
Absorption Spectra of Protonated Corroles: Two Distinct Patterns Due to Peripheral Substitution Architecture
Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
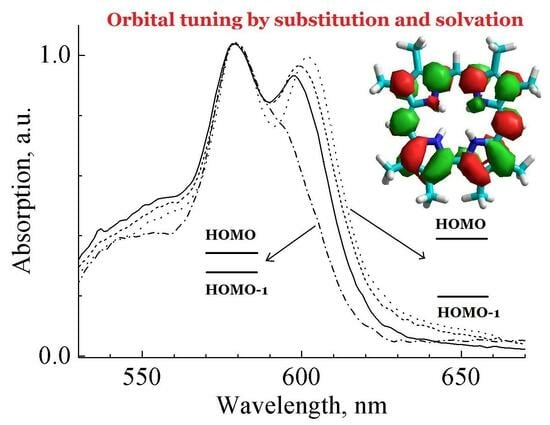
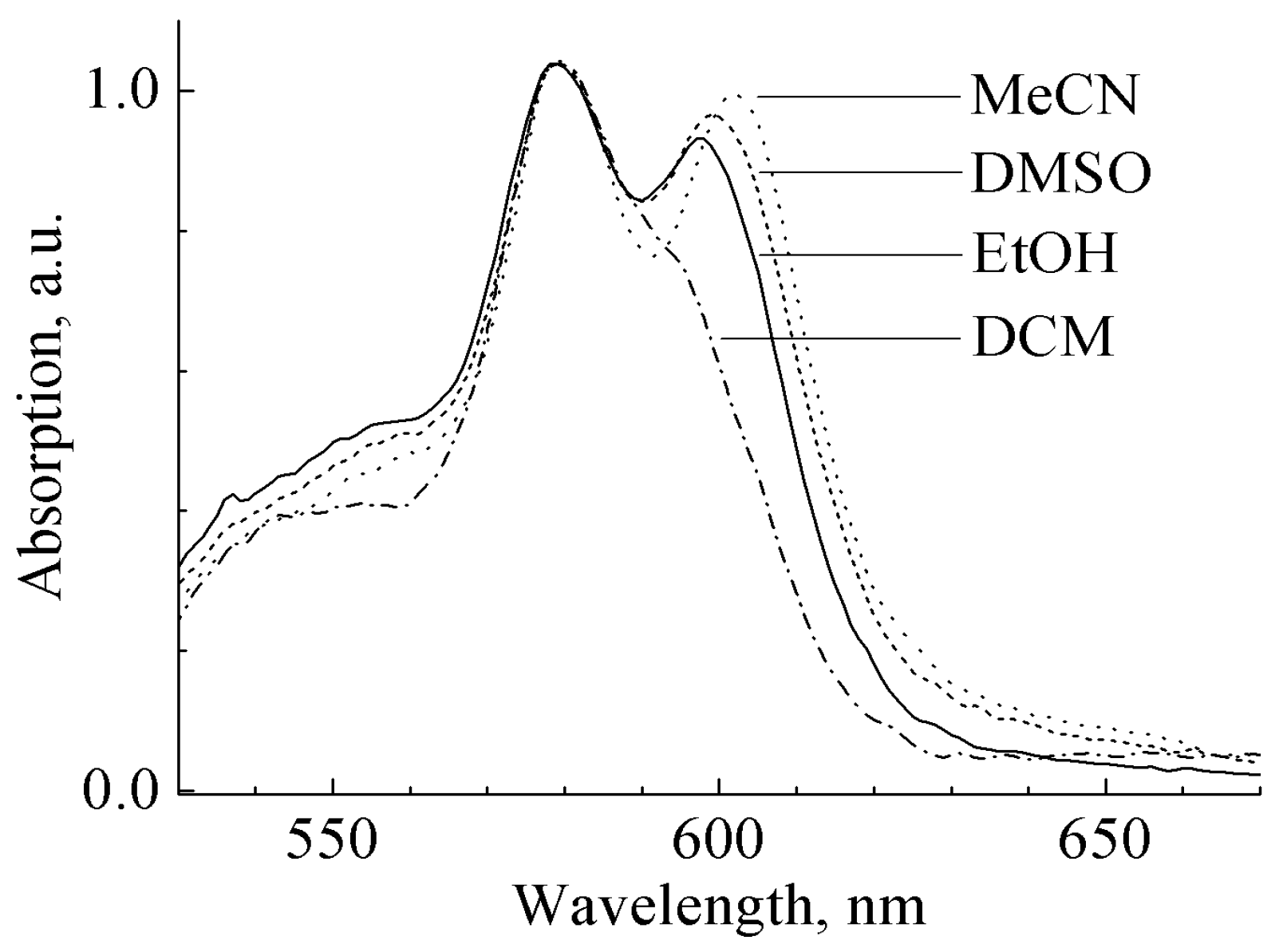
3.1. Ground State Absorption Spectra
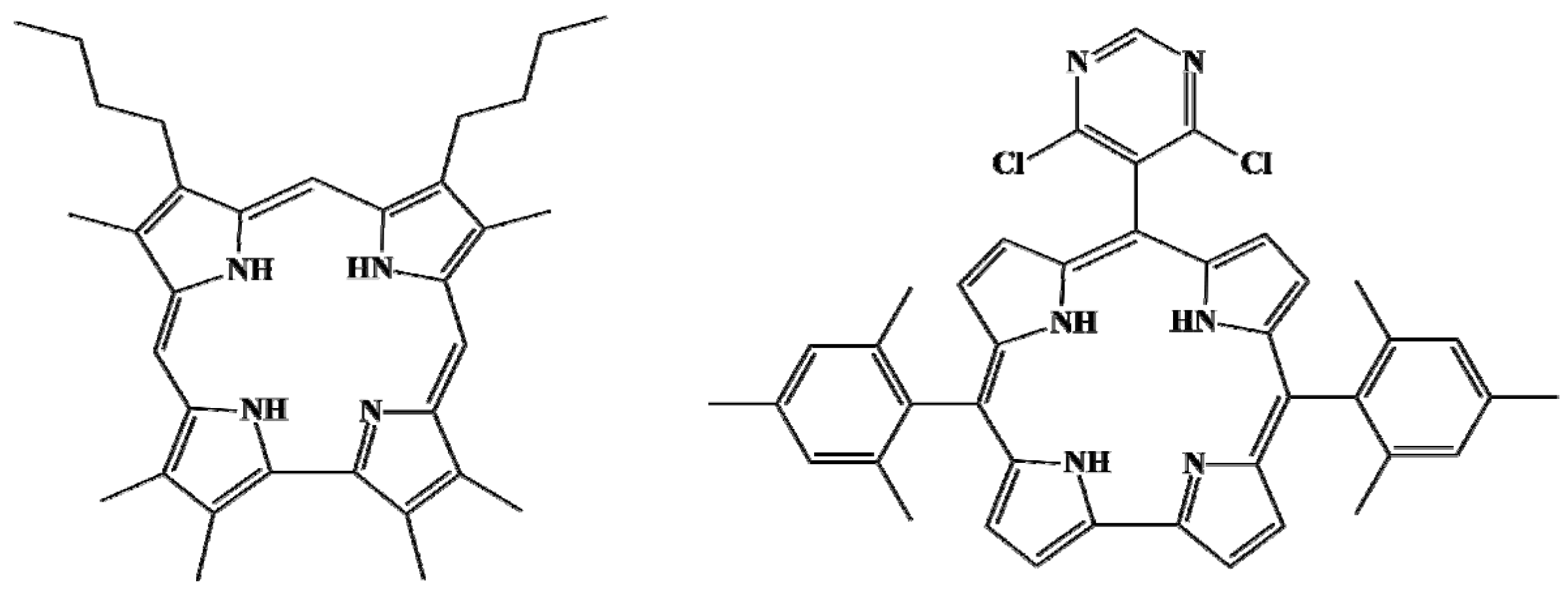
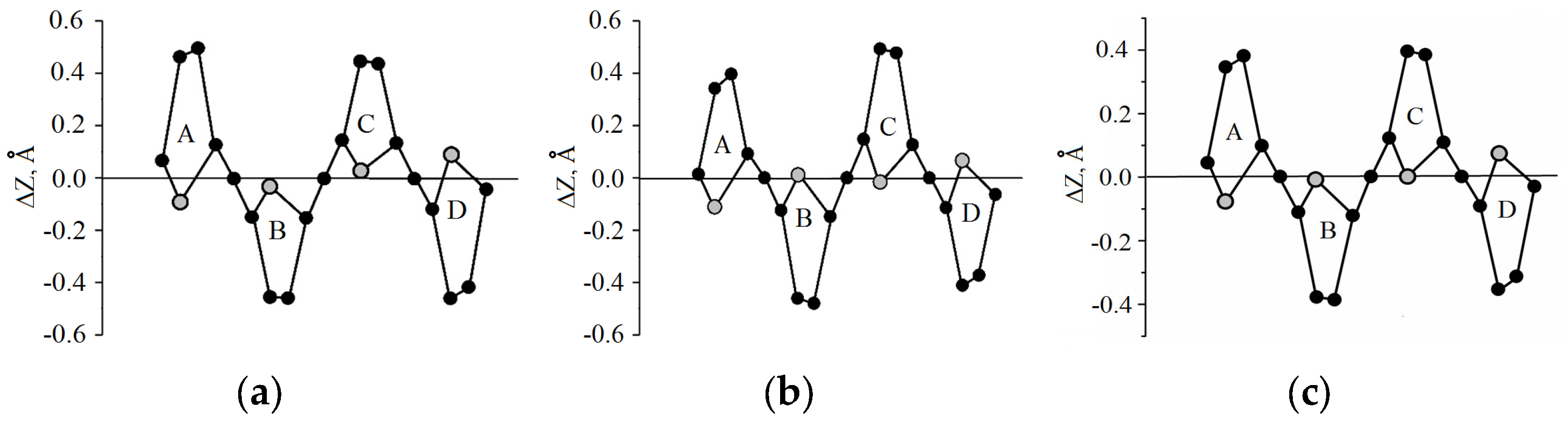
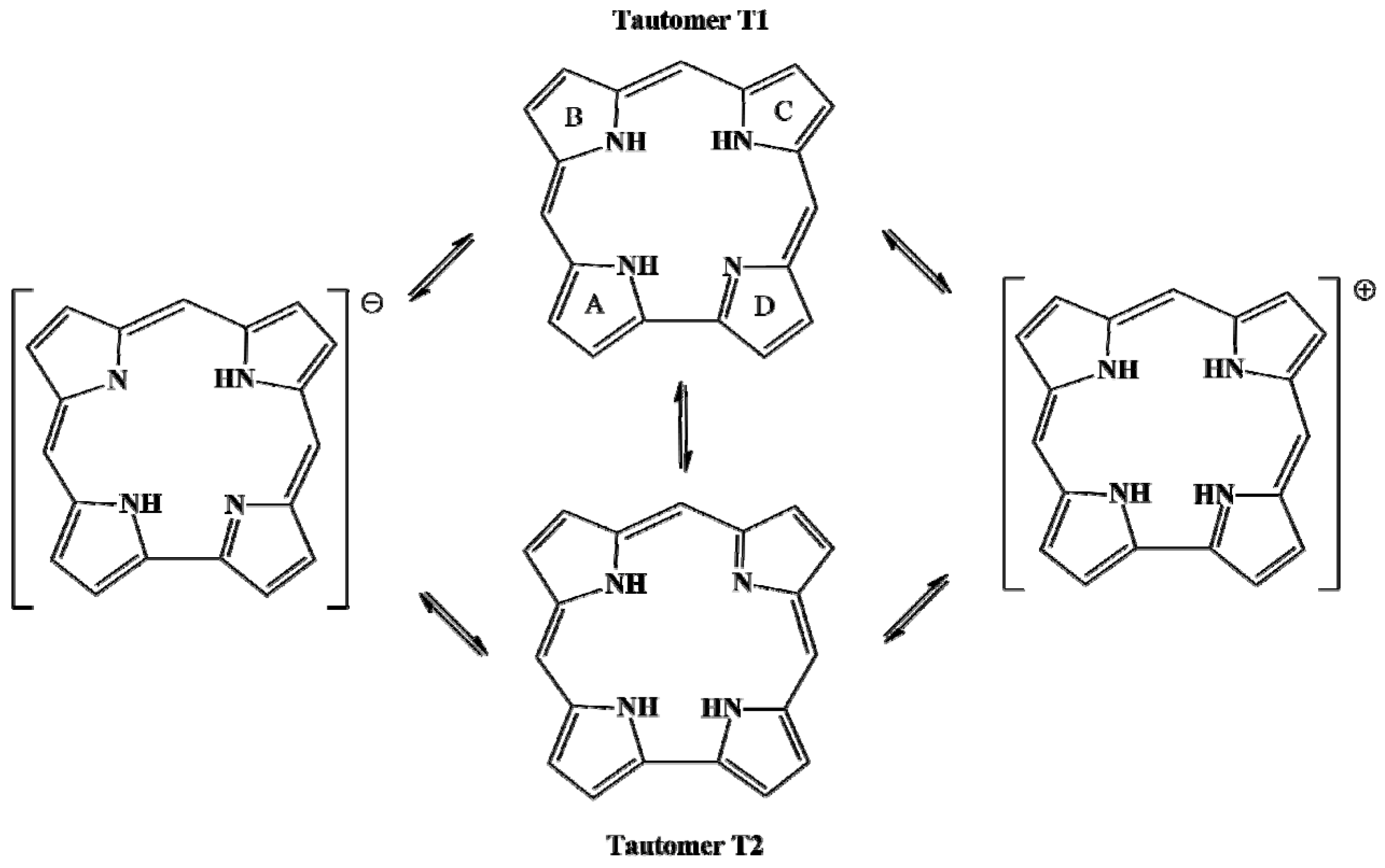
3.2. Macrocycle Conformation
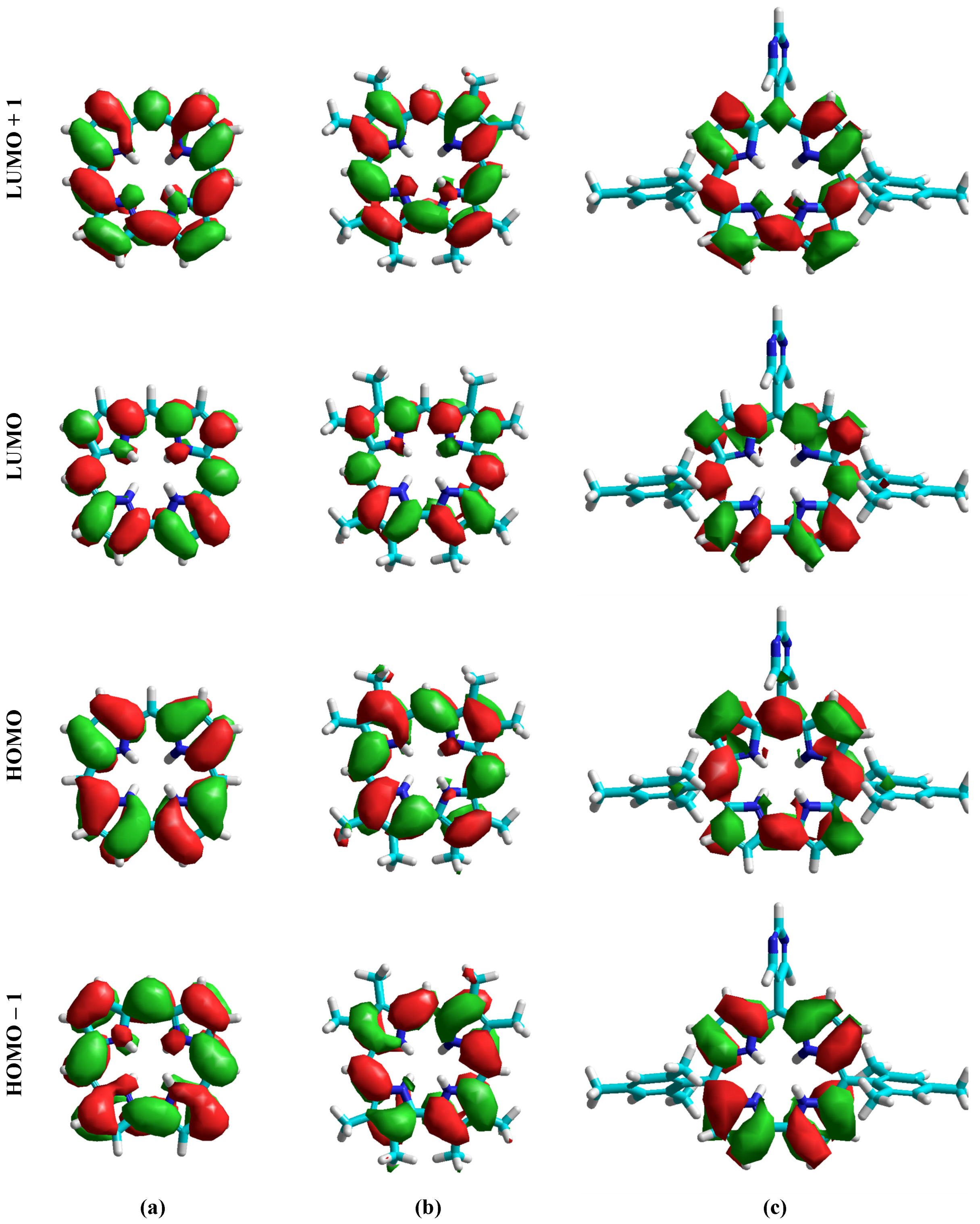
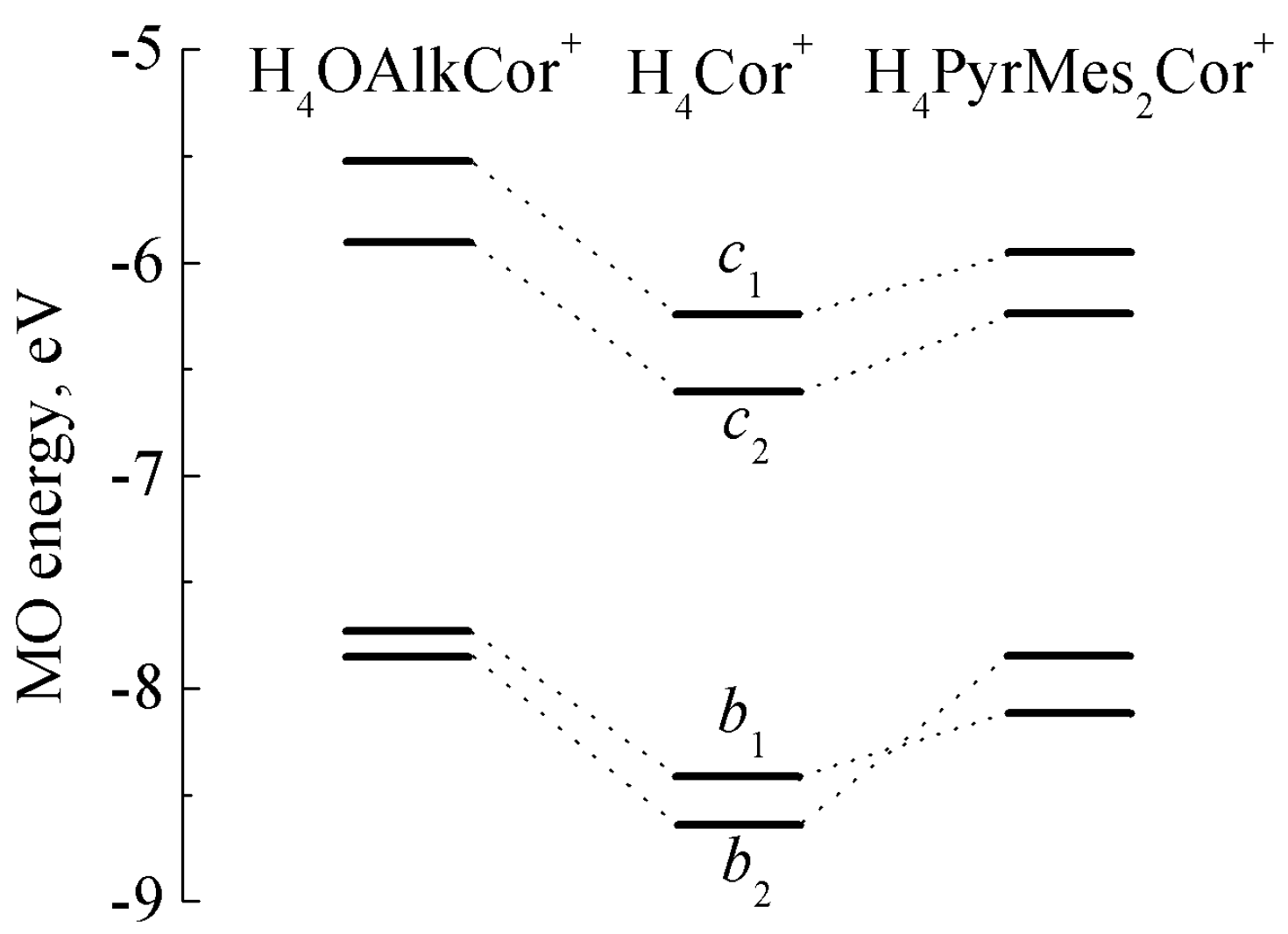
3.3. Molecular Orbitals Calculations
3.4. Calculations of the Absorption Spectra
3.5. Tuning the Configuration Interaction with Specific Solvation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senge, M.O. The conformational flexibility of tetrapyrroles—Current model studies and photobiological relevance. J. Photochem. Photobiol. B Biol. 1992, 16, 3–36. [Google Scholar] [CrossRef]
- Senge, M.O.; MacGovan, S.A.; O’Brien, J. Conformational control of cofactors in nature—The influence of protein-induced macrocycle distortion on the biological function of tetrapyrroles. Chem. Commun. 2015, 51, 17031–17063. [Google Scholar] [CrossRef]
- Kielmann, M.; Senge, M.O. Molecular engineering of free-base porphyrins as ligands—The N-H…X binding motif in tetrapyrroles. Angew. Chem. Int. Ed. 2019, 58, 418–441. [Google Scholar] [CrossRef]
- Stone, A.; Fleischer, E.B. The molecular and crystal structure of porphyrin diacids. J. Am. Chem. Soc. 1968, 90, 2735–2748. [Google Scholar] [CrossRef]
- Cheng, B.; Munro, O.Q.; Marques, H.M.; Scheidt, W.R. An analysis of porphyrin molecular flexibility—Use of porphyrin diacids. J. Am. Chem. Soc. 1997, 119, 10732–10742. [Google Scholar] [CrossRef]
- Lavallee, D.K. The Chemistry and Biochemistry of N-Substituted Porphyrins, 1st ed.; Wiley-VCH: New York, NY, USA, 1987; pp. 254–313. [Google Scholar]
- Senge, M.O. Highly substituted porphyrins. In The Porphyrin Handbook, 1st ed.; Kadish, K.M., Smith, K.M., Guillard, R., Eds.; Academic Press: New York, NY, USA; World Scientific: Singapore, 2000; Volume 1, pp. 239–347. [Google Scholar]
- Roucan, M.; Flanagan, K.J.; O’Brien, J.; Senge, M.O. Nonplanar porphyrins by N-substitution: A neglected pathway. Eur. J. Org. Chem. 2018, 46, 6432–6446. [Google Scholar] [CrossRef]
- Fang, Y.; Bhyrappa, P.; Ou, Z.; Kadish, K.M. Planar and nonplanar free-base tetrarylporphyrins: β-pyrrole substituents and geometric effects on electrochemistry, spectroelectrochemistry, and protonation/deprotonation reactions in nonaqueous media. Chem. Eur. J. 2014, 20, 524–532. [Google Scholar] [CrossRef]
- Ballester, M.; Ravotto, L.; Quirke, J.M.E.; Lopez de Vega, R.; Shelnutt, J.A.; Cheprakov, A.V.; Vinogradov, S.A.; Medforth, C.J. Protonation of planar and nonplanar porphyrins: A calorimetric and computational study. J. Phys. Chem. A 2020, 124, 8994–9003. [Google Scholar] [CrossRef]
- Kruk, M.M.; Ivanova, Y.B.; Sheinin, V.B.; Starukhin, A.S.; Mamardashvili, N.Z.; Koifman, O.I. Highly sensitive halide ions recognition with diprotonated porphyrin. Macroheterocycles 2008, 1, 50–58. [Google Scholar] [CrossRef]
- Kruk, M.M.; Starukhin, A.S.; Mamardashvili, N.Z.; Mamardashvili, G.M.; Ivanova, Y.B.; Maltseva, O.V. Tetrapyrrolic compounds as the hosts for binding of halides and alkali metal cations. J. Porph. Phthal. 2009, 13, 1148–1158. [Google Scholar] [CrossRef]
- Novaritsa, K.; Flanagan, K.J.; Gibbons, D.; Senge, M.O. Conformational Re-engineering of porphyrins as receptors with switchable N-H…X-type binding modes. Angew. Chem. Int. Ed. 2019, 58, 16553–16557. [Google Scholar] [CrossRef]
- Roucan, M.; Kielmann, M.; Connon, S.J.; Bernhard, S.S.R.; Senge, M.O. Conformational control of nonplanar freebase porphyrins: Toward bifunctional catalysts of tunable basicity. Chem. Comm. 2018, 54, 26–29. [Google Scholar] [CrossRef]
- Ding, Y.; Zhu, W.-H.; Xie, Y. Development of ion chemosensors based on porphyrin analogues. Chem. Rev. 2017, 117, 2203–2256. [Google Scholar] [CrossRef]
- Paolesse, R.; Nardis, S.; Monti, D.; Stefanelli, M.; Di Natale, C. Porphyrinoids for chemical sensor applications. Chem. Rev. 2017, 117, 2517–2583. [Google Scholar] [CrossRef]
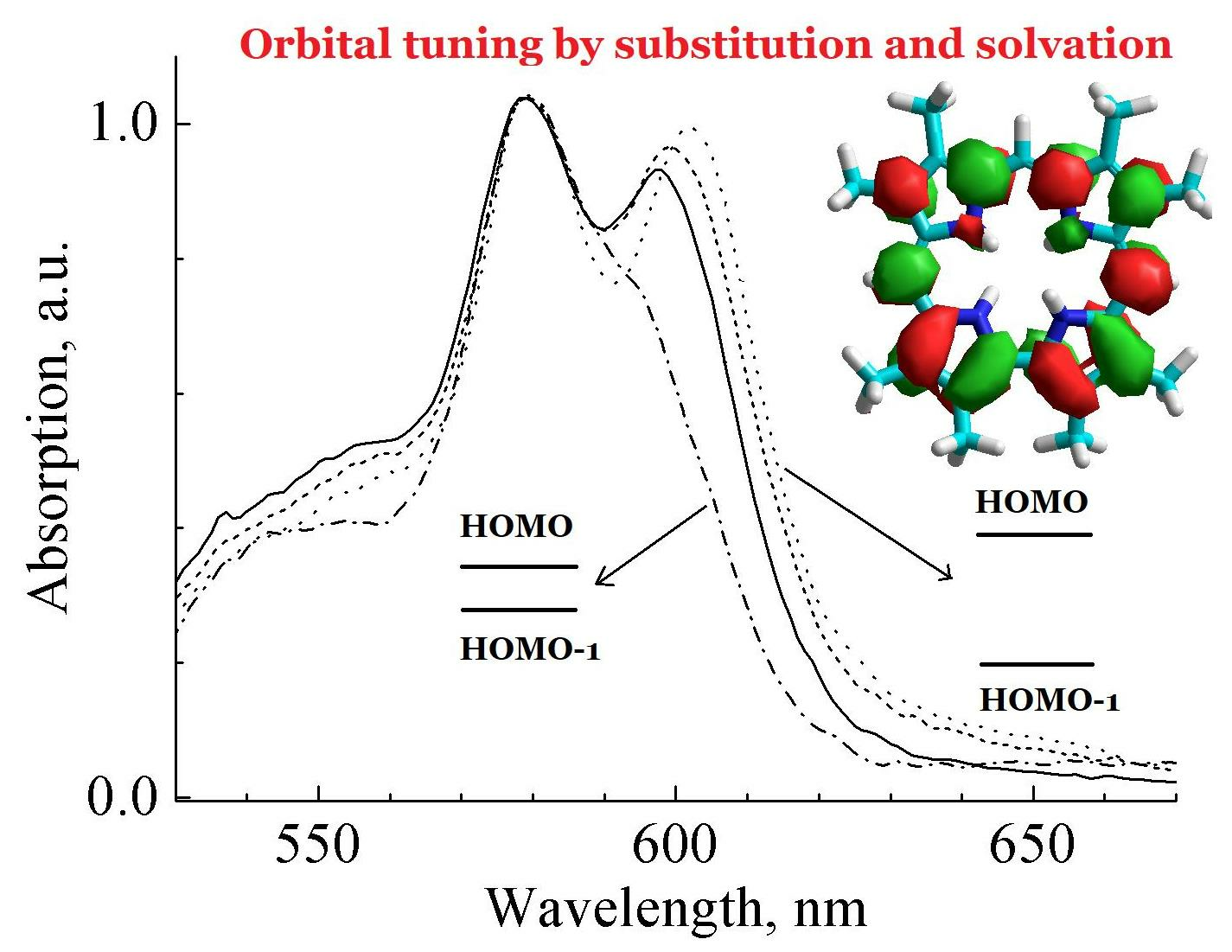
- Kruk, M.M.; Klenitsky, D.V.; Maes, W. Molecular structure and conformation of free base corroles. Macroheterocycles 2019, 12, 58–67. [Google Scholar] [CrossRef]
- Ivanova, Y.B.; Savva, V.A.; Mamardashvili, N.Z.; Starukhin, A.S.; Ngo, T.H.; Dehaen, W.; Maes, W.; Kruk, M.M. Corrole NH Tautomers: Spectral Features and Individual Protonation. J. Phys. Chem. A 2012, 116, 10683–10694. [Google Scholar] [CrossRef]
- Kruk, M.M.; Ngo, T.H.; Savva, V.A.; Starukhin, A.S.; Dehaen, W.; Maes, W. Solvent-Dependent Deprotonation ofmeso-Pyrimidinylcorroles: Absorption and Fluorescence Studies. J. Phys. Chem. A 2012, 116, 10704–10711. [Google Scholar] [CrossRef]
- Ngo, T.H.; Puntoniero, F.; Nastasi, F.; Robeyns, K.; Van Meervelt, L.; Campagna, S.; Dehaen, W.; Maes, W. Synthetic, Structural, and Photophysical Exploration of meso-Pyrimidinyl-Substituted AB2-Corroles. Chem. Eur. J. 2010, 16, 5691–5705. [Google Scholar] [CrossRef]
- Petrova, D.V.; Semeikin, A.S.; Berezina, N.M.; Berezin, M.B.; Bazanov, M.I. Synthesis and Some Physical-Chemical Properties of meso-Aryl- and Alkyl Substituted Corroles and their Metal Complexes. Macroheterocycles 2019, 12, 119–128. [Google Scholar] [CrossRef]
- Laikov, D.N. Fast evaluation of density functional exchange-correlation terms using the expansion of the electron density in auxiliary basis sets. Chem. Phys. Lett. 1997, 281, 151–156. [Google Scholar] [CrossRef]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russian Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Bresch, S.; Gomes, G.P. Orbital hybridization: A key electronic factor in control of structure and reactivity. J. Phys. Org. Chem. 2015, 28, 147–162. [Google Scholar] [CrossRef]
- Beenken, W.J.D.; Presselt, M.; Ngo, T.H.; Dehaen, W.; Maes, W.; Kruk, M.M. Molecular Structures and Absorption Spectra Assignmentof Corrole NH Tautomers. J. Phys. Chem. A 2014, 118, 862–871. [Google Scholar] [CrossRef]
- Ajeeb, Y.H.; Klenitsky, D.V.; Vershilovskaya, I.V.; Petrova, D.V.; Semeikin, A.S.; Maes, W.; Gladkov, L.L.; Kruk, M.M. Spectral and luminescent properties and NH-tautomerism of alkylated corrole free bases. J. Appl. Spectr. 2020, 87, 421–428. [Google Scholar] [CrossRef]
- Kruk, M.M.; Starukhin, A.S.; Maes, W. Influence of macrocycle protonation on the photophysical properties of porphyrins. Macroheterocycles 2011, 4, 69–79. [Google Scholar] [CrossRef]
- Rosa, A.; Ricciardi, G.; Baerends, E.J. Synergism of porphyrin-core saddling and twisting of meso-aryl substituents. J. Phys. Chem. A 2006, 110, 5180–5190. [Google Scholar] [CrossRef]
- Klenitsky, D.V.; Gladkov, L.L.; Vershilovskaya, I.V.; Maes, W.; Kruk, M.M. Inversion of aromaticity of NH-tautomers of the free base corroles in the lowest triplet T1 state. J. Appl. Spectr. 2022, 89, 426–432. [Google Scholar] [CrossRef]
- Gao, H.-L.; Yao, G.-H.; Chen, F.; Wang, W.-L.; Chen, D.-M. Density Functional Theory Investigation of Structures and Electronic Spectra of N-protonated Corroles. Chin. J. Chem. Phys. 2012, 25, 281–290. [Google Scholar] [CrossRef]
- Kruk, M.M.; Gladkov, L.L.; Klenitsky, D.V.; Krylov, A.B. Molecular conformation and aromaticity of N-substituted porphine derivatives. Proc. BSTU Issue 3 Phys. Math. Inform. 2023, 266, 34–41. (In Russian) [Google Scholar] [CrossRef]
- Gouterman, M. Optical spectra and electronic structure of porphyrins and related rings. In The Porphyrins, 1st ed.; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; Volume 3, pp. 1–165. [Google Scholar]
- Kruk, M.M. Solvatochromism of the free base corroles. J. Appl. Spectr. 2022, 89, 624–630. [Google Scholar] [CrossRef]
- Gladkov, L.L.; Klenitsky, D.V.; Kruk, M.M. Mechanisms of bathochromic band shifts in the absorption spectra of the N-substituted porphine derivatives. J. Appl. Spectr. 2023. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Δ23,Å | φ(A),° | φ(B),° | φ(C),° | φ(D),° | C1–C19,Å |
|---|---|---|---|---|---|---|
| H4OAlkCor+ | 0.247 | 14.4 | 9.9 | 9.8 | 13.5 | 1.428 |
| H4PyrMes2Cor+ | 0.265 | 13.6 | 12.9 | 13.5 | 12.8 | 1.427 |
| H4Cor+ | 0.225 | 12.5 | 10.0 | 10.4 | 11.7 | 1.424 |
| Molecule | N21(pyrrole A) | N22(pyrrole B) | N23(pyrrole C) | N24(pyrrole D) |
|---|---|---|---|---|
| H4OAlkCor+ | 2.228 | 2.306 | 2.300 | 2.225 |
| H4PyrMes2Cor+ | 2.166 | 2.297 | 2.289 | 2.154 |
| H4Cor+ | 2.202 | 2.297 | 2.293 | 2.203 |
| Molecule | Qx(0,0)/Qy(0,0), nm | fx/fy |
|---|---|---|
| H4OAlkCor+ | 676.0/632.2 | 0.060/0.045 |
| H4PyrMes2Cor+ | 669.6/589.0 | 0.115/0.005 |
| H4Cor+ | 666.4/634.1 | 0.047/0.032 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gladkov, L.L.; Klenitsky, D.V.; Kruk, M.M. Absorption Spectra of Protonated Corroles: Two Distinct Patterns Due to Peripheral Substitution Architecture. Organics 2023, 4, 490-502. https://doi.org/10.3390/org4040034
Gladkov LL, Klenitsky DV, Kruk MM. Absorption Spectra of Protonated Corroles: Two Distinct Patterns Due to Peripheral Substitution Architecture. Organics. 2023; 4(4):490-502. https://doi.org/10.3390/org4040034
Chicago/Turabian StyleGladkov, Lev L., Dmitry V. Klenitsky, and Mikalai M. Kruk. 2023. "Absorption Spectra of Protonated Corroles: Two Distinct Patterns Due to Peripheral Substitution Architecture" Organics 4, no. 4: 490-502. https://doi.org/10.3390/org4040034