
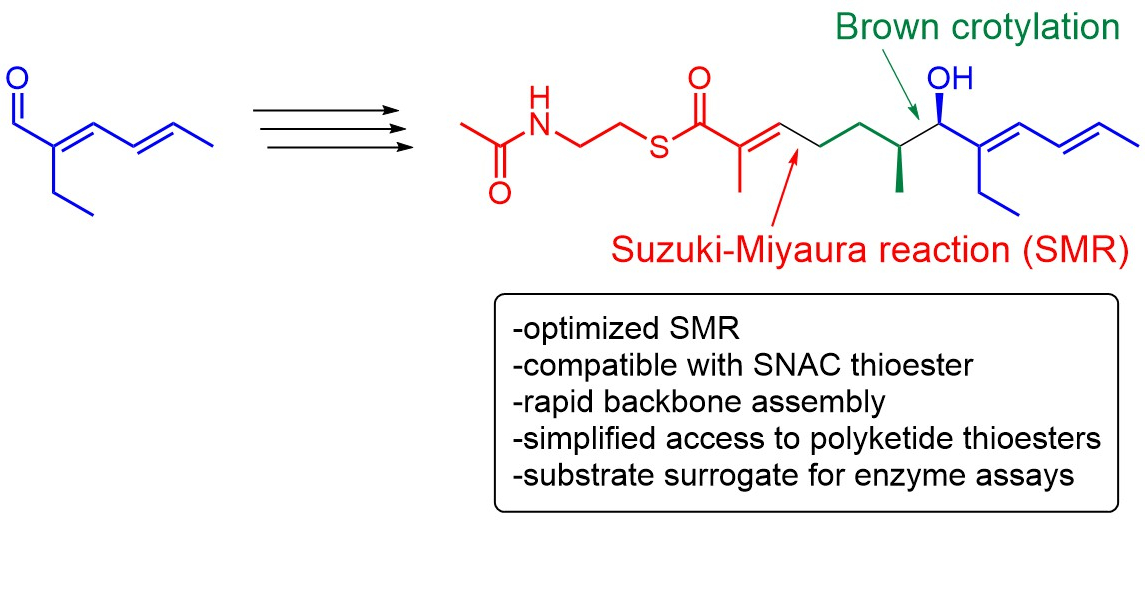
Suzuki–Miyaura Reaction in the Presence of N-Acetylcysteamine Thioesters Enables Rapid Synthesis of Biomimetic Polyketide Thioester Surrogates for Biosynthetic Studies

Abstract
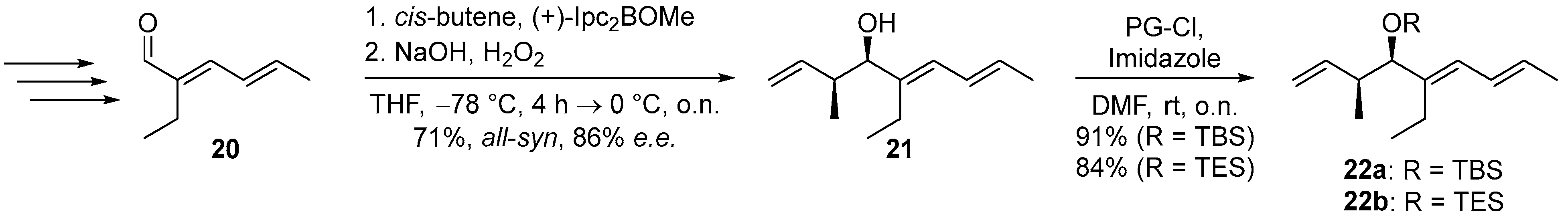
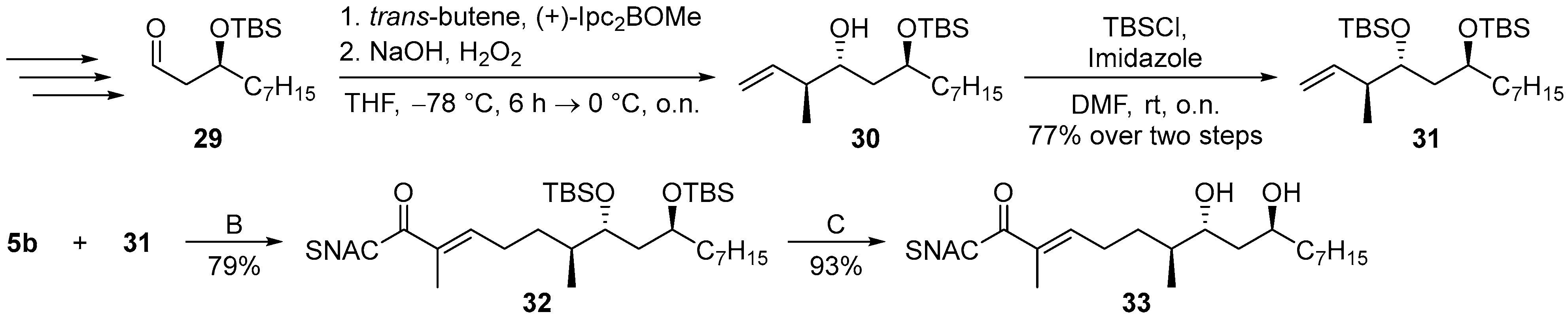
:
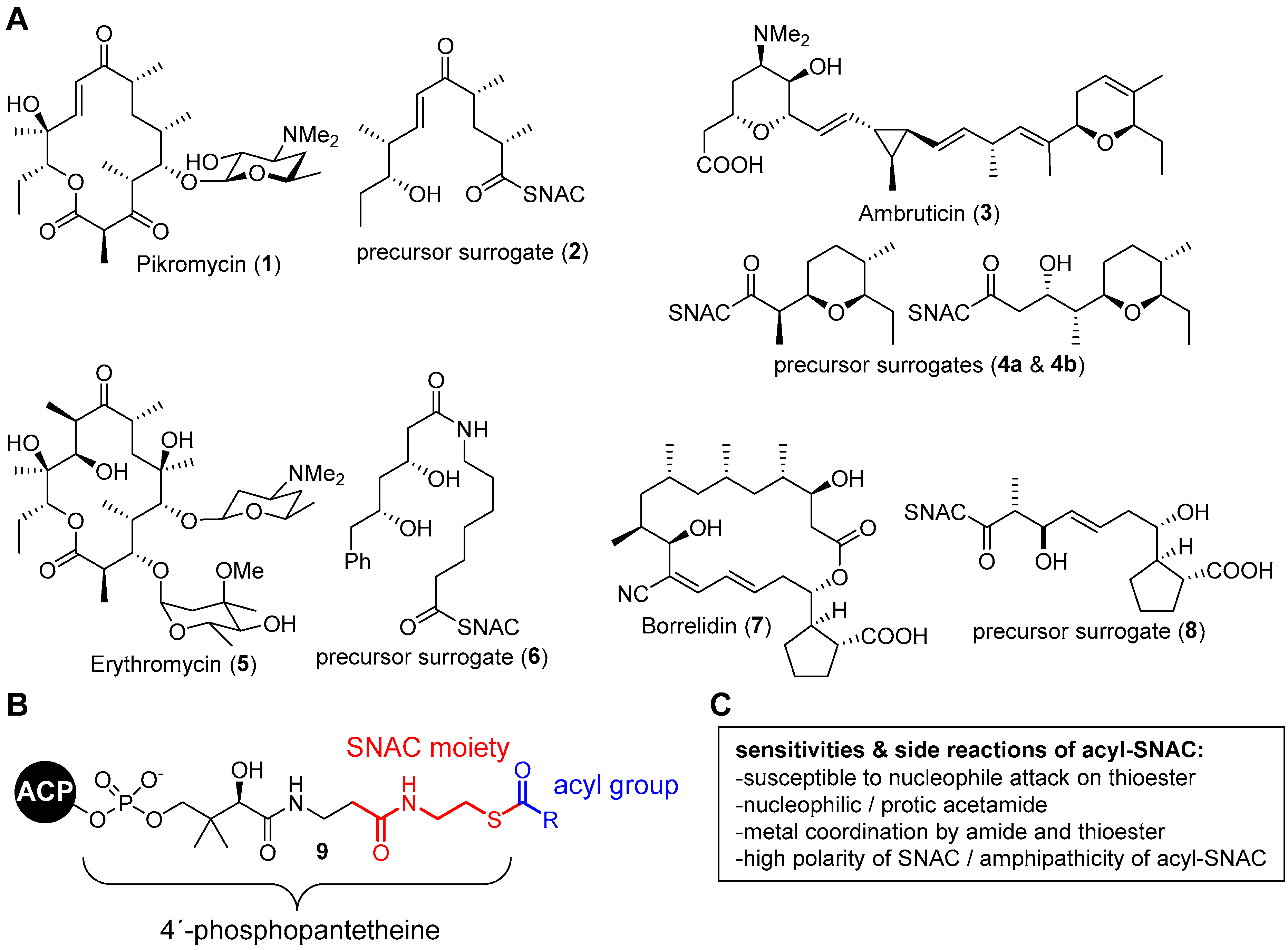
1. Introduction
2. Materials and Methods
2.1. General Methods and Materials
2.2. General Procedures
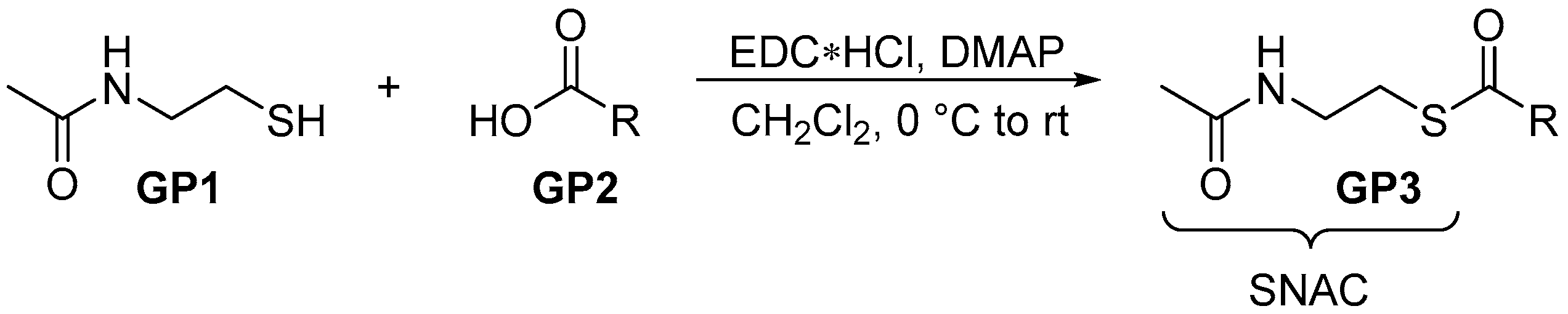
2.2.1. General Procedure 1: Steglich Esterification for SNAC Thioester Formation

2.2.2. General Procedure 2: Protection of Hydroxyls as Silylethers

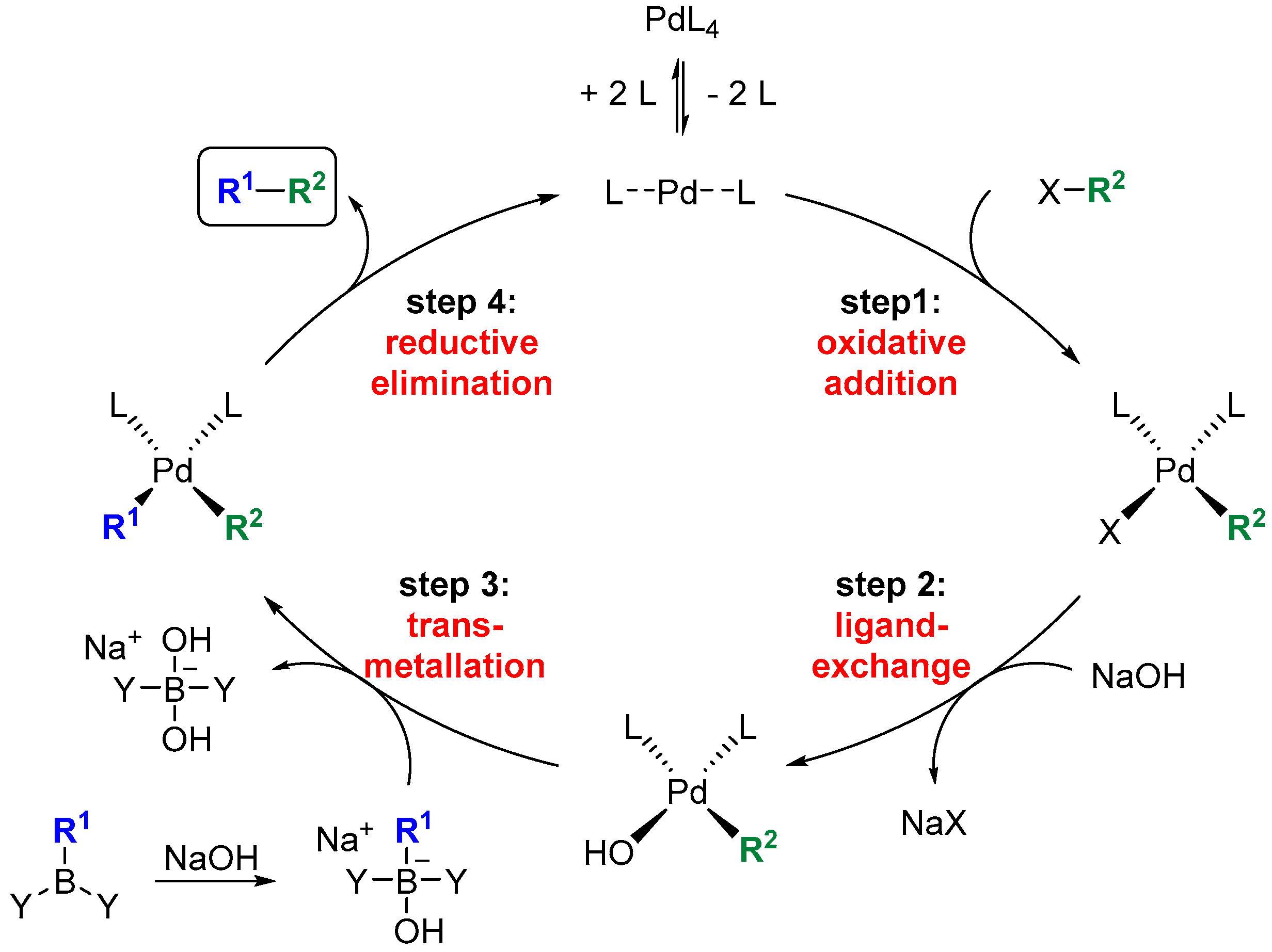
2.2.3. General Procedure 3: Hydroboration-Suzuki-Miyauri Reaction

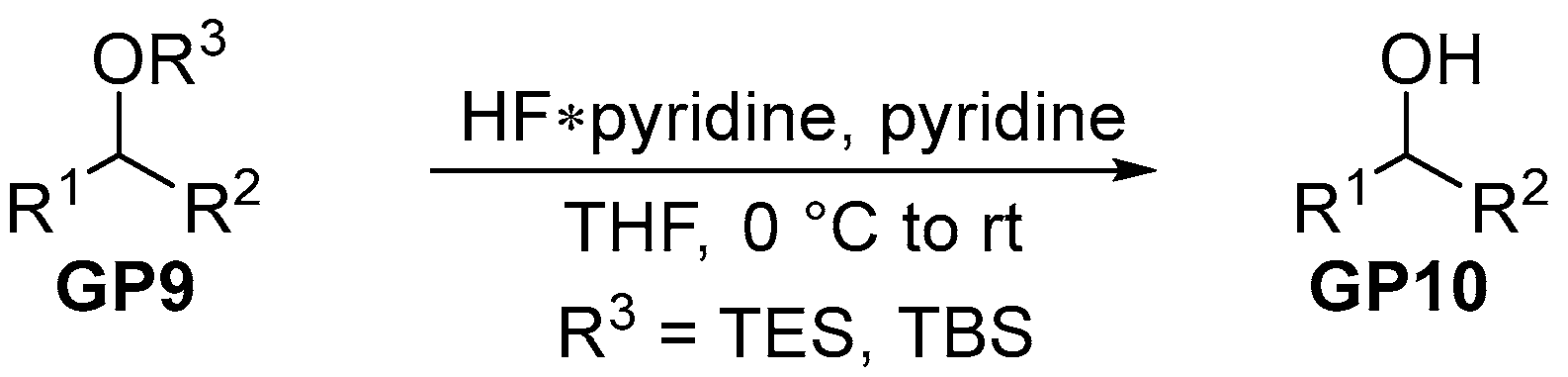
2.2.4. General Procedure 4: Deprotection

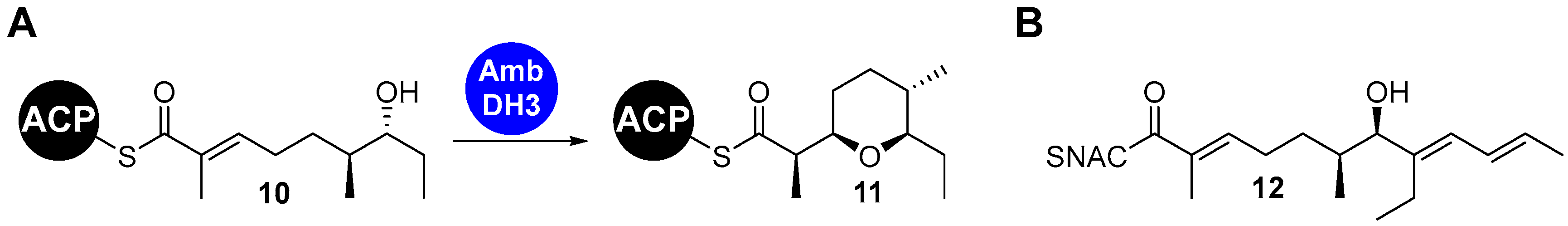
3. Results


4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weissman, K.J.; Müller, R. Protein–Protein Interactions in Multienzyme Megasynthetases. ChemBioChem 2008, 9, 826–848. [Google Scholar] [CrossRef]
- Grininger, M. Enzymology of Assembly Line Synthesis by Modular Polyketide Synthases. Nat. Chem. Biol. 2023, 19, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.-M.; Huang, T.; Rudolf, J.D.; Lohman, J.R.; Huang, S.-X.; Guo, X.; Shen, B. Enediyne Polyketide Synthases Stereoselectively Reduce the β-Ketoacyl Intermediates to β-d-Hydroxyacyl Intermediates in Enediyne Core Biosynthesis. Org. Lett. 2014, 16, 3958–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahner, J.H.; Sucipto, H.; Wenzel, S.C.; Groh, M.; Hartmann, R.W.; Müller, R. Advanced Mutasynthesis Studies on the Natural α-Pyrone Antibiotic Myxopyronin from Myxococcus Fulvus. ChemBioChem 2015, 16, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Wang, M.; Horsman, M.; Boddy, C.N. 6-Deoxyerythronolide B Synthase Thioesterase-Catalyzed Macrocyclization Is Highly Stereoselective. Org. Lett. 2012, 14, 2278–2281. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.A.; Rath, C.M.; Eisman, E.B.; Narayan, A.R.H.; Kittendorf, J.D.; Mortison, J.D.; Yoon, Y.J.; Sherman, D.H. Biocatalytic Synthesis of Pikromycin, Methymycin, Neomethymycin, Novamethymycin, and Ketomethymycin. J. Am. Chem. Soc. 2013, 135, 11232–11238. [Google Scholar] [CrossRef] [Green Version]
- Franke, J.; Hertweck, C. Biomimetic Thioesters as Probes for Enzymatic Assembly Lines: Synthesis, Applications, and Challenges. Cell Chem. Biol. 2016, 23, 1179–1192. [Google Scholar] [CrossRef] [Green Version]
- Hahn, F.; Kandziora, N.; Friedrich, S.; Leadlay, P.F. Synthesis of Complex Intermediates for the Study of a Dehydratase from Borrelidin Biosynthesis. Beilstein J. Org. Chem. 2014, 10, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Berkhan, G.; Merten, C.; Holec, C.; Hahn, F. The Interplay between a Multifunctional Dehydratase Domain and a C-Methyltransferase Effects Olefin Shift in Ambruticin Biosynthesis. Angew. Chem. Int. Ed. 2016, 55, 13589–13592. [Google Scholar] [CrossRef]
- Schröder, M.; Roß, T.; Hemmerling, F.; Hahn, F. Studying a Bottleneck of Multimodular Polyketide Synthase Processing: The Polyketide Structure-Dependent Performance of Ketoreductase Domains. ACS Chem. Biol. 2022, 17, 1030–1037. [Google Scholar] [CrossRef]
- Wunderlich, J.; Roß, T.; Schröder, M.; Hahn, F. Step-Economic Synthesis of Biomimetic β-Ketopolyene Thioesters and Demonstration of Their Usefulness in Enzymatic Biosynthesis Studies. Org. Lett. 2020, 22, 4955–4959. [Google Scholar] [CrossRef]
- Sundaram, S.; Kim, H.J.; Bauer, R.; Thongkongkaew, T.; Heine, D.; Hertweck, C. On-Line Polyketide Cyclization into Diverse Medium-Sized Lactones by a Specialized Ketosynthase Domain. Angew. Chem. Int. Ed. 2018, 57, 11223–11227. [Google Scholar] [CrossRef]
- Hooshmand, S.E.; Heidari, B.; Sedghi, R.; Varma, R.S. Recent Advances in the Suzuki–Miyaura Cross-Coupling Reaction Using Efficient Catalysts in Eco-Friendly Media. Green Chem. 2019, 21, 381–405. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Zeysing, B.; Gosch, C.; Terfort, A. Protecting Groups for Thiols Suitable for Suzuki Conditions. Org. Lett. 2000, 2, 1843–1845. [Google Scholar] [CrossRef]
- Liebeskind, L.S.; Srogl, J. Thiol Ester−Boronic Acid Coupling. A Mechanistically Unprecedented and General Ketone Synthesis. J. Am. Chem. Soc. 2000, 122, 11260–11261. [Google Scholar] [CrossRef]
- Cheng, H.-G.; Chen, H.; Liu, Y.; Zhou, Q. The Liebeskind–Srogl Cross-Coupling Reaction and Its Synthetic Applications. Asian J. Org. Chem. 2018, 7, 490–508. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Tang, G.-L.; Pan, H.-X. Enzymatic Formation of Oxygen-Containing Heterocycles in Natural Product Biosynthesis. ChemBioChem 2018, 19, 2002–2022. [Google Scholar] [CrossRef]
- Hemmerling, F.; Hahn, F. Biosynthesis of Oxygen and Nitrogen-Containing Heterocycles in Polyketides. Beilstein J. Org. Chem. 2016, 12, 1512–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pöplau, P.; Frank, S.; Morinaka, B.I.; Piel, J. An Enzymatic Domain for the Formation of Cyclic Ethers in Complex Polyketides. Angew. Chem. Int. Ed. 2013, 52, 13215–13218. [Google Scholar] [CrossRef]
- Berkhan, G.; Hahn, F. A Dehydratase Domain in Ambruticin Biosynthesis Displays Additional Activity as a Pyran-Forming Cyclase. Angew. Chem. Int. Ed. 2014, 53, 14240–14244. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, T.; Berkhan, G.; Wagner, L.; Sung, K.H.; Kolb, S.; Geise, H.; Hahn, F. Biocatalysts from Biosynthetic Pathways: Enabling Stereoselective, Enzymatic Cycloether Formation on a Gram Scale. ACS Catal. 2020, 10, 4973–4982. [Google Scholar] [CrossRef] [Green Version]
- Wagner, L.; Stang, J.; Derra, S.; Hollmann, T.; Hahn, F. Towards Understanding Oxygen Heterocycle-Forming Biocatalysts: A Selectivity Study of the Pyran Synthase PedPS7. Org. Biomol. Chem. 2022, 20, 9645–9649. [Google Scholar] [CrossRef]
- Sung, K.H.; Berkhan, G.; Hollmann, T.; Wagner, L.; Blankenfeldt, W.; Hahn, F. Insights into the Dual Activity of a Bifunctional Dehydratase-Cyclase Domain. Angew. Chem. Int. Ed. 2018, 57, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Wagner, L.; Roß, T.; Hollmann, T.; Hahn, F. Cross-Linking of a Polyketide Synthase Domain Leads to a Recyclable Biocatalyst for Chiral Oxygen Heterocycle Synthesis. RSC Adv. 2021, 11, 20248–20251. [Google Scholar] [CrossRef]
- Wagner, D.T.; Zhang, Z.; Meoded, R.A.; Cepeda, A.J.; Piel, J.; Keatinge-Clay, A.T. Structural and Functional Studies of a Pyran Synthase Domain from a Trans-Acyltransferase Assembly Line. ACS Chem. Biol. 2018, 13, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Ueoka, R.; Uria, A.R.; Reiter, S.; Mori, T.; Karbaum, P.; Peters, E.E.; Helfrich, E.J.N.; Morinaka, B.I.; Gugger, M.; Takeyama, H.; et al. Metabolic and Evolutionary Origin of Actin-Binding Polyketides from Diverse Organisms. Nat. Chem. Biol. 2015, 11, 705–712. [Google Scholar] [CrossRef]
- Luhavaya, H.; Dias, M.V.B.; Williams, S.R.; Hong, H.; de Oliveira, L.G.; Leadlay, P.F. Enzymology of Pyran Ring A Formation in Salinomycin Biosynthesis. Angew. Chem. Int. Ed. 2015, 54, 13622–13625. [Google Scholar] [CrossRef] [Green Version]
- Woo, A.J.; Strohl, W.R.; Priestley, N.D. Nonactin Biosynthesis: The Product of NonS Catalyzes the Formation of the Furan Ring of Nonactic Acid. Antimicrob. Agents Chemother. 1999, 43, 1662–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaura, N.; Ishiyama, T.; Sasaki, H.; Ishikawa, M.; Sato, M.; Suzuki, A. Palladium-Catalyzed Inter- and Intramolecular Cross-Coupling Reactions of B-Alkyl-9-Borabicyclo [3.3.1] Nonane Derivatives with 1-Halo-1-Alkenes or Haloarenes. Syntheses of Functionalized Alkenes, Arenes, and Cycloalkenes via a Hydroboration-Coupling Sequence. J. Am. Chem. Soc. 1989, 111, 314–321. [Google Scholar] [CrossRef]
- Farina, V.; Krishnan, B. Large Rate Accelerations in the Stille Reaction with Tri-2-Furylphosphine and Triphenylarsine as Palladium Ligands: Mechanistic and Synthetic Implications. J. Am. Chem. Soc. 1991, 113, 9585–9595. [Google Scholar] [CrossRef]
- Chishiro, A.; Konishi, M.; Inaba, R.; Yumura, T.; Imoto, H.; Naka, K. Tertiary Arsine Ligands for the Stille Coupling Reaction. Dalton Trans. 2021, 51, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Carreira, E.M.; Du Bois, J. (+)-Zaragozic Acid C: Synthesis and Related Studies. J. Am. Chem. Soc. 1995, 117, 8106–8125. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, W.; Zhang, H.; Tian, W.; Wu, L.; Wang, S.; Zheng, M.; Zhang, J.; Sun, C.; Deng, Z.; et al. Structural Basis of a Broadly Selective Acyltransferase from the Polyketide Synthase of Splenocin. Angew. Chem. 2018, 130, 5925–5929. [Google Scholar] [CrossRef]
- Campbell, N.E.; Sammis, G.M. Single-Electron/Pericyclic Cascade for the Synthesis of Dienes. Angew. Chem. Int. Ed. 2014, 53, 6228–6231. [Google Scholar] [CrossRef]
- Whittaker, A.M.; Lalic, G. Monophasic Catalytic System for the Selective Semireduction of Alkynes. Org. Lett. 2013, 15, 1112–1115. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Moloney, M.; Parsons, A.F. An intramolecular cobalt cyclisation for the construction of substituted pyrrolidines. Tetrahedron 1992, 48, 9373–9384. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
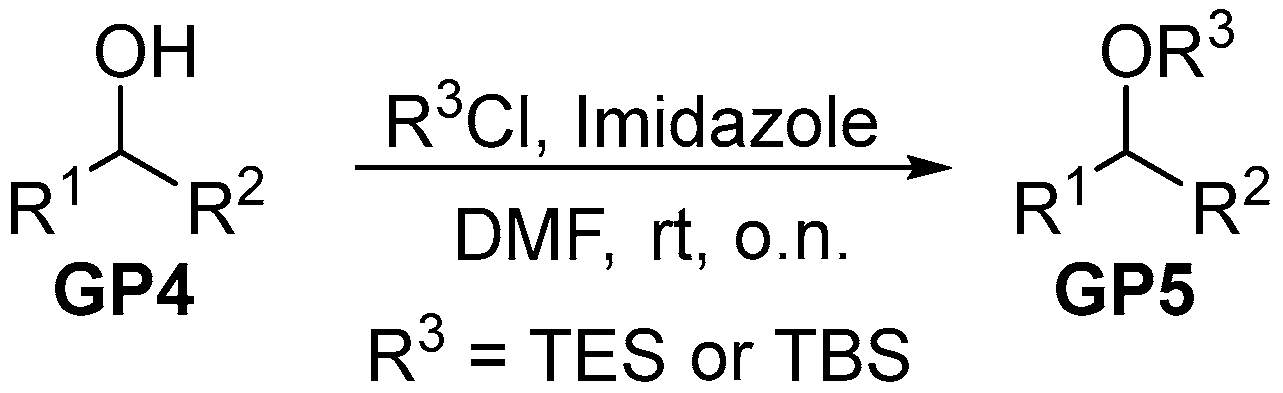{kind=link}
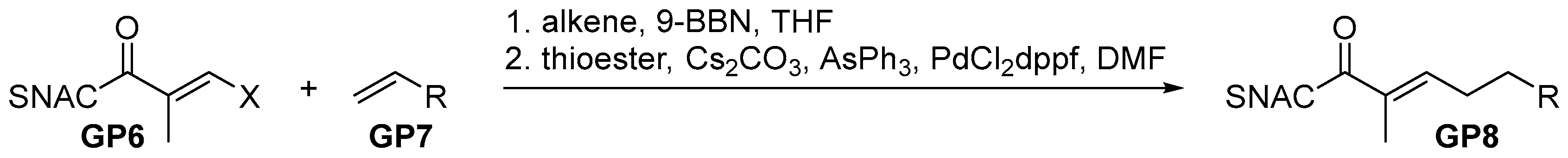{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Entry | X | Base | Additive | Temperature [°C] | Isolated Yield [%] |
| a | Br | K2CO3 | - | 50 | 54 |
| b | Br | K2CO3 | P(o-furyl)3 | 50 | 23 |
| c | Br | K2CO3 | AsPh3 | 50 | 55 |
| d | Br | Cs2CO3 | - | 50 | 55 |
| e | Br | K2CO3 | - | 20 | 13 |
| f | I | K2CO3 | - | 50 | 55 |
| g | I | Cs2CO3 | AsPh3 | 50 | 34 |
| h | I | Cs2CO3 | AsPh3 | 65 | - |
| i | I | Cs2CO3 | AsPh3 | 20 | 78 |
 | ||||||
| Entry | X | PG | Base | Additive | Temperature [° C] | Isolated Yield [%] |
| a | Br | TBS | 2 eq. K2CO3 | - | 50 | 27 |
| b | Br | TBS | 3 eq. K3PO4 | - | 50 | 17 |
| c | Br | TES | 2 eq. K2CO3 | - | 50 | 25 |
| d | Br | TES | 3 eq. K3PO4 | - | 50 | 12 |
| e | Br | TES | 2 eq. K2CO3 | - | 20 | 15 |
| f | I | TES | 2 eq. K2CO3 | - | 50 | 49 |
| g | Br | TES | 2 eq. Cs2CO3 | - | 50 | 80 |
| h | Br | TES | 2 eq. K2CO3 | AsPh3 | 50 | 77 |
| i | I | TES | 2 eq. K2CO3 | - | 20 | 63 |
| j | I | TES | 2 eq. Cs2CO3 | AsPh3 | 20 | 87 |
 | ||||
| Entry | PG | Reagent | Conditions | Result |
| a | TBS | PPTS | DMSO, 50 °C, o.n. | Decomposition |
| b | TBS | TBAF | THF, 0 °C, 1 h | No reaction |
| c | TBS | TBAF | THF, 0 → 20 °C, o.n. | Decomposition |
| d | TES | TBAF | THF, 0 °C, 1 h | Decomposition |
| e | TBS | HF*pyridine | THF, 0 °C, 3 h | Decomposition |
| f | TBS | HF*pyridine, pyridine | THF, 0 → 20 °C, 3 h | 51% |
| g | TES | HF*pyridine, pyridine | THF, 0 → 20 °C, 3 h | 81% |
 | ||||||
| Entry | X | PG | Conditions | Coupling Yield [%] | Deprotection Yield [%] | Overall Yield [%] |
| a | Br | TBS | A | 30 | 53 | 16 |
| b | Br | TES | A | 51 | 86 | 44 |
| c | I | TES | B | 74 | 81 | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derra, S.; Schlotte, L.; Hahn, F. Suzuki–Miyaura Reaction in the Presence of N-Acetylcysteamine Thioesters Enables Rapid Synthesis of Biomimetic Polyketide Thioester Surrogates for Biosynthetic Studies. Chemistry 2023, 5, 1407-1418. https://doi.org/10.3390/chemistry5020096
Derra S, Schlotte L, Hahn F. Suzuki–Miyaura Reaction in the Presence of N-Acetylcysteamine Thioesters Enables Rapid Synthesis of Biomimetic Polyketide Thioester Surrogates for Biosynthetic Studies. Chemistry. 2023; 5(2):1407-1418. https://doi.org/10.3390/chemistry5020096
Chicago/Turabian StyleDerra, Sebastian, Luca Schlotte, and Frank Hahn. 2023. "Suzuki–Miyaura Reaction in the Presence of N-Acetylcysteamine Thioesters Enables Rapid Synthesis of Biomimetic Polyketide Thioester Surrogates for Biosynthetic Studies" Chemistry 5, no. 2: 1407-1418. https://doi.org/10.3390/chemistry5020096