
Synthesis of 3-Hydroxy-9H-fluorene-2-carboxylates via Michael Reaction, Robinson Annulation, and Aromatization

Abstract
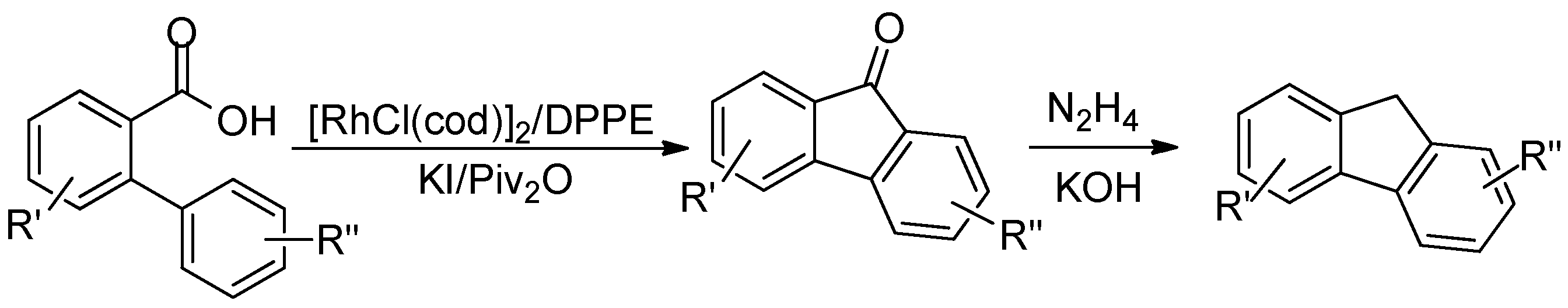
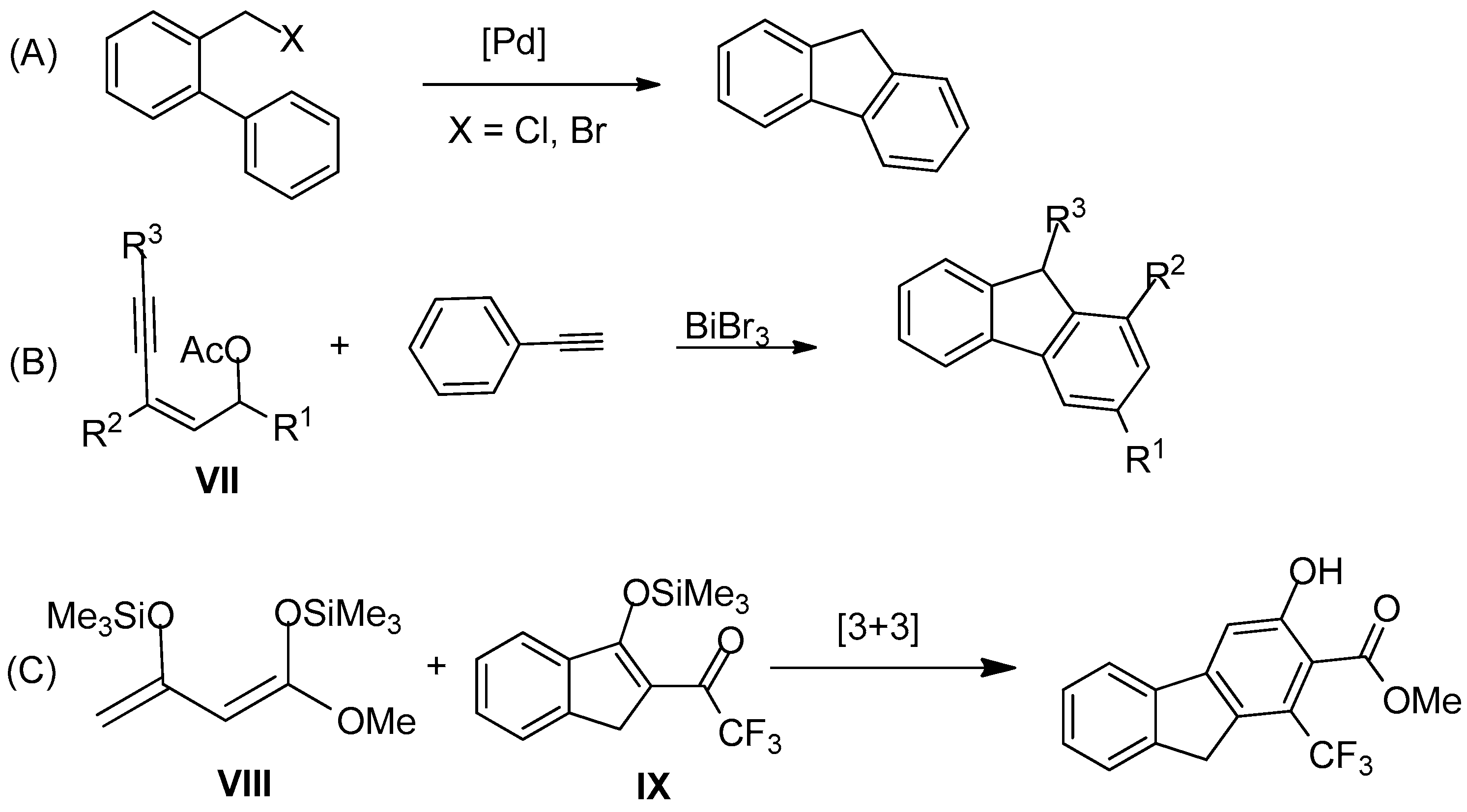
:1. Introduction
2. Materials and Methods
2.1. Materials and Instrumentation
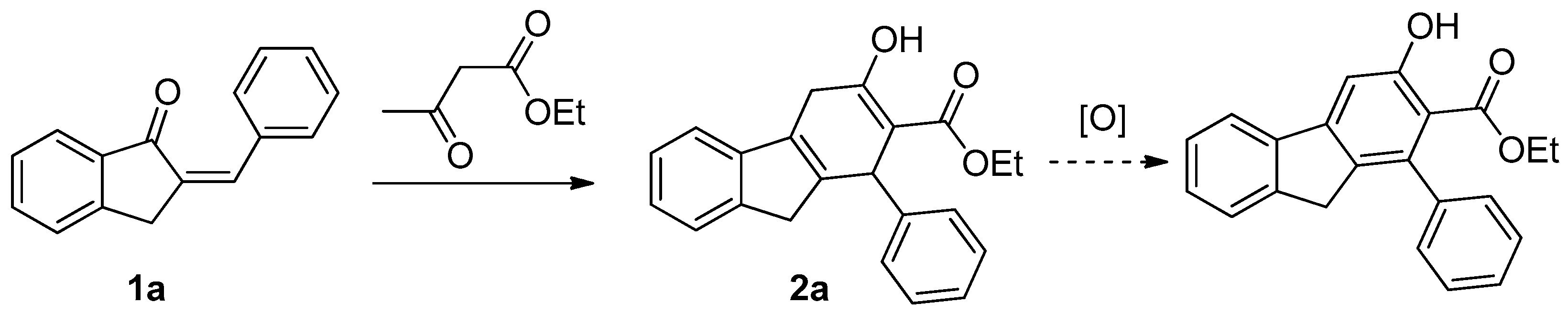
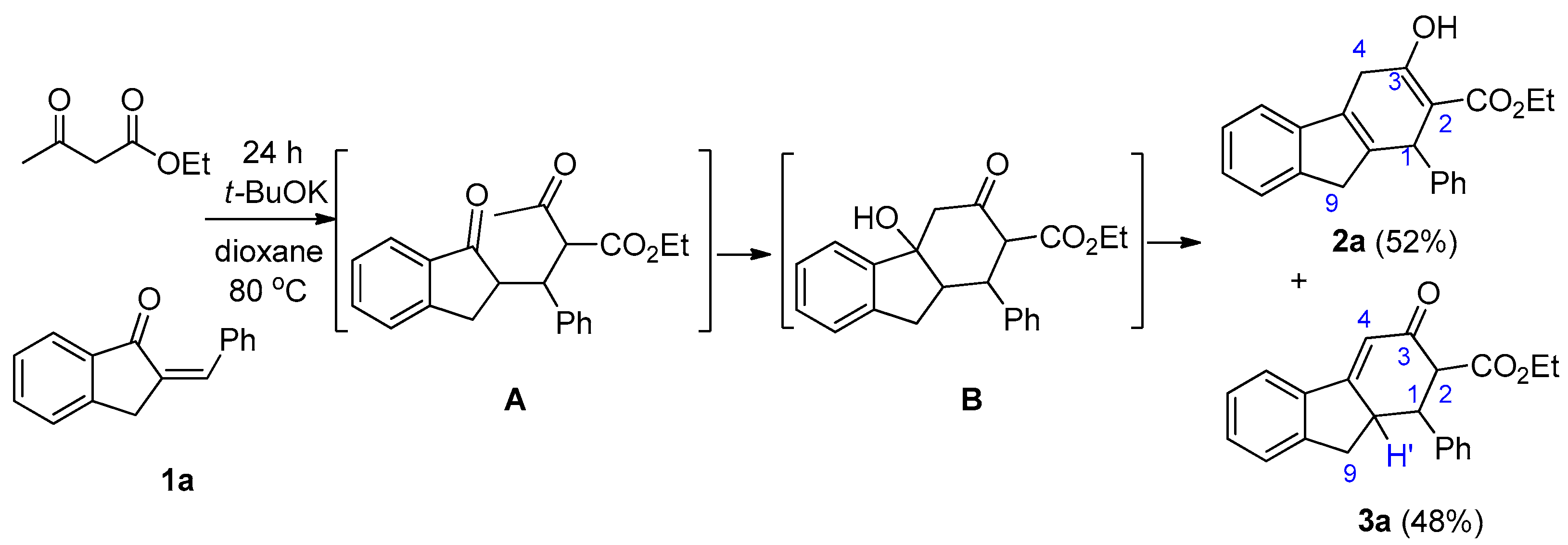
2.2. Reaction of 1a with Ethyl Acetoacetate
2.3. Direct Preparation of Fluorenes without Isolation
2.4. Crystallogarphy
3. Results and Discussion
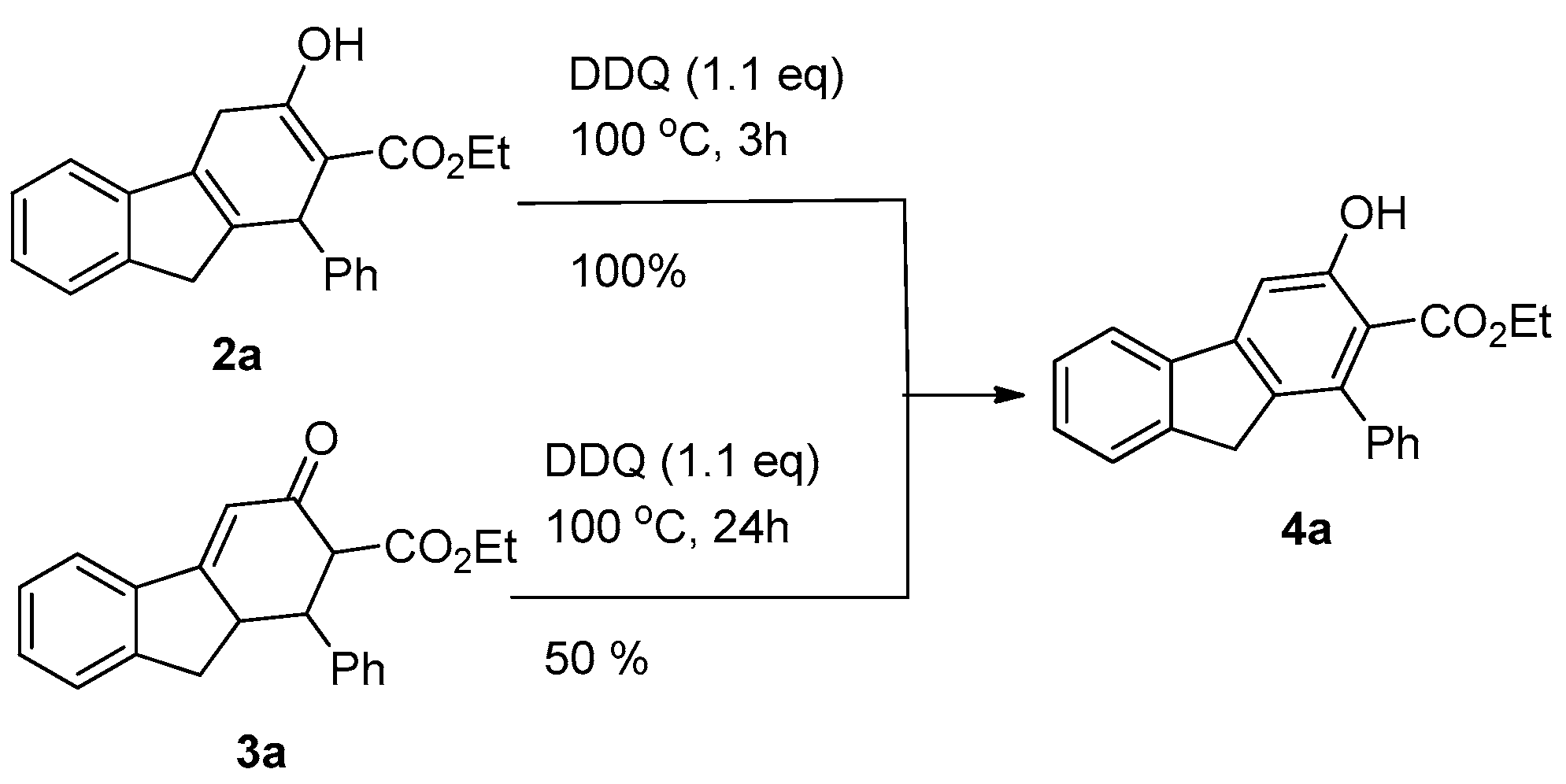
3.1. Preliminary study and Optimization
3.2. Reaction of 1a with Acetoacetate—Selectivity of Formation of 2a Versus 3a
3.3. Preparation of Fluorenes via a Two-Step Reaction without Isolation of 2 and 3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, H.; Chou, G.-X.; Wang, Z.-T.; Guo, Y.-W.; Hu, Z.-B.; Xu, L.-S. Two New Compounds from Dendrobium chrysotoxum. Helv. Chim. Acta 2004, 87, 394–399. [Google Scholar] [CrossRef]
- Ye, Q.-H.; Zhao, W.-M.; Qin, G.-W. New fluorenone and phenanthrene derivatives from Dendrobium chrysanthum. Nat. Prod. Res. 2003, 17, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, H.-B.; Huang, Y.-Y.; Bao, J.-M.; Tang, G.-H.; Chen, Y.-Y.; Wang, J.; Yin, S. Selaginpulvilins A–D, New Phosphodiesterase-4 Inhibitors with an Unprecedented Skeleton from Selaginella pulvinate. Org. Lett. 2014, 16, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Chui, C.-H.; Wong, R.S.-M.; Gambari, R.; Cheng, G.Y.-M.; Yuen, M.C.-W.; Chan, K.-W.; Tong, S.-W.; Lau, F.-Y.; Lai, P.B.-S.; Lam, K.-H.; et al. Antitumor activity of diethynylfluorene derivatives of gold(I). Bioorg. Med. Chem. 2009, 17, 7872–7877. [Google Scholar] [CrossRef] [PubMed]
- Misaki, K.; Matsui, S.; Matsuda, T. Metabolic Enzyme Induction by HepG2 Cells Exposed to Oxygenated and Nonoxygenated Polycyclic Aromatic Hydrocarbons. Chem. Res. Toxicol. 2007, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Mehta, G. Natural products as modulators of the cyclic-AMP pathway: Evaluation and synthesis of lead compounds. Org. Biomol. Chem. 2018, 16, 6372–6390. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Pradines, B.; Helle, F.; Dassonville-Klimpt, A.; Taudon, N.; Sonnet, P. Enantiopure aminoaryl-alcohols with fluorene core and their antimalarial activities. In 4th International Electronic Conference on Medicinal Chemistry; MDPI: Basel, Switzerland, 2018. [Google Scholar] [CrossRef] [Green Version]
- Beije, B.; Moeller, L. Correlation between induction of unscheduled DNA synthesis in the liver and excretion of mutagenic metabolites in the urine of rats exposed to the carcinogenic air pollutant 2-nitrofluorene. Carcinogenesis 1988, 9, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, H.R.; Galitski, S.B.; Foley, W.A. Carcinogenicity of the o-methoxy derivatives of N-2-fluorenylacetamide and of related compounds in the rat. Cancer Res. 1968, 28, 234–244. [Google Scholar] [PubMed]
- Mondal, E.; Hung, W.-Y.; Dai, H.-C.; Wong, K.-T. Fluorene-based asymmetric biopolar universial hosts for white organic light emitting devices. Adv. Funct. Mater. 2013, 23, 3096–3105. [Google Scholar] [CrossRef]
- Oyston, S.; Wang, C.; Hughes, G.; Batsanov, A.S.; Perepichka, I.F.; Bryce, M.R.; Ahn, J.H.; Pearson, C.; Petty, M.C. New 2,5-diaryl-1,3,4-oxadiazolefluorene hybrids as electron transporting materials for blended-layer organic light emitting diodes. J. Mater. Chem. 2005, 15, 194–203. [Google Scholar] [CrossRef]
- Wong, W.-Y. Metallated molecular materials of fluorene derivatives and their analogues. Coord. Chem. Rev. 2005, 249, 971–997. [Google Scholar] [CrossRef]
- Fukuyama, T.; Maetani, S.; Miyagawa, K.; Ryu, I. Synthesis of Fluorenones through Rhodium-Catalyzed Intramolecular Acylation of Biarylcarboxylic Acids. Org. Lett. 2014, 16, 3216–3219. [Google Scholar] [CrossRef] [PubMed]
- Kashulin, I.A.; Nifant’ev, I.E. Efficient Method for the Synthesis of Hetarenoindanones Based on 3-Arylhetarenes and Their Conversion into Hetarenoindenes. J. Org. Chem. 2004, 69, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.P.; Caivano, I.; Kotora, M. Transition-metal-catalyzed methods for synthesis of fluorenes. Tetrahedron 2019, 75, 2981–2992. [Google Scholar] [CrossRef]
- Hwang, S.J.; Kim, H.J.; Chang, S. Highly efficient and versatile synthesis of poly aryl fluorenes via Pd-catalyzed C-H bond activation. Org. Lett. 2009, 11, 4588–4591. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Yan, R.L.; Zhong, M.J.; Liang, Y.M. Bi(III)-Catalyzed Intermolecular Reactions of (Z)-Pent-2-en-4-yl Acetates with Ethynylarenes for the Construction of Multisubstituted Fluorene Skeletons through a Cascade Electrophilic Addition/Cycloisomerization Sequence. J. Org. Chem. 2012, 77, 2064–2068. [Google Scholar] [CrossRef] [PubMed]
- Buettner, S.; Kelzhanova, N.K.; Abilov, Z.A.; Villinger, A.; Langer, P. [3+3] Cyclizations of 1,3-bis(trimethylsilyloxy)-1,3-butadienes-a new approach to diverse CF3-substituted fluorenes, dibenzofurans, 9,10-dihydrophenanthrenes and 6H-benzo[c]chromenes. Tetrahedron 2012, 68, 3654–3668. [Google Scholar] [CrossRef]
- Anderson, D.M.W.; Leaver, D. Stereochemistry and isomerization of some hydrofluorene and hydrophenanthrene β-oxo esters. J. Chem. Soc. 1962, 450–456. [Google Scholar] [CrossRef]
- Lantano, B.; Aguirre, J.M.; Drago, E.V.; Bollini, M.; de la Faba, D.J.; Mufato, J.D. Synthesis of benzylidenecycloalkan-1-ones and 1,5-diketones under Claisen-Schmidt reaction: Influence of the temperature and electronic nature of arylaldehydes. Synth. Commun. 2017, 47, 2202–2214. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS-97 Phase annealing in SHELX-90: Direct methods for larger structures, Acta Crystallogr. Sect. A Found. Crystallogr. 1990, 46, 467–473. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Base | Solvent | Temp | Time | 2a 2 | 3a 2 |
|---|---|---|---|---|---|---|
| 1 | DABCO | Dioxane | 80 °C | 24 h | 0 | 0 |
| 2 | DBU | Dioxane | 80 °C | 24 h | complex mixture | |
| 3 | t-BuOK | Dioxane | 80 °C | 24 h | 52% | 48% |
| 4 | t-BuOK | Dioxane | 50 °C | 24 h | 7% | 48% |
| 5 | t-BuOK | Dioxane | 50 °C | 48 h | 24% | 75% |
| 6 | t-BuOK | Dioxane | 40 °C | 48 h | 6% | 45% |
| 7 3 | t-BuOK | Dioxane | 80 °C | 24 h | Trace | Trace |
| 8 | t-BuOK | Toluene | 80 °C | 24 h | 60% | 40% |
| 9 | t-BuOK | THF | 50 °C | 48 h | 20% | 40% |
| 10 | t-BuOK | t-BuOH | 50 °C | 48 h | 30% | 55% |
| Entry | Oxidant | Temp | Yield |
|---|---|---|---|
| 1 | O2/CF3COOH | 100 °C | trace |
| 2 | H2O2 (1.0 eq) | 80 °C | trace |
| 3 | Cu(OTf)(10 mol%)/bipy/NHPI/KI (1.0 eq) 2 | 80 °C | trace |
| 4 | CuBr(10 mol%)/LiBr(2.0 eq) 2 | 80 °C | 56% |
| 5 | DDQ (1.1 eq) | RT | 41% |
| 6 3 | DDQ (1.1 eq) | 100 °C | 75% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-M.; Liu, Y.-H.; Liu, S.-T. Synthesis of 3-Hydroxy-9H-fluorene-2-carboxylates via Michael Reaction, Robinson Annulation, and Aromatization. Organics 2022, 3, 481-490. https://doi.org/10.3390/org3040031
Wang Y-M, Liu Y-H, Liu S-T. Synthesis of 3-Hydroxy-9H-fluorene-2-carboxylates via Michael Reaction, Robinson Annulation, and Aromatization. Organics. 2022; 3(4):481-490. https://doi.org/10.3390/org3040031
Chicago/Turabian StyleWang, Yu-Min, Yi-Hung Liu, and Shiuh-Tzung Liu. 2022. "Synthesis of 3-Hydroxy-9H-fluorene-2-carboxylates via Michael Reaction, Robinson Annulation, and Aromatization" Organics 3, no. 4: 481-490. https://doi.org/10.3390/org3040031