019, 141, 10556–10564. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Wu, Z.; Zhu, C. Visible-Light-Induced Consecutive C-C Bond Fragmentation and Formation for the Synthesis of Elusive Unsymmetric 1,8-Dicarbonyl Compounds. Chem. Commun. 2019, 55, 2368–2371. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Liu, S.; Huang, L.; Liu, Z.Q. Surgical Cleavage of Unstrained C(Sp3)−C(Sp3) Bonds in General Alcohols for Heteroaryl C−H Alkylation and Acylation. Adv. Synth. Catal. 2019, 361, 4568–4574. [Google Scholar] [CrossRef]
- Santana, A.G.; González, C.C. Tandem Radical Fragmentation/Cyclization of Guanidinylated Monosaccharides Grants Access to Medium-Sized Polyhydroxylated Heterocycles. Org. Lett. 2020, 22, 8492–8495. [Google Scholar] [CrossRef]
- Zhu, C.; Shao, X.; Wu, X.; Wu, S. Metal-Free Radical-Mediated C(Sp3)-H Heteroarylation of Alkanes. Org. Lett. 2020, 22, 7450–7454. [Google Scholar]
- Chen, Y.; Du, J.; Zuo, Z. Selective C-C Bond Scission of Ketones via Visible-Light-Mediated Cerium Catalysis. Chem 2020, 6, 266–279. [Google Scholar] [CrossRef]
- Zheng, M.; Hou, J.; Zhan, L.W.; Huang, Y.; Chen, L.; Hua, L.L.; Li, Y.; Tang, W.Y.; Li, B.D. Visible-Light-Driven, Metal-Free Divergent Difunctionalization of Alkenes Using Alkyl Formates. ACS Catal. 2021, 11, 542–553. [Google Scholar] [CrossRef]
- Wu, L.; Wang, L.; Chen, P.; Guo, Y.L.; Liu, G. Enantioselective Copper-Catalyzed Radical Ring-Opening Cyanation of Cyclopropanols and Cyclopropanone Acetals. Adv. Synth. Catal. 2020, 362, 2189–2194. [Google Scholar] [CrossRef]
- Huang, L.; Ji, T.; Zhu, C.; Yue, H.; Zhumabay, N.; Rueping, M. Bioinspired Desaturation of Alcohols Enabled by Photoredox Proton-Coupled Electron Transfer and Cobalt Dual Catalysis. Nat. Commun. 2022, 13, 809. [Google Scholar] [CrossRef]
- Vellakkaran, M.; Kim, T.; Hong, S. Visible-Light-Induced C4-Selective Functionalization of Pyridinium Salts with Cyclopropanols. Angew. Chem. Int. Ed. 2022, 61, e202113658. [Google Scholar] [CrossRef] [PubMed]
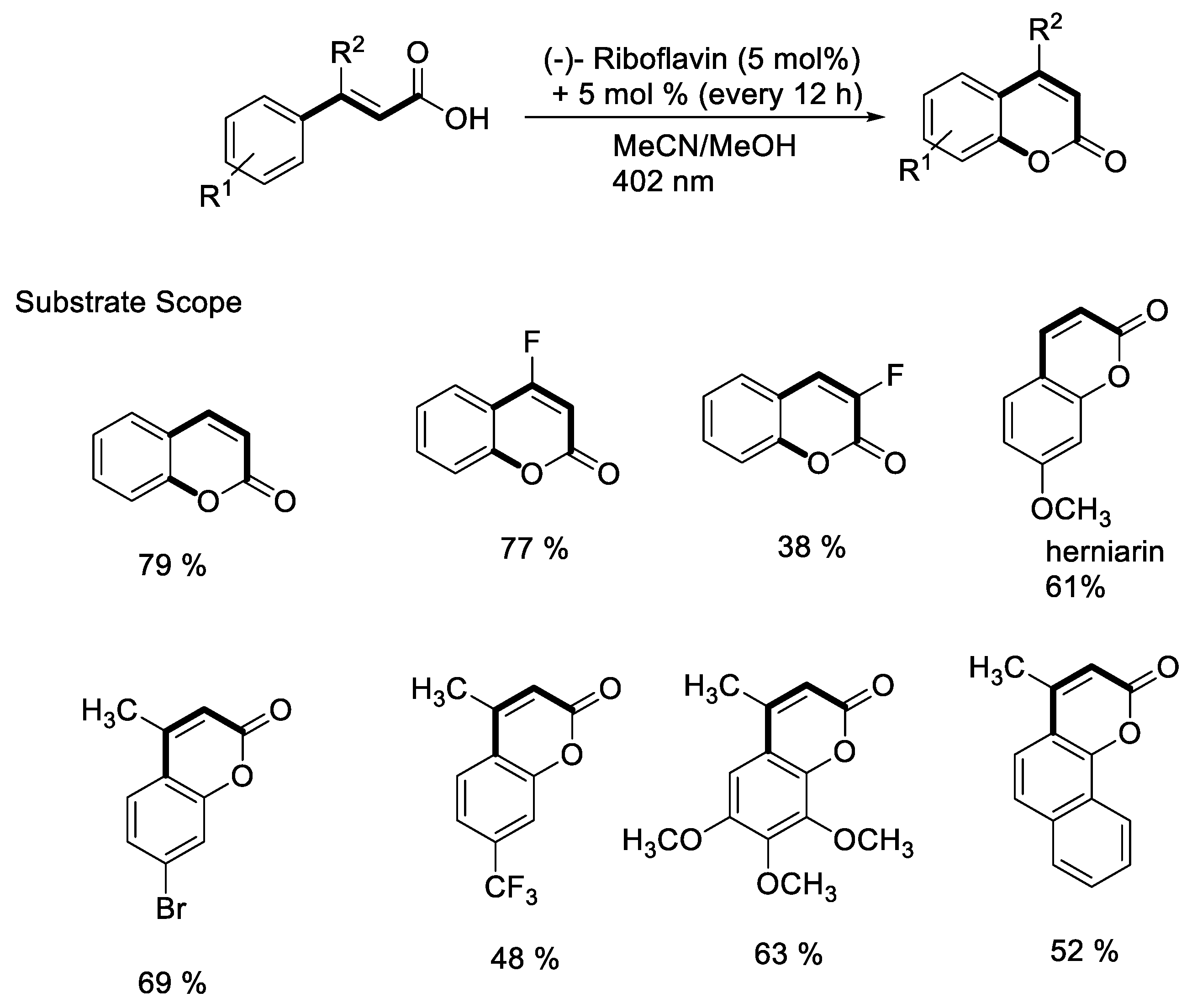
- Metternich, J.B.; Gilmour, R. One Photocatalyst, n Activation Modes Strategy for Cascade Catalysis: Emulating Coumarin Biosynthesis with (-)-Riboflavin. J. Am. Chem. Soc. 2016, 138, 1040–1045. [Google Scholar] [CrossRef]
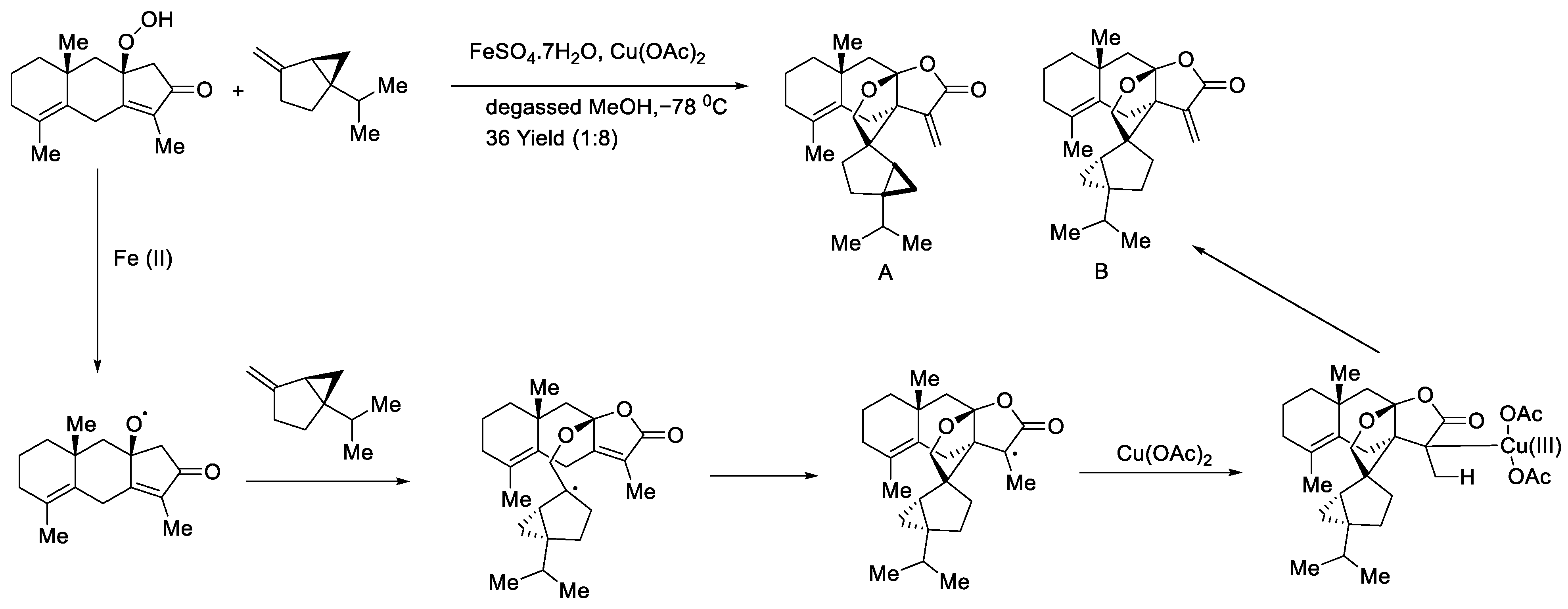
- Li, X.; Zhai, T.; He, P.; Wei, Z.; Wang, Z. Biomimetic Synthesis of Hitorins A and B via an Intermolecular Alkoxy Radical-Olefin Coupling Cascade. In ChemRxiv; Cambridge Open Engage: Cambridge, UK, 2019. [Google Scholar] [CrossRef]
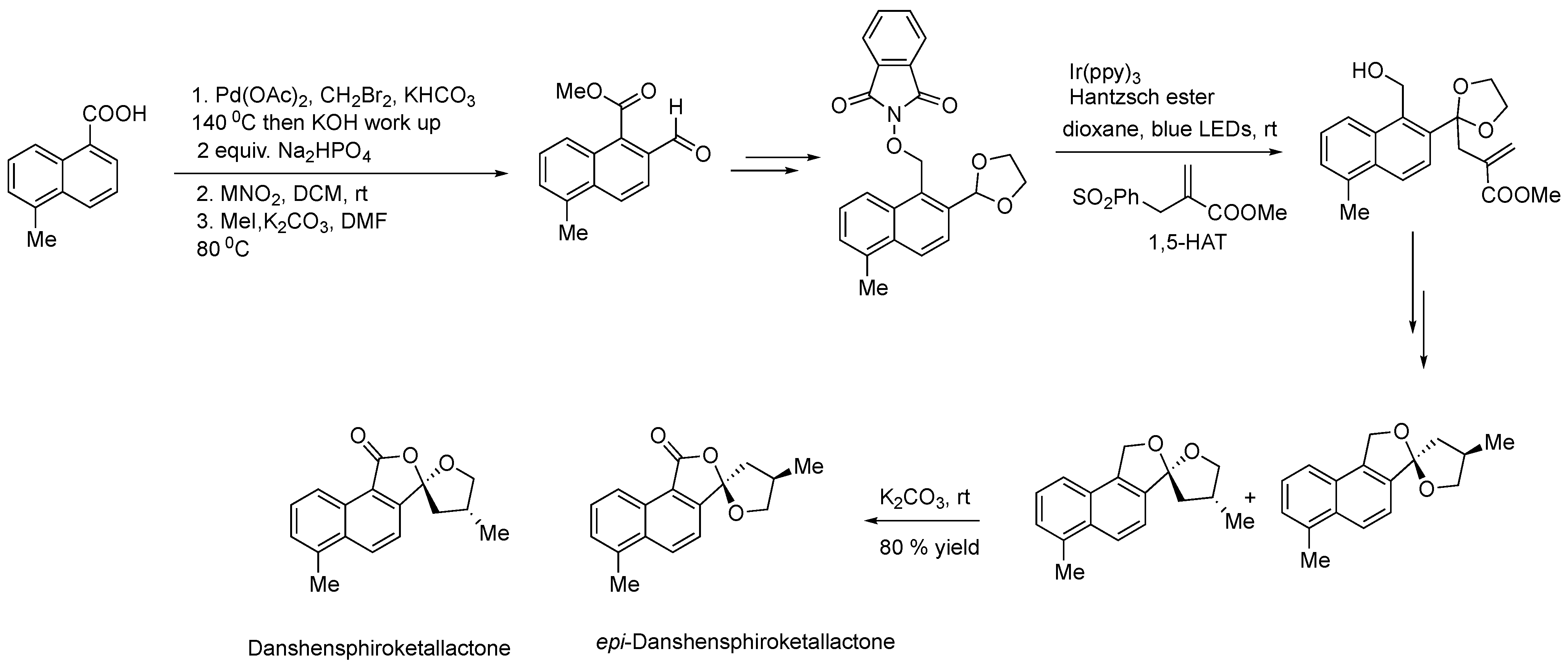
- Deng, Y.; Nguyen, M.D.; Zou, Y.; Houk, K.N.; Smith, A.B. Generation of Dithianyl and Dioxolanyl Radicals Using Photoredox Catalysis: Application in the Total Synthesis of the Danshenspiroketallactones via Radical Relay Chemistry. Org. Lett. 2019, 21, 1708–1712. [Google Scholar] [CrossRef] [PubMed]
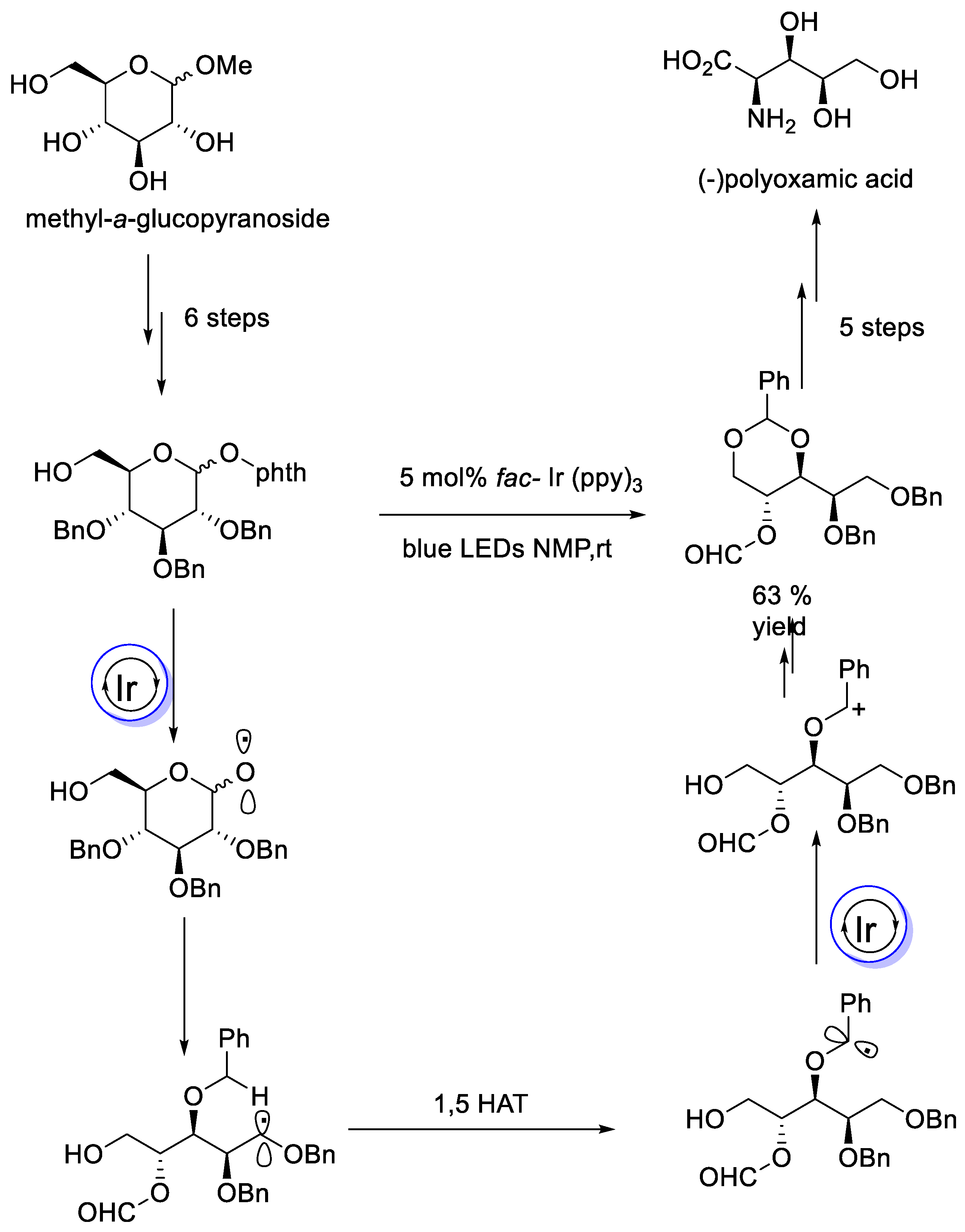
- Matsuoka, T.; Inuki, S.; Miyagawa, T.; Oishi, S.; Ohno, H. Total Synthesis of (+)-Polyoxamic Acid via Visible-Light-Mediated Photocatalytic β-Scission and 1,5-Hydrogen Atom Transfer of Glucose Derivative. J. Org. Chem. 2020, 85, 8271–8278. [Google Scholar] [CrossRef] [PubMed]




































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.; Sewell, S.; Li, J.; Wang, T. Recent Advances in Application of Alkoxy Radical in Organic Synthesis. Organics 2023, 4, 459-489. https://doi.org/10.3390/org4040033
Ali M, Sewell S, Li J, Wang T. Recent Advances in Application of Alkoxy Radical in Organic Synthesis. Organics. 2023; 4(4):459-489. https://doi.org/10.3390/org4040033
Chicago/Turabian StyleAli, Munsaf, Shi Sewell, Juncheng Li, and Ting Wang. 2023. "Recent Advances in Application of Alkoxy Radical in Organic Synthesis" Organics 4, no. 4: 459-489. https://doi.org/10.3390/org4040033