
How Do Positions of Phosphito Units on a Calix[4]Arene Platform Affect the Enantioselectivity of a Catalytic Reaction?
Abstract
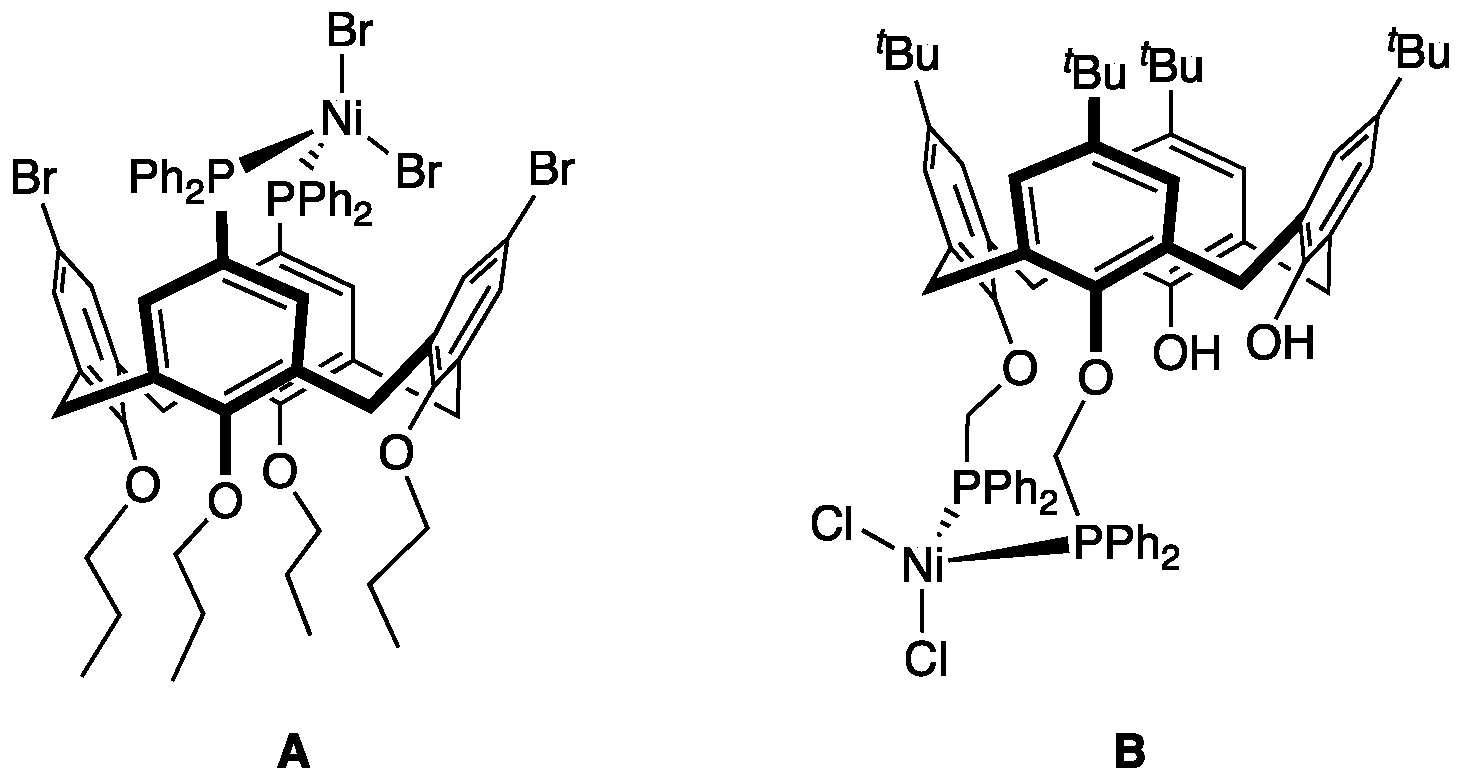
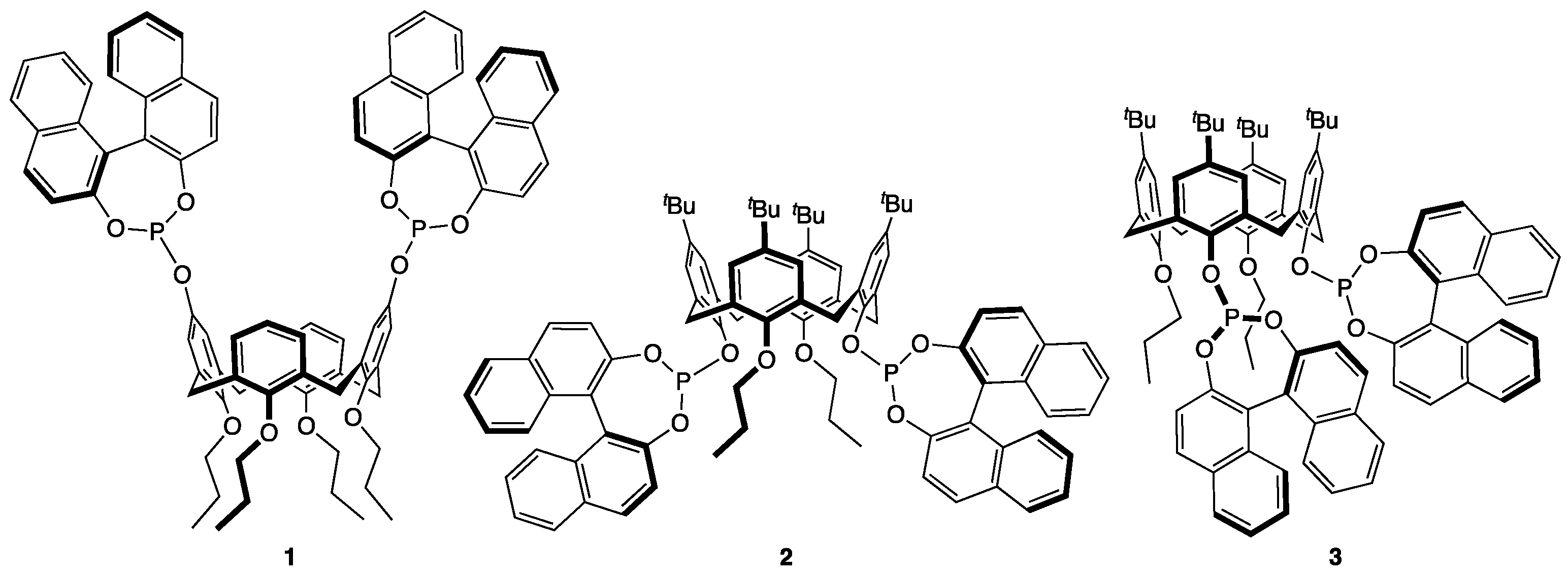
:1. Introduction
2. Materials and Methods
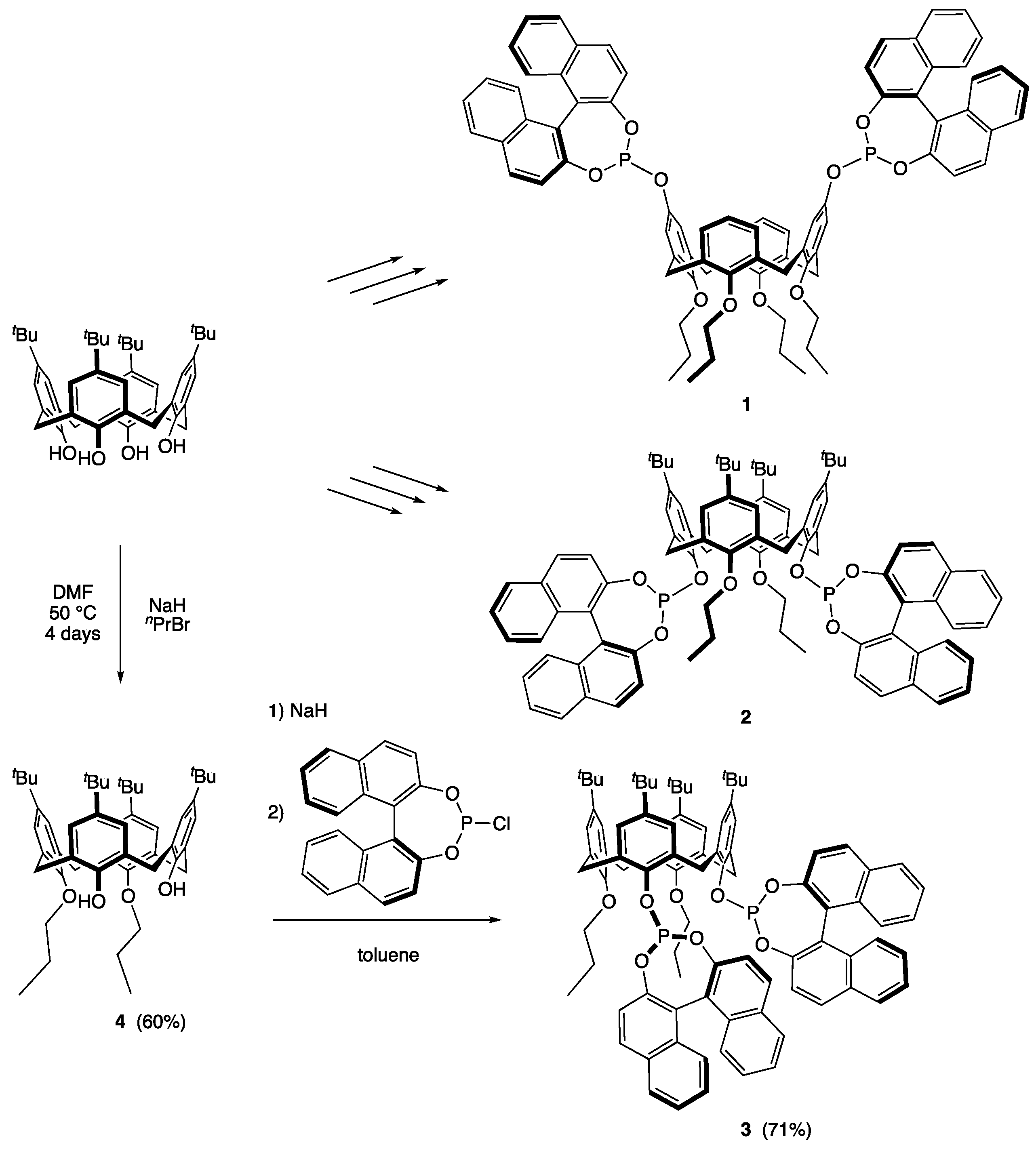
2.1. Synthesis of 5,11,17,23-Tetra-tert-butyl-25,26-dipropyloxy-27,28-dihydroxycalix[4]arene (4) [47]
2.2. Synthesis of (S,S)-5,11,17,23-Tetra-tert-butyl-25,26-dipropoxy-27,28-bis(1,1′-binaphthyl-2,2′-dioxyphosphanyloxy)calix[4]arene (3)
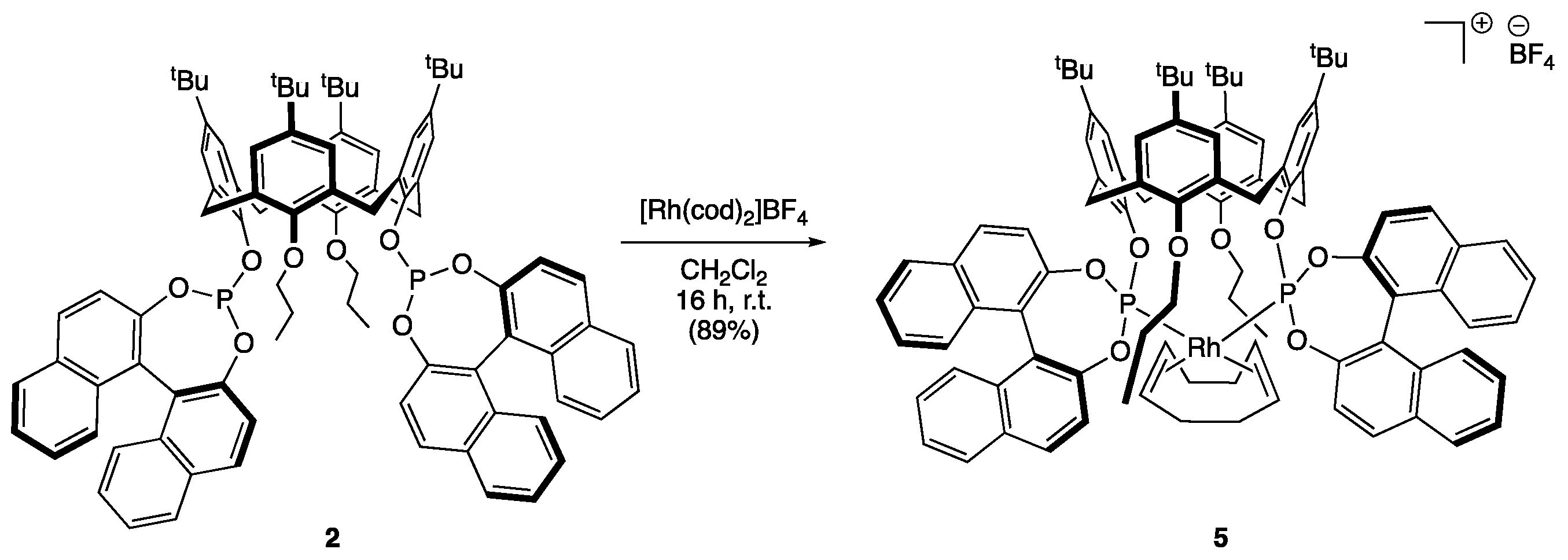
2.3. Synthesis of cis-P,P’-{[(S,S)-5,11,17,23-Tetra-tert-butyl-25,27-dipropyloxy-26,28-bis (1,1′-binaphtyl-phosphite)calix[4]arene]-1,5-cyclooctadiene}rhodium(I) Tetrafluoroborate (5)
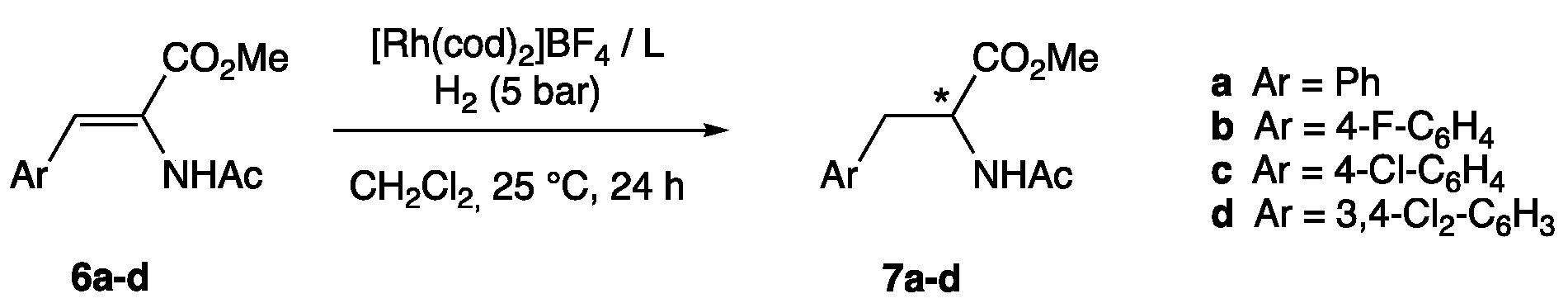
2.4. General Procedure for the Hydrogenation Experiments
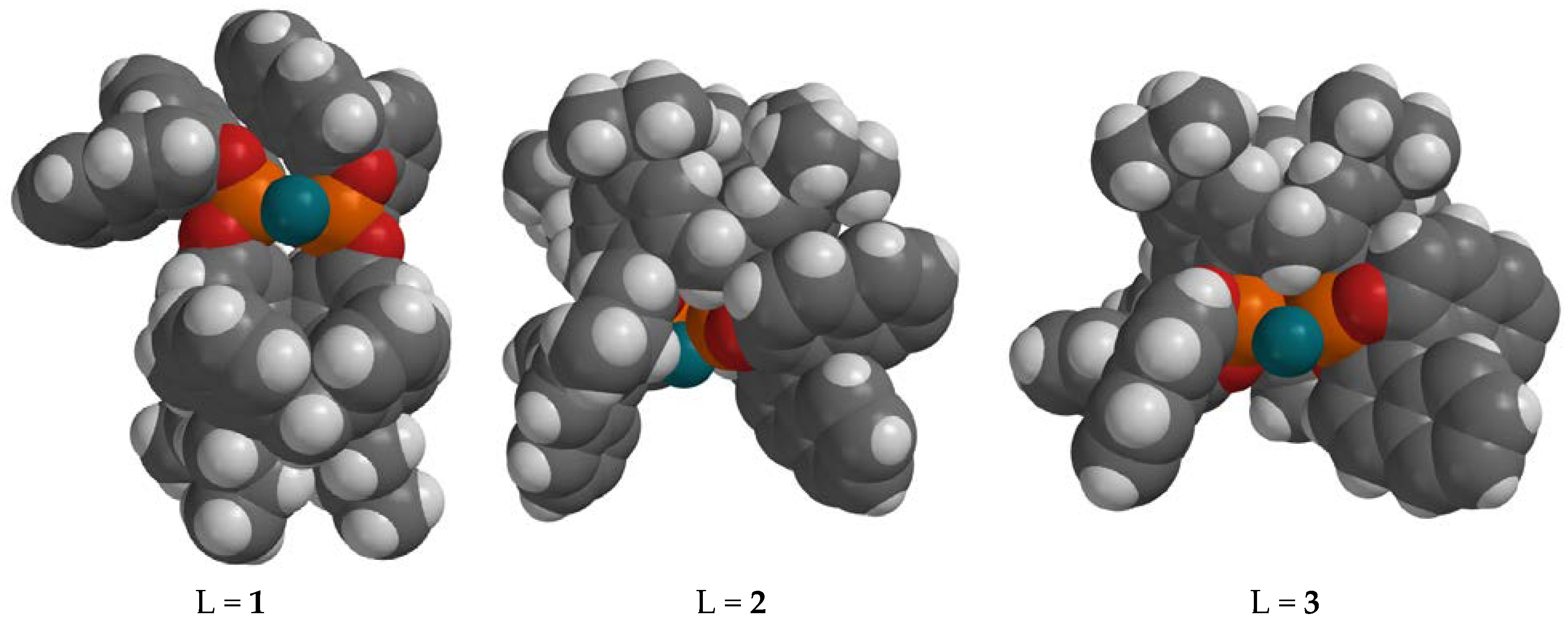
3. Results
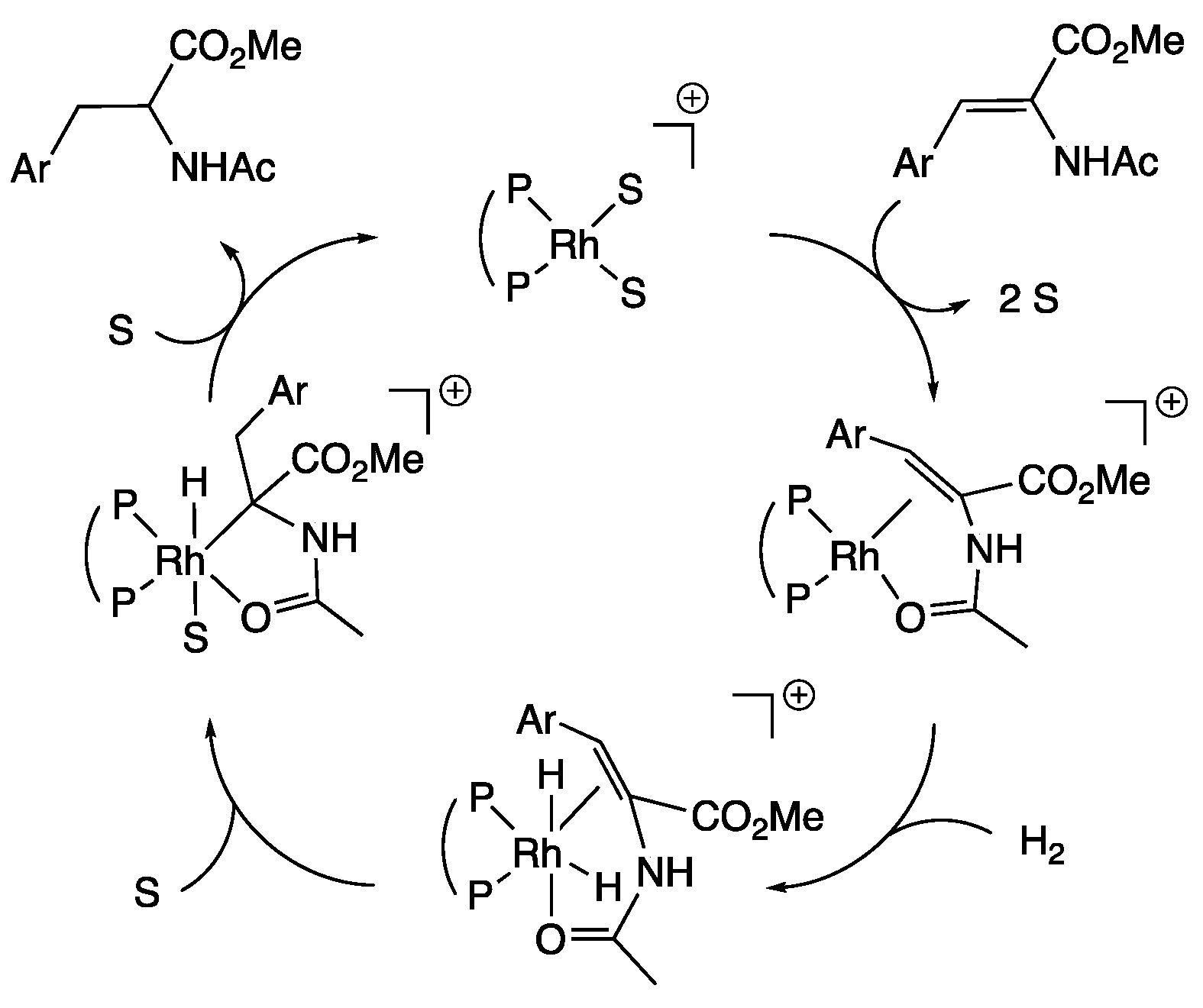
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gutsche, C.D.; Muthukrishnan, R. Calixarenes. 1. Analysis of the product mixtures produced by the base-catalyzed condensation of formaldehyde with para-substituted phenols. J. Org. Chem. 1978, 43, 4905–4906. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Dhawan, B.; No, K.H.; Muthukrishnan, R. Calixarenes. 4. The synthesis, characterization, and properties of the calixarenes from p-tert-butylphenol. J. Am. Chem. Soc. 1981, 103, 3782–3792. [Google Scholar] [CrossRef]
- Gutsche, C.D. Calixarenes. Acc. Chem. Res. 1983, 16, 161–170. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Iqbal, M.; Stewart, D. Calixarenes. 18. Synthesis procedures for p-tert-butylcalix[4]arene. J. Org. Chem. 1986, 51, 742–745. [Google Scholar] [CrossRef]
- Wieser, C.; Dieleman, C.B.; Matt, D. Calixarene and resorcinarene ligands in transition metal chemistry. Coord. Chem. Rev. 1997, 165, 93–161. [Google Scholar] [CrossRef]
- Sliwa, W. Calixarene complexes with transition metal, lanthanide and actinide ions. Croat. Chem. Acta 2002, 75, 131–153. [Google Scholar]
- Sliwa, W. Calixarene complexes with transition metal ions. J. Incl. Phenom. Macrocycl. Chem. 2005, 52, 13–37. [Google Scholar] [CrossRef]
- Homden, D.M.; Redshaw, C. The use of calixarenes in metal-based catalysis. Chem. Rev. 2008, 108, 5086–5130. [Google Scholar] [CrossRef]
- Santoro, O.; Redshaw, C. Metallocalix[n]arenes in catalysis: A 13-year update. Coord. Chem. Rev. 2021, 448, 214173. [Google Scholar] [CrossRef]
- Sémeril, D.; Matt, D. Synthesis and catalytic relevance of P(III) and P(V)-functionalised calixarenes and resorcinarenes. Coord. Chem. Rev. 2014, 279, 58–95. [Google Scholar] [CrossRef]
- Bauder, C.; Sémeril, D. Styrene hydroformylation with cavity-shaped ligands. Eur. J. Inorg. Chem. 2019, 47, 4951–4965. [Google Scholar] [CrossRef]
- Hamada, F.; Fukugaki, T.; Murai, K.; Orr, G.W.; Atwood, J.L. Liquid-liquid extraction of transition and alkali metal cations by a new calixarene: Diphenylphosphino calix[4]arene methyl ether. J. Incl. Phenom. Macrocycl. Chem. 1991, 10, 57–61. [Google Scholar] [CrossRef]
- Shimizu, S.; Shirakawa, S.; Sasaki, Y.; Hirai, C. Novel Water-soluble calix[4]arene ligands with phosphane-containing groups for dual functional metal-complex catalysts: The biphasic hydroformylation of water-insoluble olefins. Angew. Chem. Int. Ed. 2000, 39, 1256–1259. [Google Scholar] [CrossRef]
- Fang, X.; Scott, B.L.; Watkin, J.G.; Carter, C.A.G.; Kubas, G.J. Metal complexes based on an upper-rim calix[4]arene phosphine ligand. Inorg. Chim. Acta 2001, 317, 276–281. [Google Scholar] [CrossRef]
- Mongrain, P.; Harvey, P.D. An original calix[4]arene-containing oligomer/polymer catalyst for homogeneous hydroformylation. Macromol. Rapid Commun. 2008, 29, 1752–1757. [Google Scholar] [CrossRef]
- Khiri, N.; Bertrand, E.; Ondel-Eymin, M.-J.; Rousselin, Y.; Bayardon, J.; Harvey, P.D.; Jugé, S. Enantioselective hydrogenation catalysis aided by a σ-bonded calix[4]arene to a P-chirogenic aminophosphane phosphinite rhodium complex. Organometallics 2010, 29, 3622–3631. [Google Scholar] [CrossRef]
- Monnereau, L.; Sémeril, D.; Matt, D. High efficiency of cavity-based triaryl-phosphines in nickel-catalysed Kumada-Tamao-Corriu cross-coupling. Chem. Commun. 2011, 47, 6626–6628. [Google Scholar] [CrossRef] [Green Version]
- Khiri-Meribout, N.; Bertrand, E.; Bayardon, J.; Eymin, M.-J.; Rousselin, Y.; Cattey, H.; Fortin, D.; Harvey, P.D.; Jugé, S. P-Chirogenic phosphines supported by calix[4]arene: New insight into palladium-catalyzed asymmetric allylic substitution. Organometallics 2013, 32, 2827–2839. [Google Scholar] [CrossRef]
- Elaieb, F.; Hedhli, A.; Sémeril, D.; Matt, D. Arylcalixarenyl phosphines in palladium-catalyzed Suzuki-Miyaura cross-coupling reactions. Eur. J. Org. Chem. 2016, 10, 1867–1873. [Google Scholar] [CrossRef]
- Elaieb, F.; Sémeril, D.; Matt, D.; Pfeffer, M.; Bouit, P.-A.; Hissler, M.; Gourlaouen, C.; Harrowfield, J. Calix[4]arene-fused phospholes. Dalton Trans. 2017, 46, 9833–9845. [Google Scholar] [CrossRef] [Green Version]
- Csók, Z.; Szalontai, G.; Czira, G.; Kollár, L. Carbonylation (hydroformylation and hydrocarbalkoxylation) reactions in the presence of transition metal: p-tert-Butyl-calix[4]arene-based phosphine and phosphinite systems. J. Organomet. Chem. 1998, 570, 23–29. [Google Scholar] [CrossRef]
- Paciello, R.; Siggel, L.; Röper, M. Chelated bisphosphites with a calix[4]arene backbone: New ligands for rhodium-catalyzed low-pressure hydroformylation with controlled regioselectivity. Angew. Chem. Int. Ed. 1999, 38, 1920–1923. [Google Scholar] [CrossRef]
- Jeunesse, C.; Dieleman, C.; Steyer, S.; Matt, D. Calix[4]arene-derived diphosphines, diphosphinites and diphosphites as chelating ligands for transition metal ions. Encapsulation of silver(I) in a calix-crown diphosphite. J. Chem. Soc. Dalton Trans. 2001, 6, 881–892. [Google Scholar] [CrossRef]
- Kunze, C.; Selent, D.; Neda, I.; Freytag, M.; Jones, P.G.; Schmutzler, R.; Baumann, W.; Börner, A. Calix[4]arene-based bis-phosphonites, bis-phosphites, and bis-O-acyl-phosphites as ligands in the rhodium(I)-catalyzed hydroformylation of 1-octene. Z. Anorg. Allg. Chem. 2002, 628, 779–787. [Google Scholar] [CrossRef]
- Marson, A.; Freixa, Z.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. Chiral calix[4]arene-based diphosphites as ligands in the asymmetric hydrogenation of prochiral olefins. Eur. J. Inorg. Chem. 2007, 29, 4587–4591. [Google Scholar] [CrossRef]
- Sémeril, D.; Matt, D.; Toupet, L. Highly regioselective hydroformylation with hemispherical chelators. Chem. Eur. J. 2008, 14, 7144–7155. [Google Scholar] [CrossRef]
- Maji, P.; Mahalakshmi, L.; Krishnamurthy, S.S.; Nethaji, M. Cyclometalated complexes derived from calix[4]arene bisphosphites and their catalytic applications in cross-coupling reactions. J. Organomet. Chem. 2011, 696, 3169–3179. [Google Scholar] [CrossRef]
- Cobley, C.J.; Pringle, P.G. A water-soluble phosphite derived from sulfonated calix[4]arene. The remarkable stability of its rhodium complexes and two phase hydroformylation studies. Catal. Sci. Technol. 2011, 1, 239–242. [Google Scholar] [CrossRef]
- Hirasawa, K.; Tanaka, S.; Horiuchi, T.; Kobayashi, T.; Sato, T.; Morohashi, N.; Hattori, T. Pd(II) Complexes ligated by 1,3-bis(diphenylphosphino)calix[4]arene: Preparation, X-ray structures, and catalyses. Organometallics 2016, 35, 420–427. [Google Scholar] [CrossRef]
- Karpus, A.; Yesypenko, O.; Boiko, V.; Poli, R.; Daran, J.-C.; Voitenko, Z.; Kalchenko, V.; Manoury, E. Chiral phosphinoferrocenyl-calixarenes. Eur. J. Org. Chem. 2016, 20, 3386–3394. [Google Scholar] [CrossRef]
- Kuhn, P.; Sémeril, D.; Jeunesse, C.; Matt, D.; Lutz, P.J.; Louis, R.; Neuburger, M. Catalytic applications of keto-stabilised phosphorus ylides based on a macrocyclic scaffold: Calixarenes with one or two pendant Ni(P,O)-subunits as ethylene oligomerisation and polymerisation catalysts. Dalton Trans. 2006, 30, 3647–3659. [Google Scholar] [CrossRef] [PubMed]
- Frediani, M.; Sémeril, D.; Comucci, A.; Bettucci, L.; Frediani, P.; Rosi, L.; Matt, D.; Toupet, L.; Kaminsky, W. Ultrahigh-molecular-weight polyethylene by using a titanium calix[4]arene complex with high thermal stability under polymerization conditions. Macromol. Chem. Phys. 2007, 208, 938–945. [Google Scholar] [CrossRef] [Green Version]
- Walton, M.J.; Lancaster, S.J.; Redshaw, C. Highly selective and immortal magnesium calixarene complexes for the ring-opening polymerization of rac-Lactide. ChemCatChem 2014, 6, 1892–1898. [Google Scholar] [CrossRef]
- Sarkar, A.; Krishnamurthy, S.S.; Nethaji, M. Calix[4]arene bisphosphite ligands bearing two distal 2,20-biphenyldioxy or 2,20-binaphthyldioxy moieties: Conformational flexibility and allyl–palladium complexes. Tetrahedron 2009, 65, 374–382. [Google Scholar] [CrossRef]
- Sémeril, D.; Matt, D.; Toupet, L.; Oberhauser, W.; Bianchini, C. High-pressure investigations under CO/H2 of rhodium complexes containing hemispherical diphosphites. Chem. Eur. J. 2010, 16, 13843–13849. [Google Scholar] [CrossRef]
- Dieleman, C.; Steyer, S.; Jeunesse, C.; Matt, D. Diphosphines based on an inherently chiral calix[4]arene scaffold: Synthesis and use in enantioselective catalysis. J. Chem. Soc. Dalton Trans. 2001, 17, 2508–2517. [Google Scholar] [CrossRef]
- Karpus, A.; Yesypenko, O.; Boiko, V.; Daran, J.-C.; Voitenko, Z.; Kalchenko, V.; Manoury, E. Synthesis of an enantiomerically pure inherently chiral calix[4]arene phosphonic acid and its evaluation as an organocatalyst. J. Org. Chem. 2018, 83, 1146–1153. [Google Scholar] [CrossRef]
- Arnott, G.E. Inherently chiral calixarenes: Synthesis and applications. Chem. Eur. J. 2018, 24, 1744–1754. [Google Scholar] [CrossRef]
- Lejeune, M.; Sémeril, D.; Jeunesse, C.; Matt, D.; Lutz, P.; Toupet, L. Fast propene dimerization using upper rim-diphosphinated calix[4]arenes as chelators. Adv. Synth. Catal. 2006, 348, 881–886. [Google Scholar] [CrossRef]
- Monnereau, L.; Sémeril, D.; Matt, D.; Toupet, L.; Mota, A.J. Efficient, nickel-catalysed Kumada-Tamao-Corriu cross-coupling with a calix[4]arene-diphosphine ligand. Adv. Synth. Catal. 2009, 351, 1383–1389. [Google Scholar] [CrossRef]
- Monnereau, L.; Sémeril, D.; Matt, D.; Toupet, L. Cavity-shaped ligands: Calix[4]arene-based monophosphanes for fast Suzuki-Miyaura cross-coupling. Chem. Eur. J. 2010, 16, 9237–9247. [Google Scholar] [CrossRef] [PubMed]
- Sémeril, D.; Jeunesse, C.; Matt, D. Influence des propriétes intrinsèques de ligands calixaréniques sur des réactions de transformation catalytique de l’éthylène. Comptes Rendus Chim. 2008, 11, 583–594. [Google Scholar] [CrossRef]
- Lejeune, M.; Sémeril, D.; Jeunesse, C.; Matt, D.; Peruch, F.; Lutz, P.J.; Ricard, L. Diphosphines with expandable bite angles: Highly active ethylene dimerisation catalysts based on upper rim, distally diphosphinated calix[4]arenes. Chem. Eur. J. 2004, 10, 5354–5360. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Jeunesse, C.; Sémeril, D.; Matt, D.; Lutz, P.; Welter, R. Coordination chemistry of large diphosphanes—directional properties of a calix[4]arene proximally substituted by two−OCH2PPh2 podand arms. Eur. J. Inorg. Chem. 2004, 2004, 4602–4607. [Google Scholar] [CrossRef]
- Natarajan, N.; Pierrevelcin, M.-C.; Sémeril, D.; Bauder, C.; Matt, D.; Ramesh, R. Chiral calixarene and resorcinarene derivatives. Conical cavities substituted at their upper rim by two phosphito units and their use as ligands in Rh-catalysed hydroformylation. Catal. Commun. 2019, 118, 70–75. [Google Scholar] [CrossRef]
- Sémeril, D.; Jeunesse, C.; Matt, D.; Toupet, L. Regioselectivity with hemispherical chelators: Increasing the catalytic efficiency of complexes of diphosphanes with large bite angles. Angew. Chem. Int. Ed. 2006, 45, 5810–5814. [Google Scholar] [CrossRef]
- Gagnon, J.; Vézina, M.; Drouin, M.; Harvey, P.D. Regioselective upper-rim functionalizations of calix[4]arene by diphenylphosphino groups. Can. J. Chem. 2001, 79, 1439–1446. [Google Scholar] [CrossRef]
- Jaime, C.; de Mendoza, J.; Prados, P.; Nieto, P.M.; Sanchez, C. Carbon-13 NMR chemical shifts. A single rule to determine the conformation of calix[4]arenes. J. Org. Chem. 1991, 56, 3372–3376. [Google Scholar] [CrossRef]
- Liu, S.; Sandoval, C.A. Evaluation of calix[4]arene-based chiral diphosphite ligands in Rh-catalyzed asymmetric hydrogenation of simple dehydroamino acid derivatives. J. Mol. Catal. A Chem. 2010, 325, 65–72. [Google Scholar] [CrossRef]
- Diéguez, M.; Ruiz, A.; Claver, C. Chiral diphosphites derived from D-glucose: New highly modular ligands for the asymmetric catalytic hydrogenation. J. Org. Chem. 2002, 67, 3796–3801. [Google Scholar] [CrossRef]
- Kostas, I.D.; Vallianatou, K.A.; Holz, J.; Börner, A. Rhodium complexes with a new chiral nitrogen containing BINOL-based diphosphite or phosphonite ligand: Synthesis and application to hydroformylation of styrene and/or hydrogenation of prochiral olefins. Appl. Organomet. Chem. 2005, 19, 1090–1095. [Google Scholar] [CrossRef]
- Li, Y.; Ma, B.; He, Y.; Zhang, F.; Fan, Q.-H. Chiral metallacrown ethers for asymmetric hydrogenation: Alkali-metal ion mediated enhancement of enantioselectivity. Chem. Asian J. 2010, 5, 2454–2458. [Google Scholar] [CrossRef]
- Cai, C.; Deng, F.; Sun, W.; Xia, C. New bidentate phosphorus ligands based on a norbornane backbone for rhodium-catalyzed asymmetric hydrogenation. Synlett 2007, 19, 3007–3010. [Google Scholar] [CrossRef]
- Chan, A.S.C.; Pluth, J.J.; Halpern, J. Identification of the enantioselective step in the asymmetric catalytic hydrogenation of a prochiral olefin. J. Am. Chem. Soc. 1980, 102, 5952–5954. [Google Scholar] [CrossRef]
- Halpern, J. Mechanism and stereoselectivity of asymmetric hydrogenation. Science 1982, 217, 401–407. [Google Scholar] [CrossRef]
- Landis, C.R.; Halpern, J. Asymmetric hydrogenation of methyl-(Z)-α-acetamidocinnamate catalyzed by {l,2-bis((phenyl-o-anisoyl)phosphino)ethane}rhodium(I): Kinetics, mechanism, and origin of enantioselection. J. Am. Chem. Soc. 1987, 109, 1754–1757. [Google Scholar]
- Sarkar, A.; Nethaji, M.; Krishnamurthy, S.K. Phosphite ligands derived from distally and proximally substituted dipropyloxy calix[4]arenes and their palladium complexes: Solution dynamics, solid-state structures and catalysis. J. Organomet. Chem. 2008, 693, 2097–2110. [Google Scholar] [CrossRef]
- Liu, W.; Das, P.J.; Colquhoun, H.M.; Stoddart, J.F. Whither Second-Sphere Coordination? CCS Chem. 2022, 4, 755–784. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate (Ar) | Ligand | Conversion (%) 2 | ee (%) 3 | |
|---|---|---|---|---|---|
| 1 | 6a | (Ar = Ph) | 1 | 100 | 57 (R) |
| 2 | 6b | (Ar = 4-F-C6H4) | 1 | 83 | 48 (R) |
| 3 | 6c | (Ar = 4-Cl-C6H4) | 1 | 86 | 57 (R) |
| 4 | 6d | (Ar = 3,4-Cl2-C6H3) | 1 | 91 | 52 (R) |
| 5 | 6a | (Ar = Ph) | 2 | 100 | 91 (R) |
| 6 | 6b | (Ar = 4-F-C6H4) | 2 | 92 | 95 (R) |
| 7 | 6c | (Ar = 4-Cl-C6H4) | 2 | 97 | 94 (R) |
| 8 | 6d | (Ar = 3,4-Cl2-C6H3) | 2 | 100 | 90 (R) |
| 9 4 | 6a | (Ar = Ph) | 2 | 100 | 92 (R) |
| 10 | 6a | (Ar = Ph) | 3 | 100 | 62 (R) |
| 11 | 6b | (Ar = 4-F-C6H4) | 3 | 100 | 66 (R) |
| 12 | 6c | (Ar = 4-Cl-C6H4) | 3 | 100 | 58 (R) |
| 13 | 6d | (Ar = 3,4-Cl2-C6H3) | 3 | 100 | 63 (R) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hkiri, S.; Sémeril, D. How Do Positions of Phosphito Units on a Calix[4]Arene Platform Affect the Enantioselectivity of a Catalytic Reaction? Organics 2022, 3, 470-480. https://doi.org/10.3390/org3040030
Hkiri S, Sémeril D. How Do Positions of Phosphito Units on a Calix[4]Arene Platform Affect the Enantioselectivity of a Catalytic Reaction? Organics. 2022; 3(4):470-480. https://doi.org/10.3390/org3040030
Chicago/Turabian StyleHkiri, Shaima, and David Sémeril. 2022. "How Do Positions of Phosphito Units on a Calix[4]Arene Platform Affect the Enantioselectivity of a Catalytic Reaction?" Organics 3, no. 4: 470-480. https://doi.org/10.3390/org3040030