
Synthetic and Structural Chemistry of Uranyl-Amidoxime Complexes: Technological Implications †
 ,
,
Abstract
:
1. Introduction—Scope and Organization of This Review
2. The Current Interest in Uranium Chemistry and the Renaissance of the Uranyl Complexes
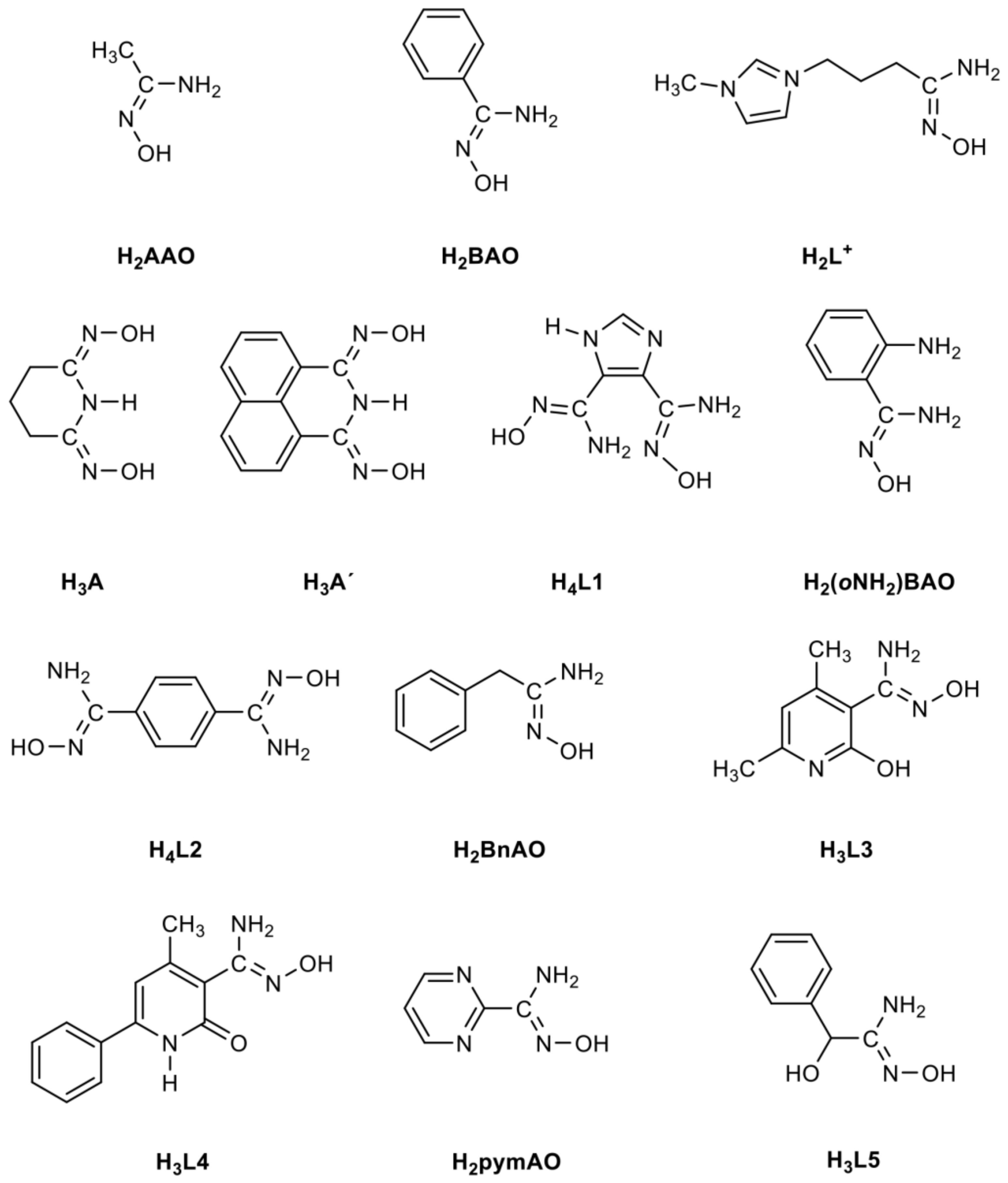
3. Amidoximes: A Class of Interesting Organic Compounds and Exciting Ligands
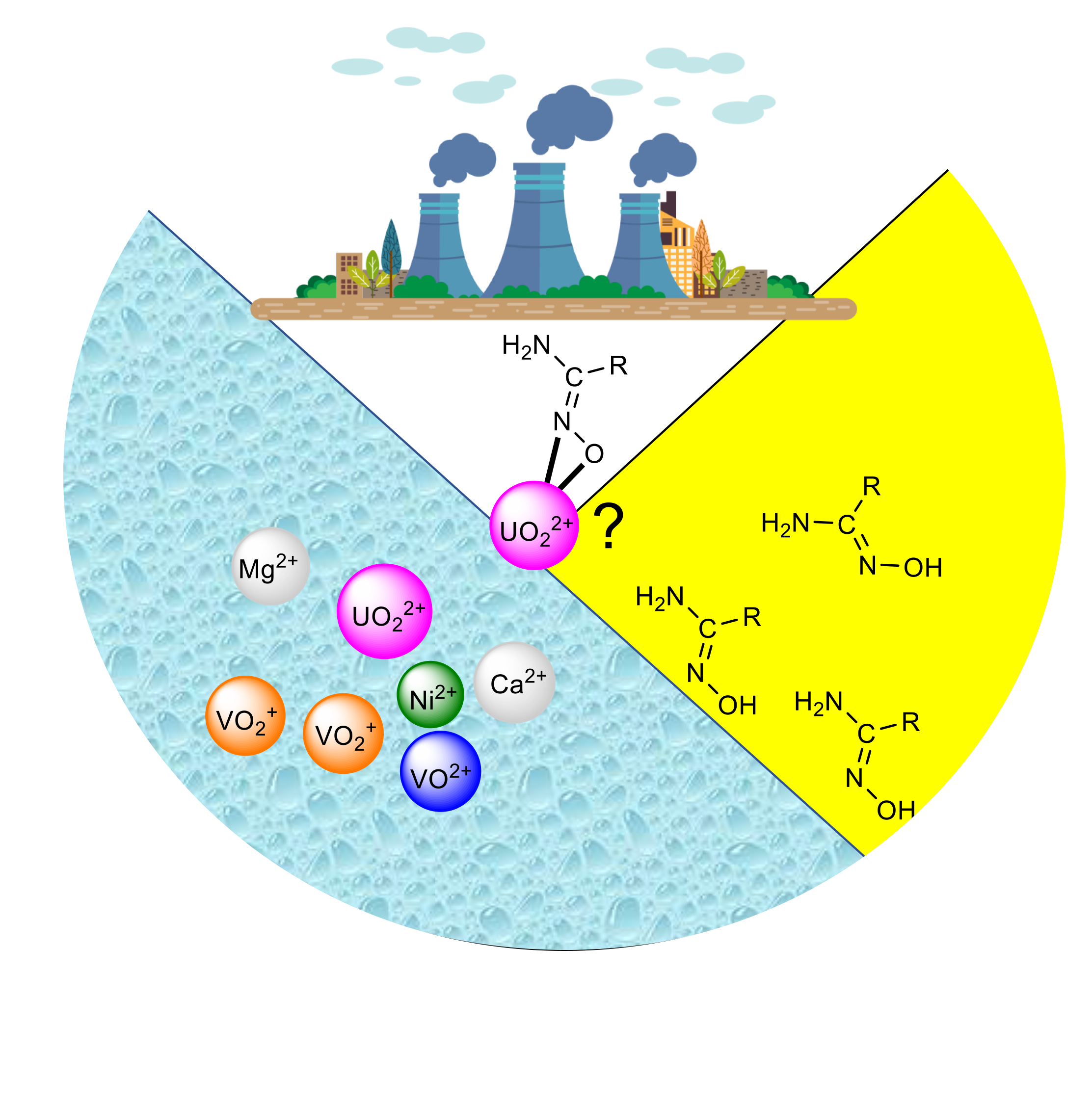
4. Extraction of Uranium from Seawater—The Importance of the Amidoxime Sorbents
5. Synthetic and Structural Studies on Uranyl-Amidoxime Complexes and Their Technological Implications
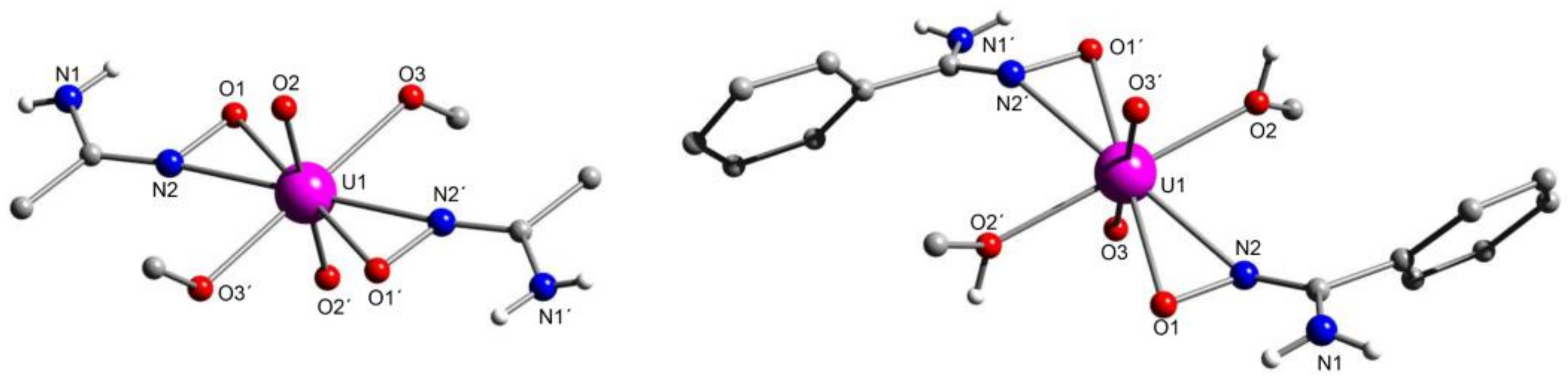
5.1. The Beginning of the Story and the Uranyl-Acetamidoxime and Uranyl-Benzamidoxime Chemistry
5.2. Incorporation of an Amidoxime Coordination Site within a Hydrophobic Ionic Liquid (IL)
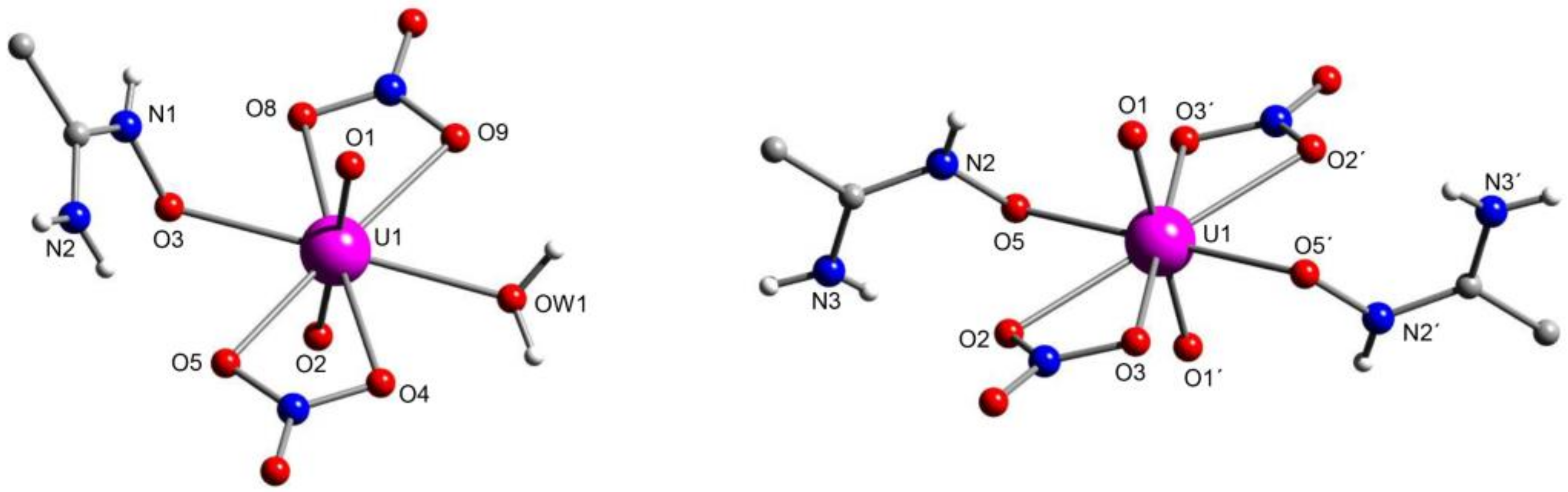
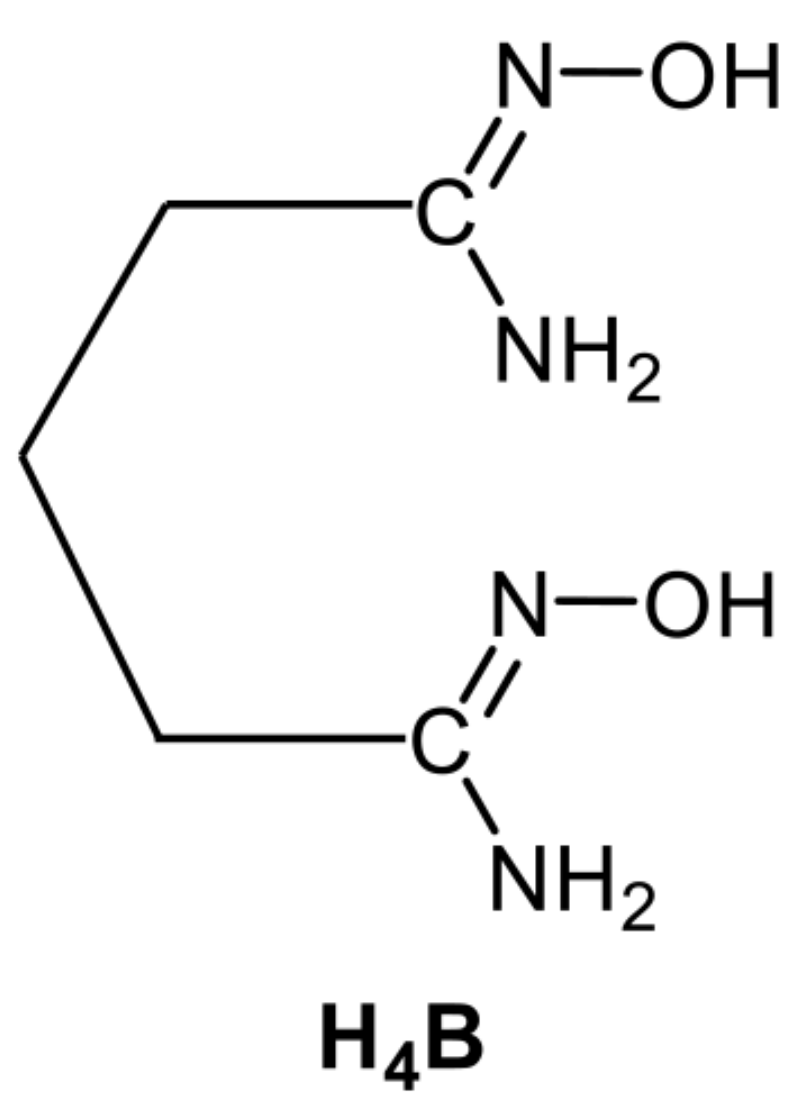
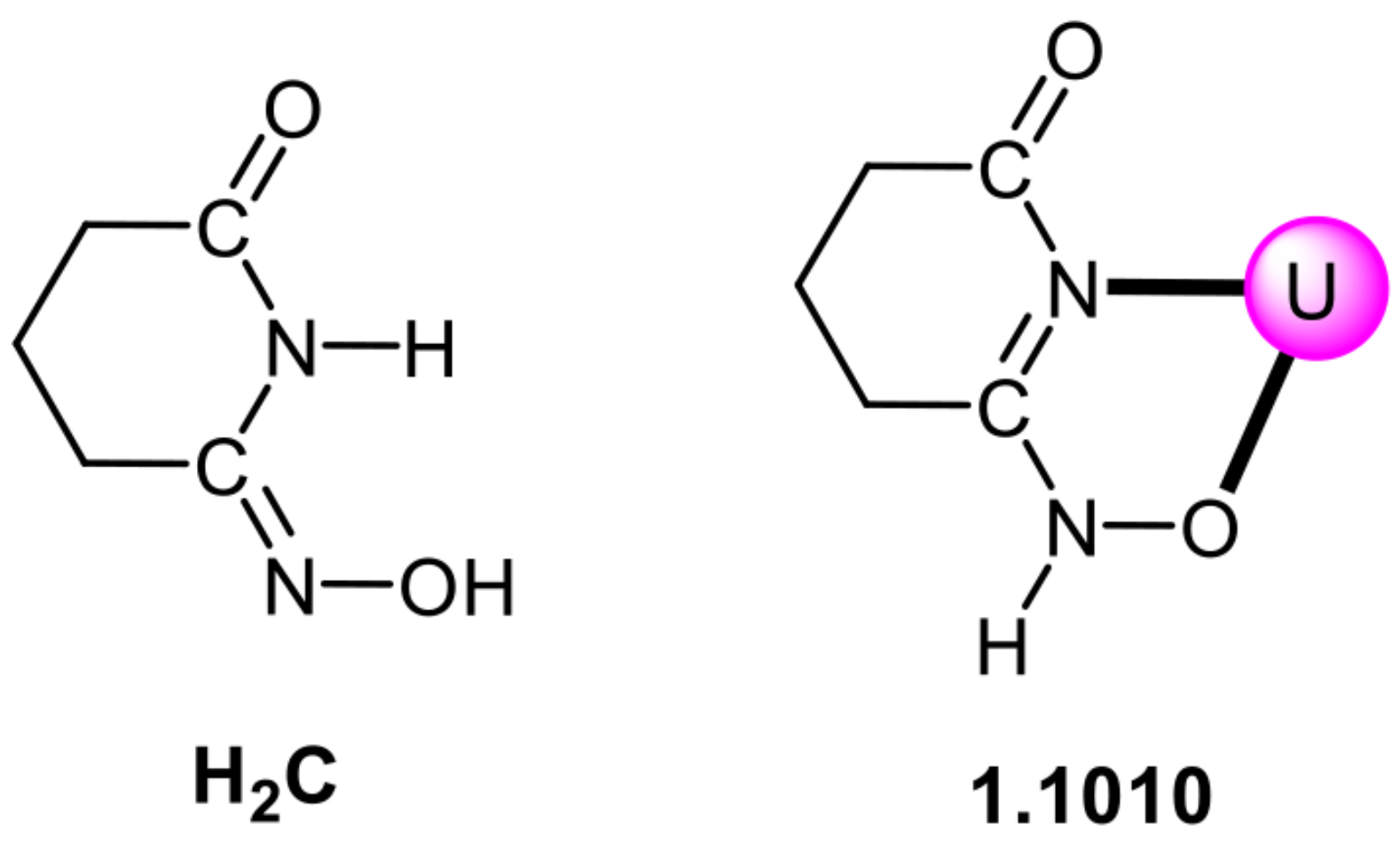
5.3. Closed-Ring Glutarimidedioxime, Closed-Ring Glutarimidoxioxime, or Open-Chain Glutardiamidoxime Motifs? Structural and Solution Studies
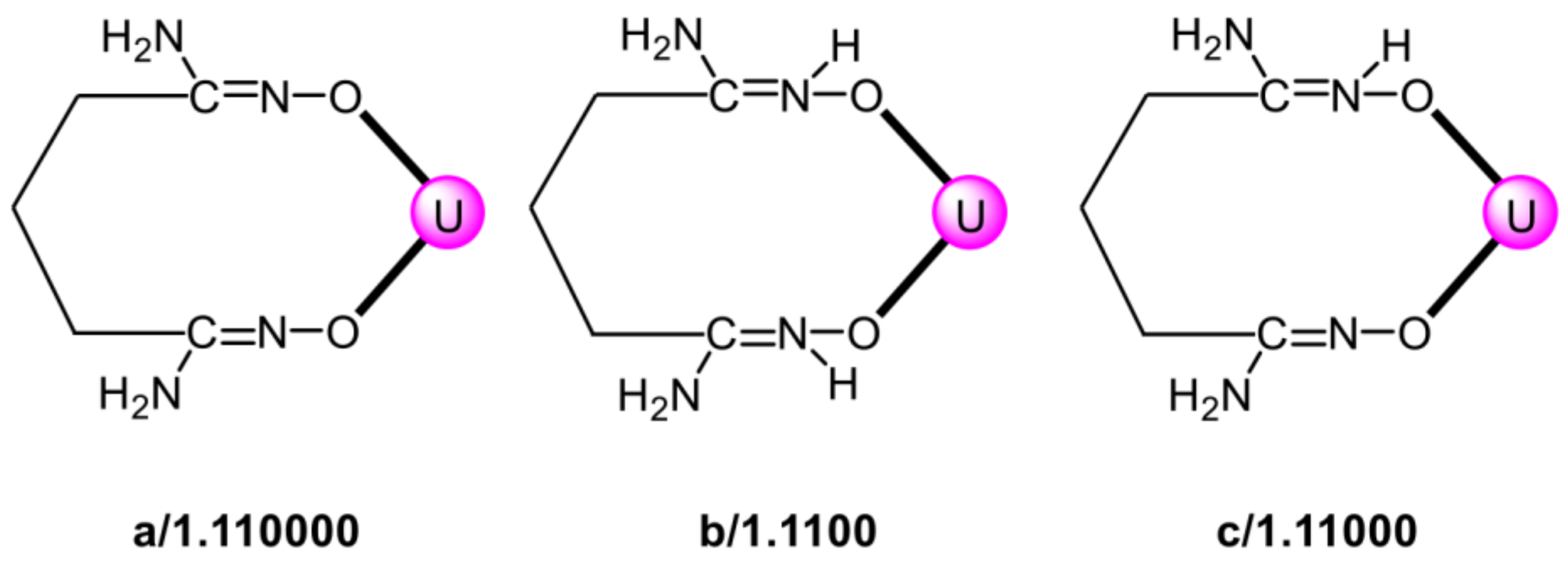
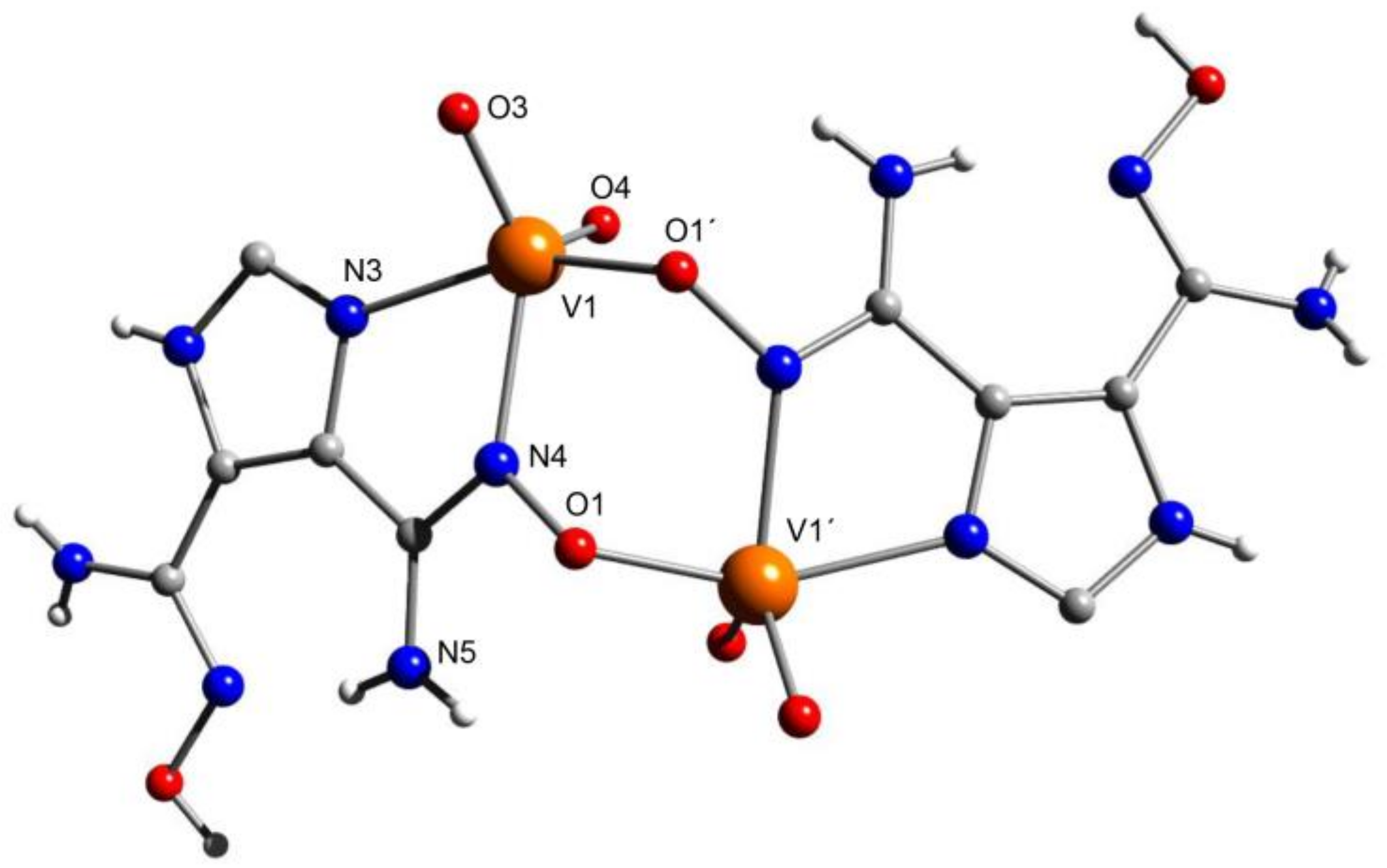
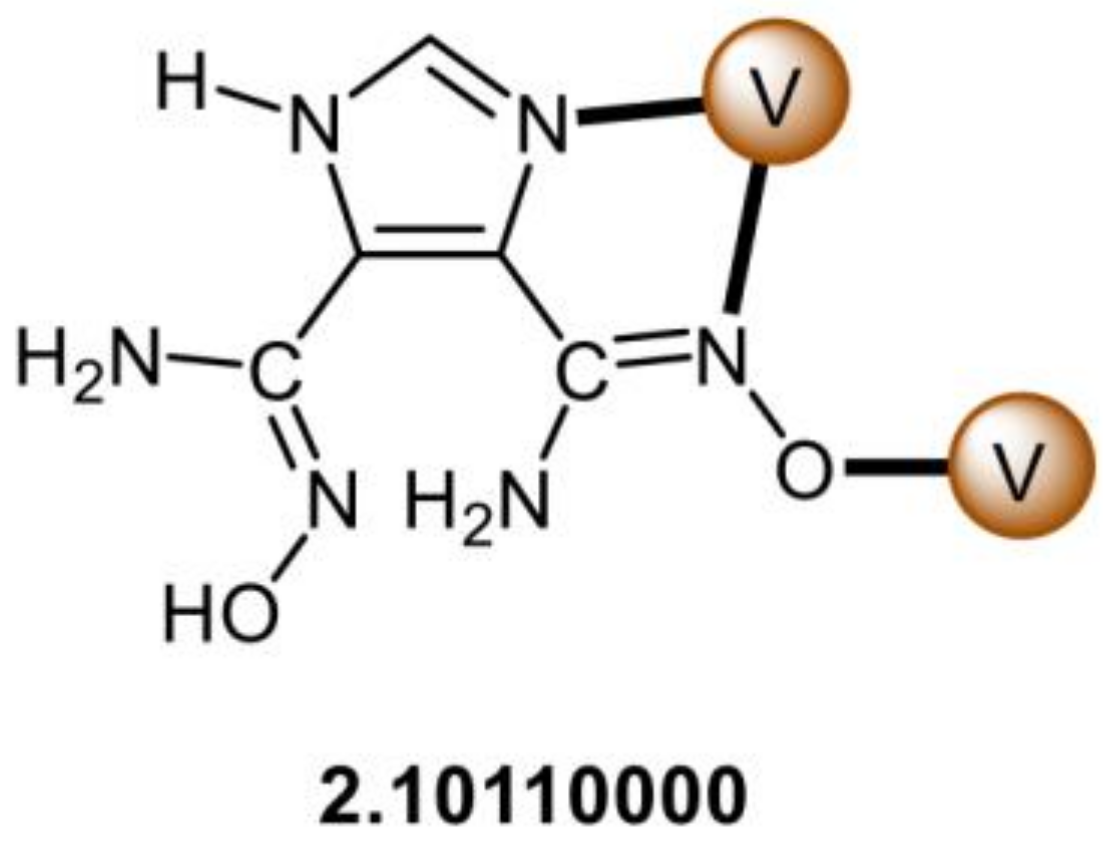
5.4. Structural Clues to {UO2}2+/{VVO2}+ Competition in Seawater Extraction Using Amidoxime-Based Sorbents
5.5. Bio-Inspired Nano-Traps for Uranyl Extraction
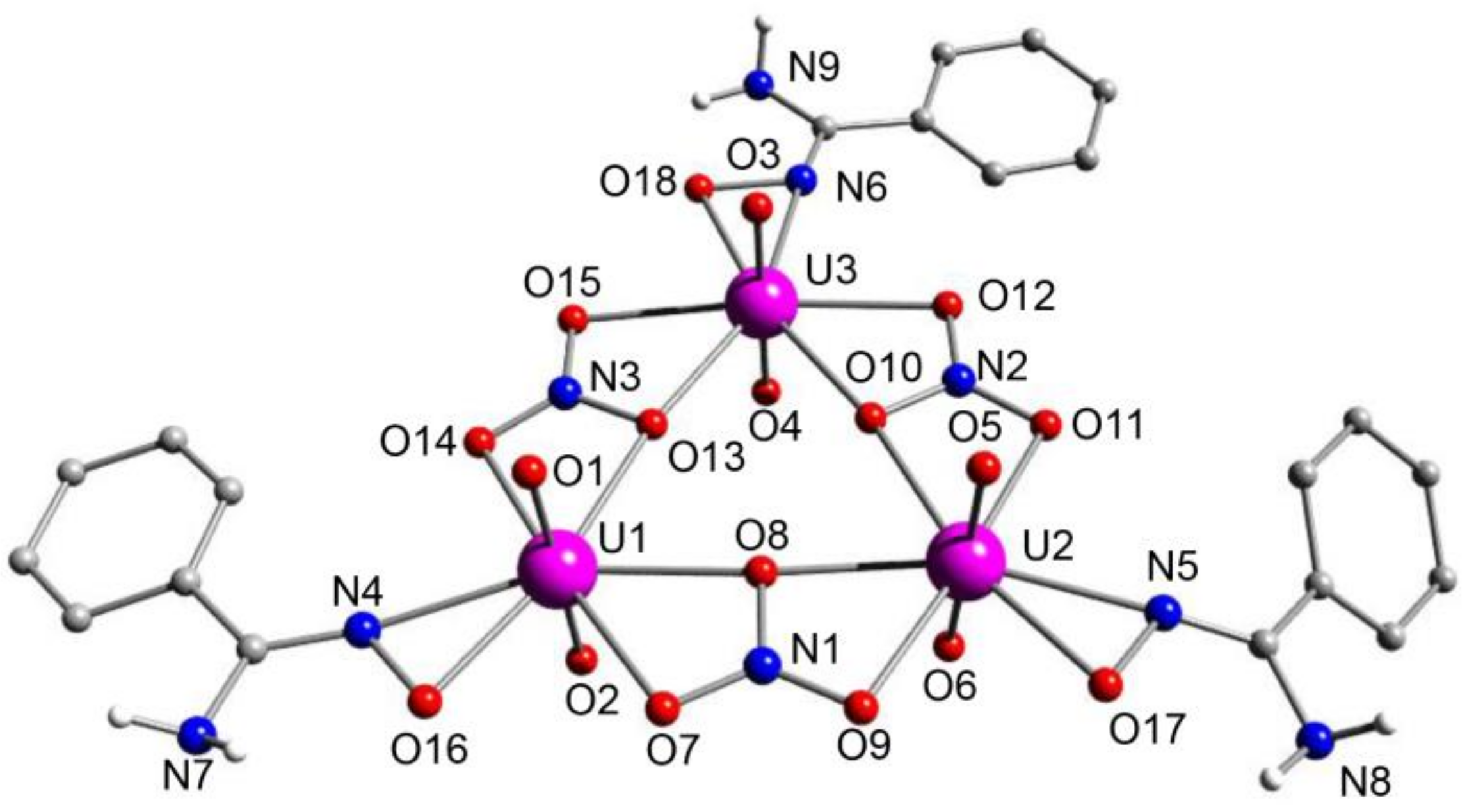
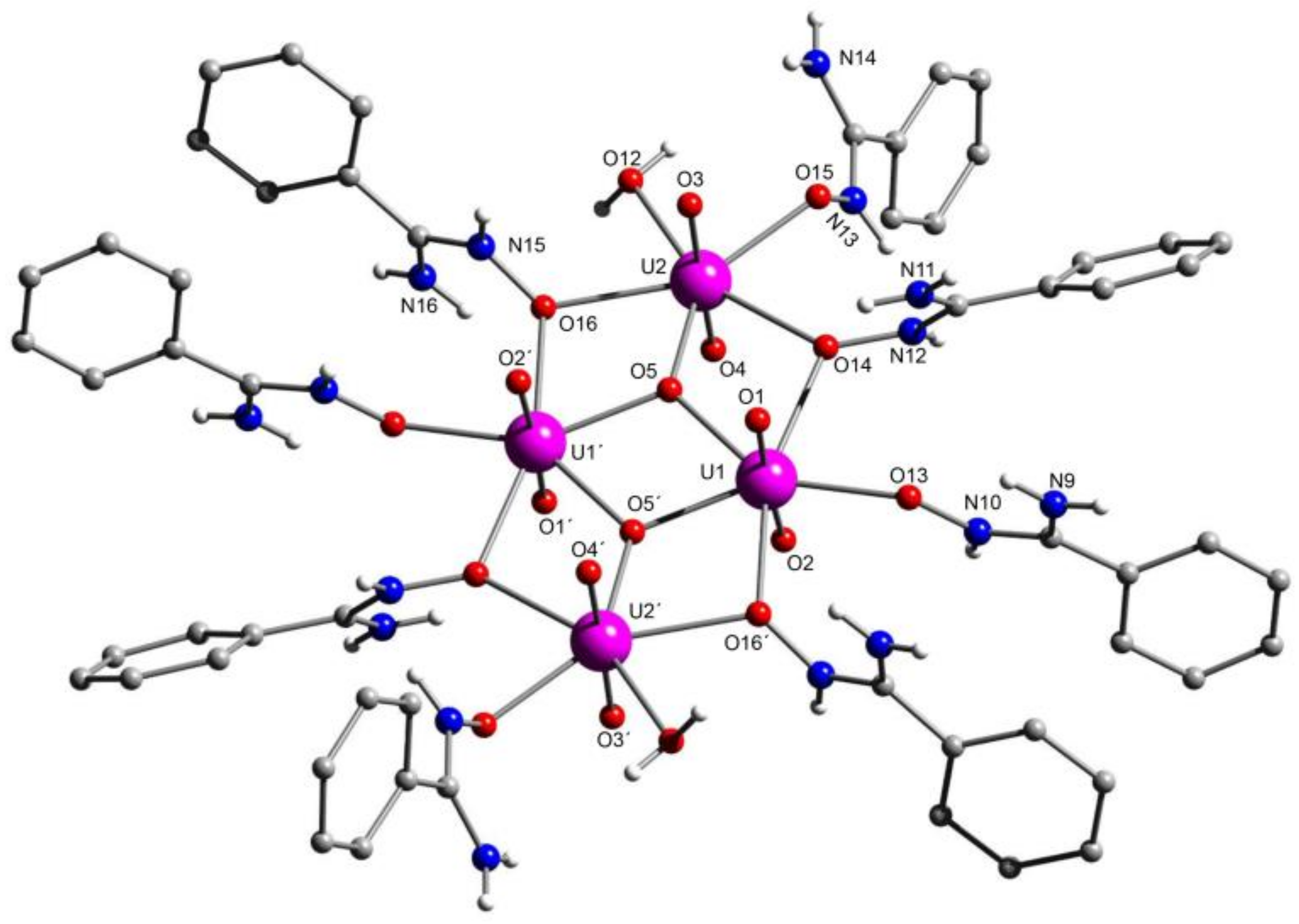
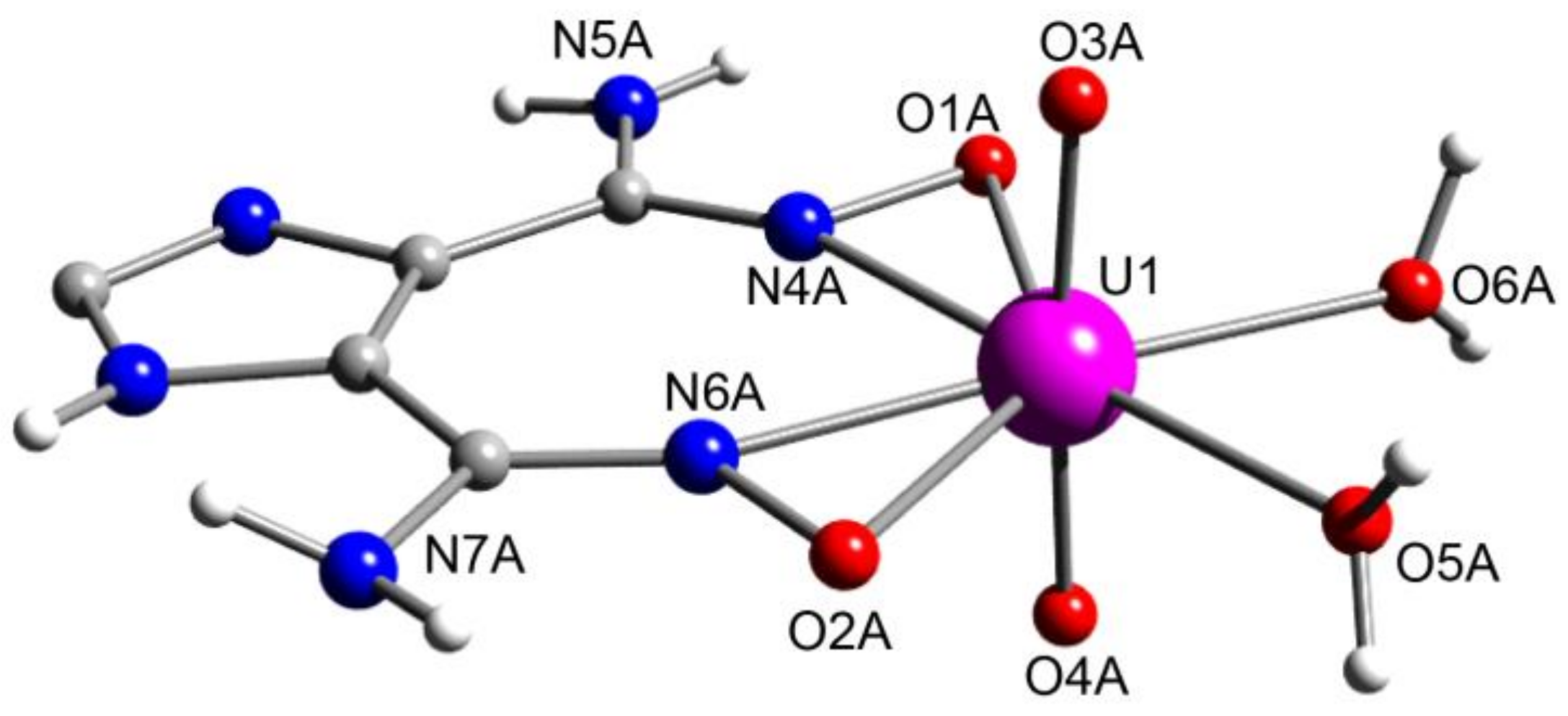
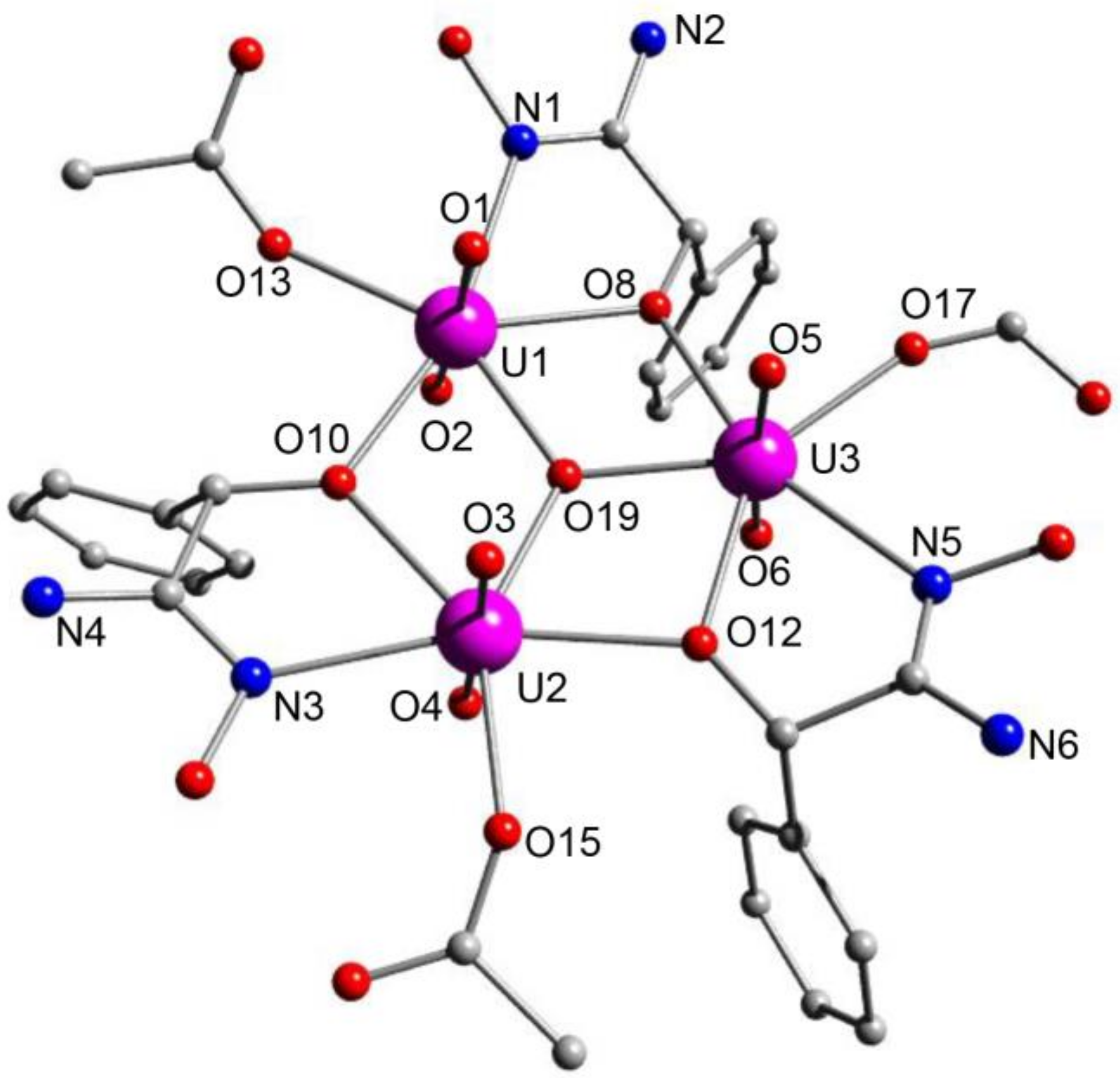
5.6. An Uranyl MOF Containing a Partially Hydrolyzed Tetranuclear Mode from the Use of a Diamidoxime Ligand
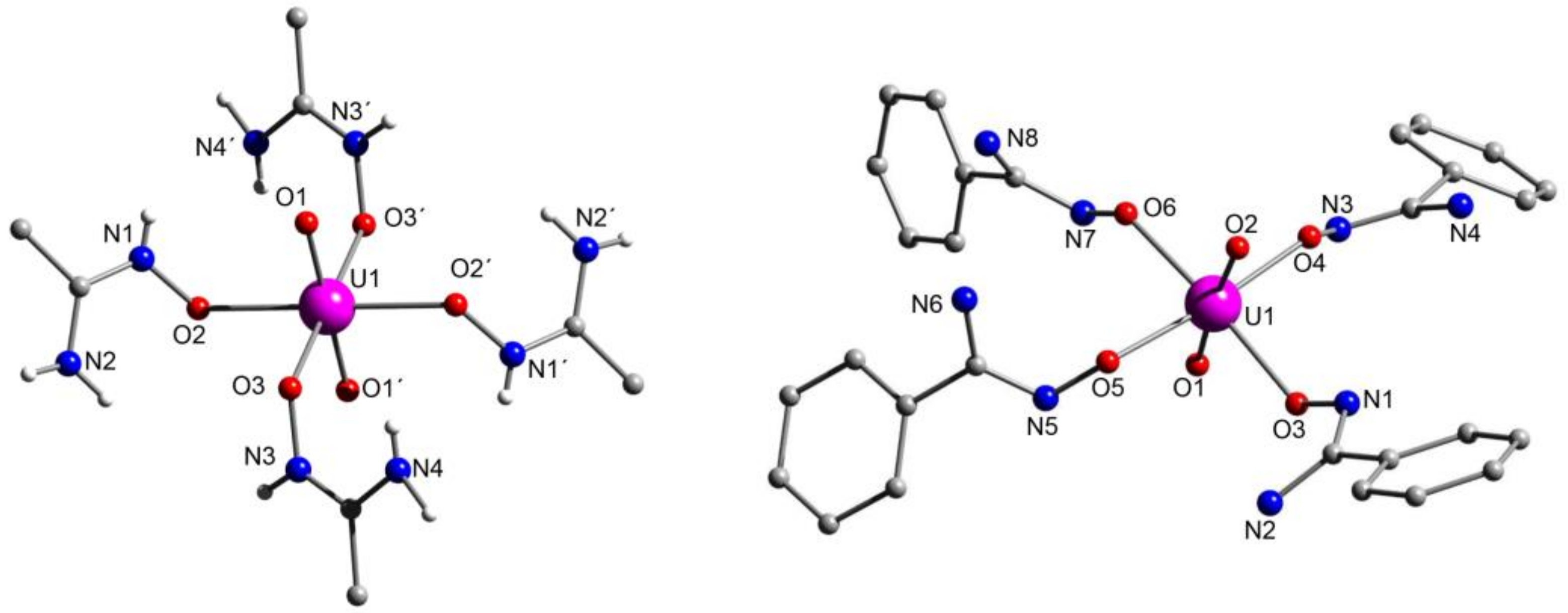
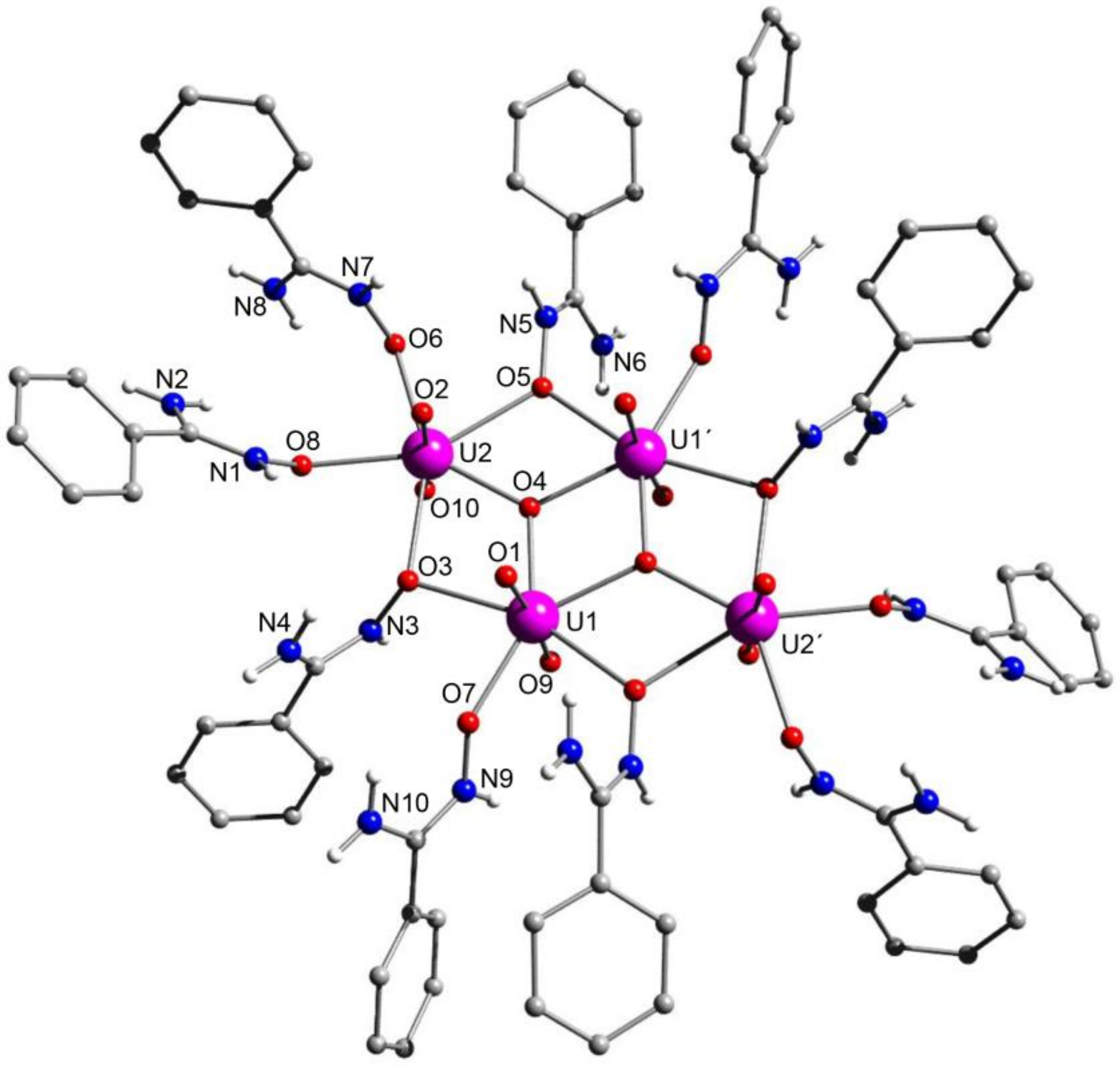
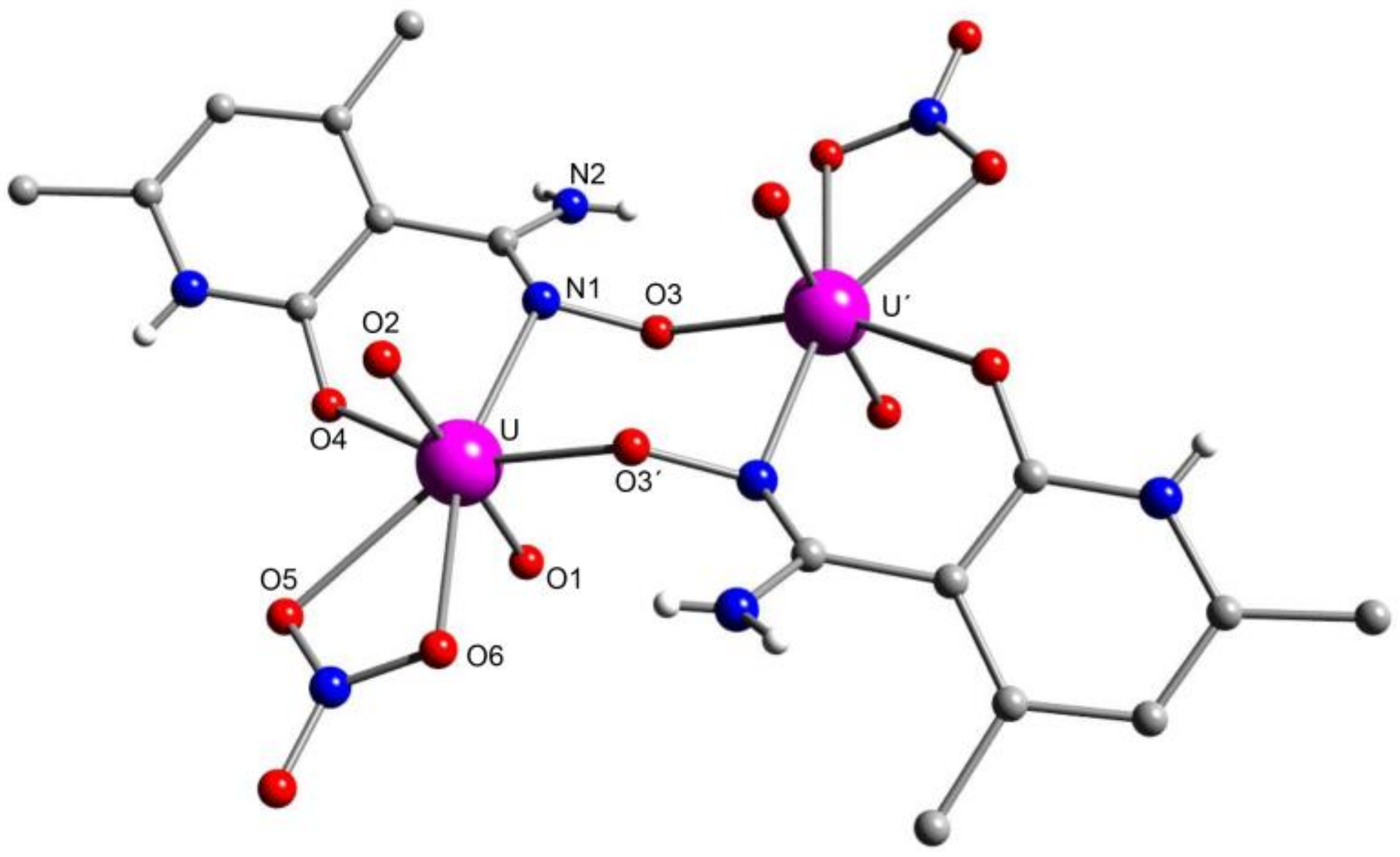
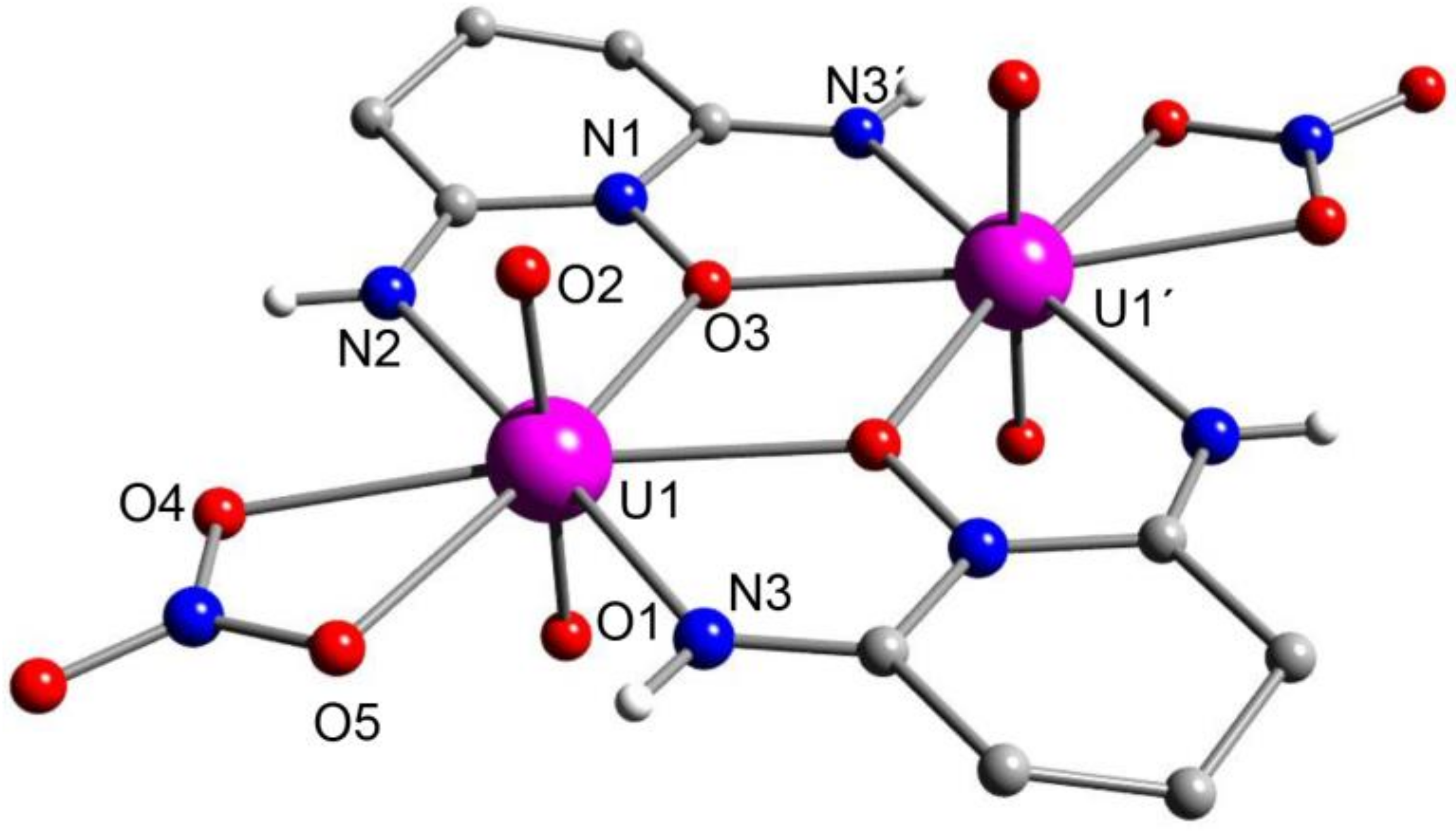
5.7. A Dinuclear Uranyl Complex with a Rare Bridging Zwitterionic-Amidoxime Group
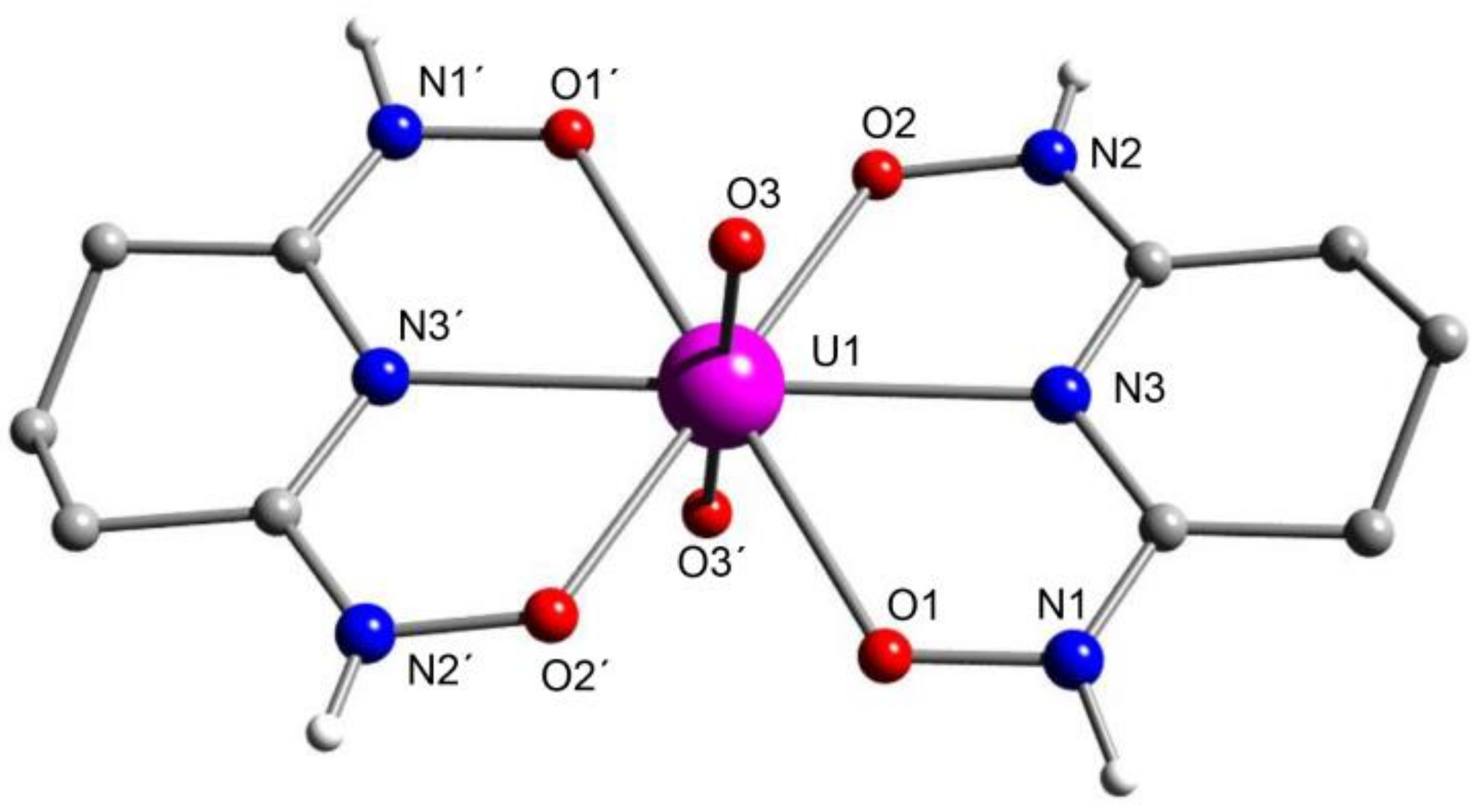
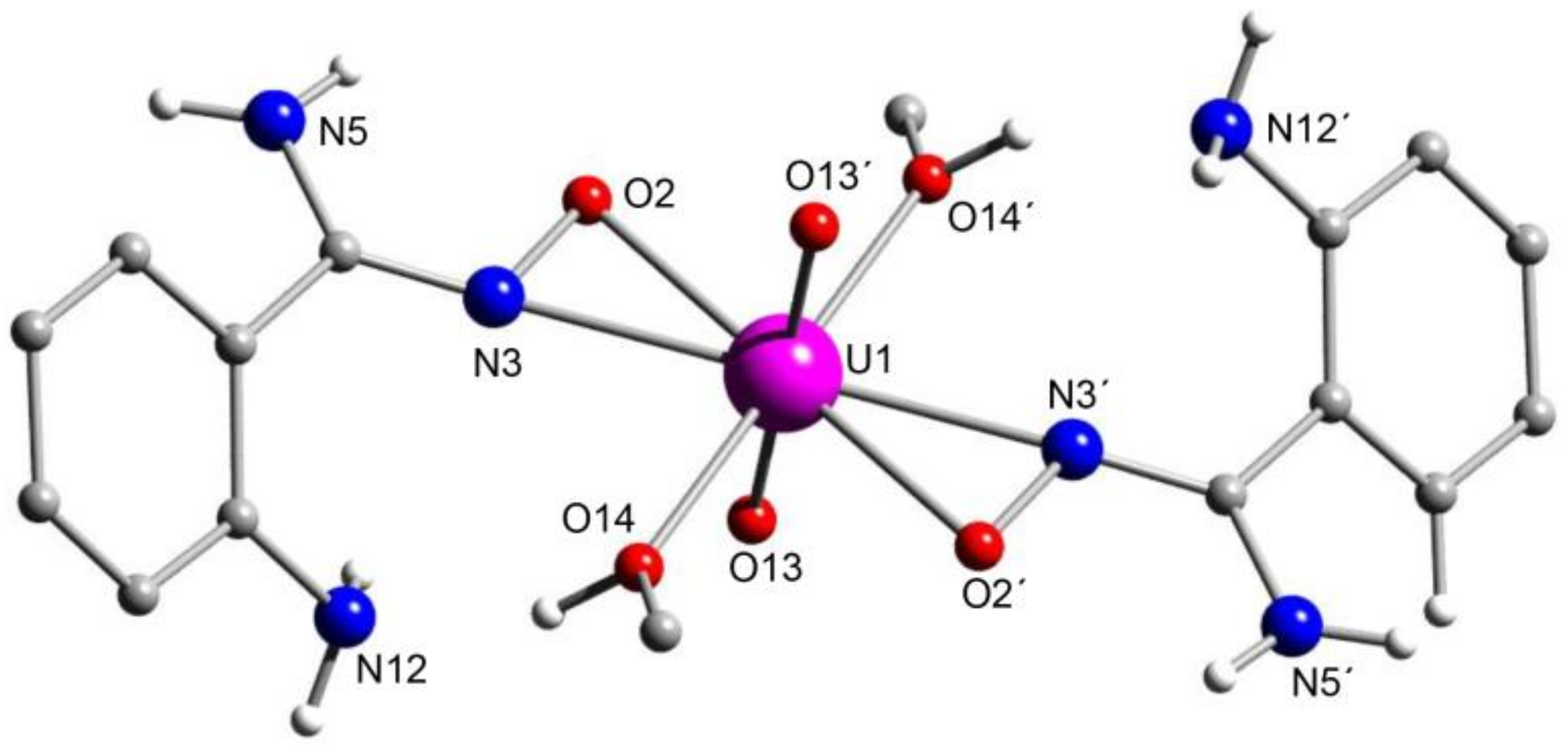
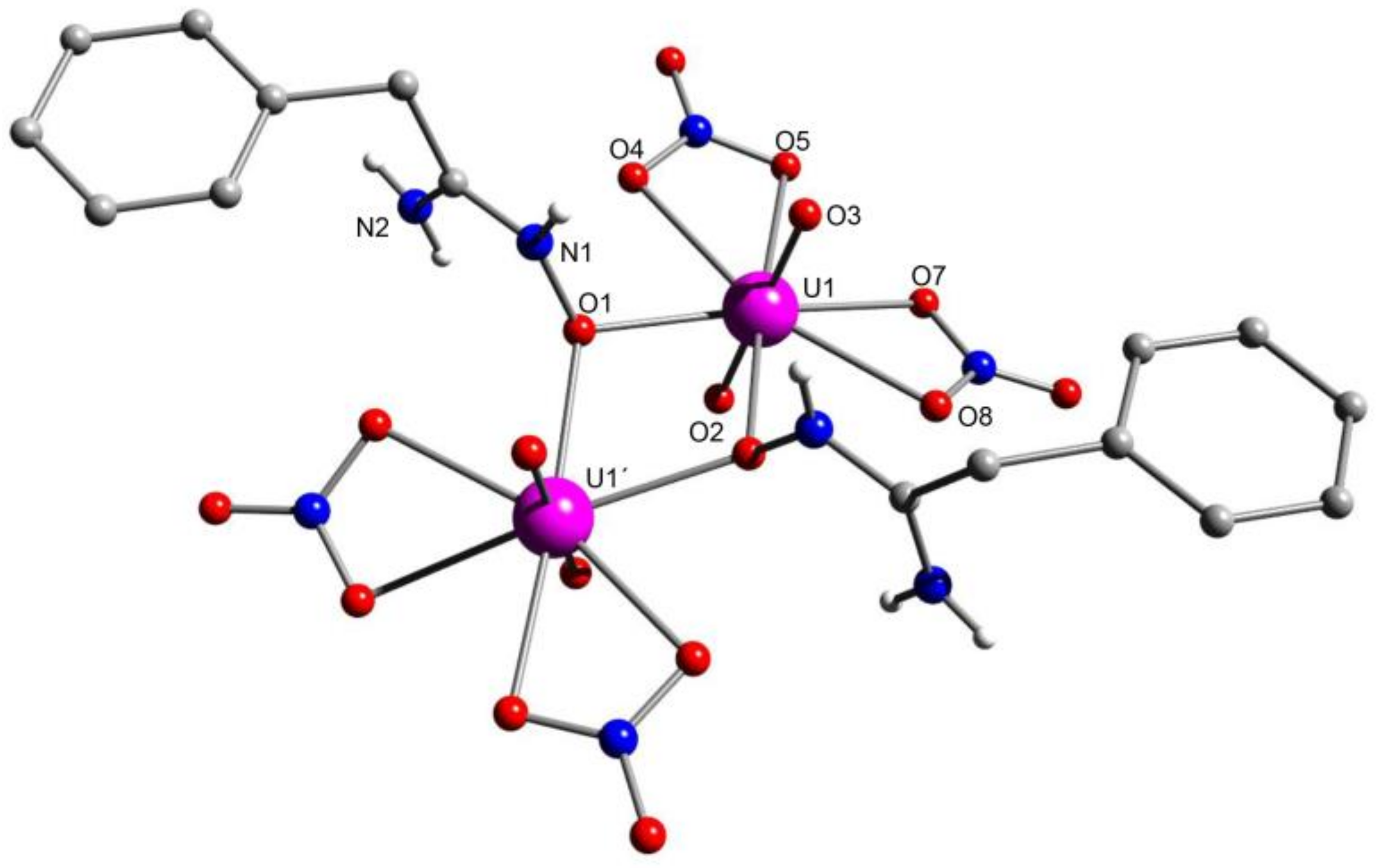
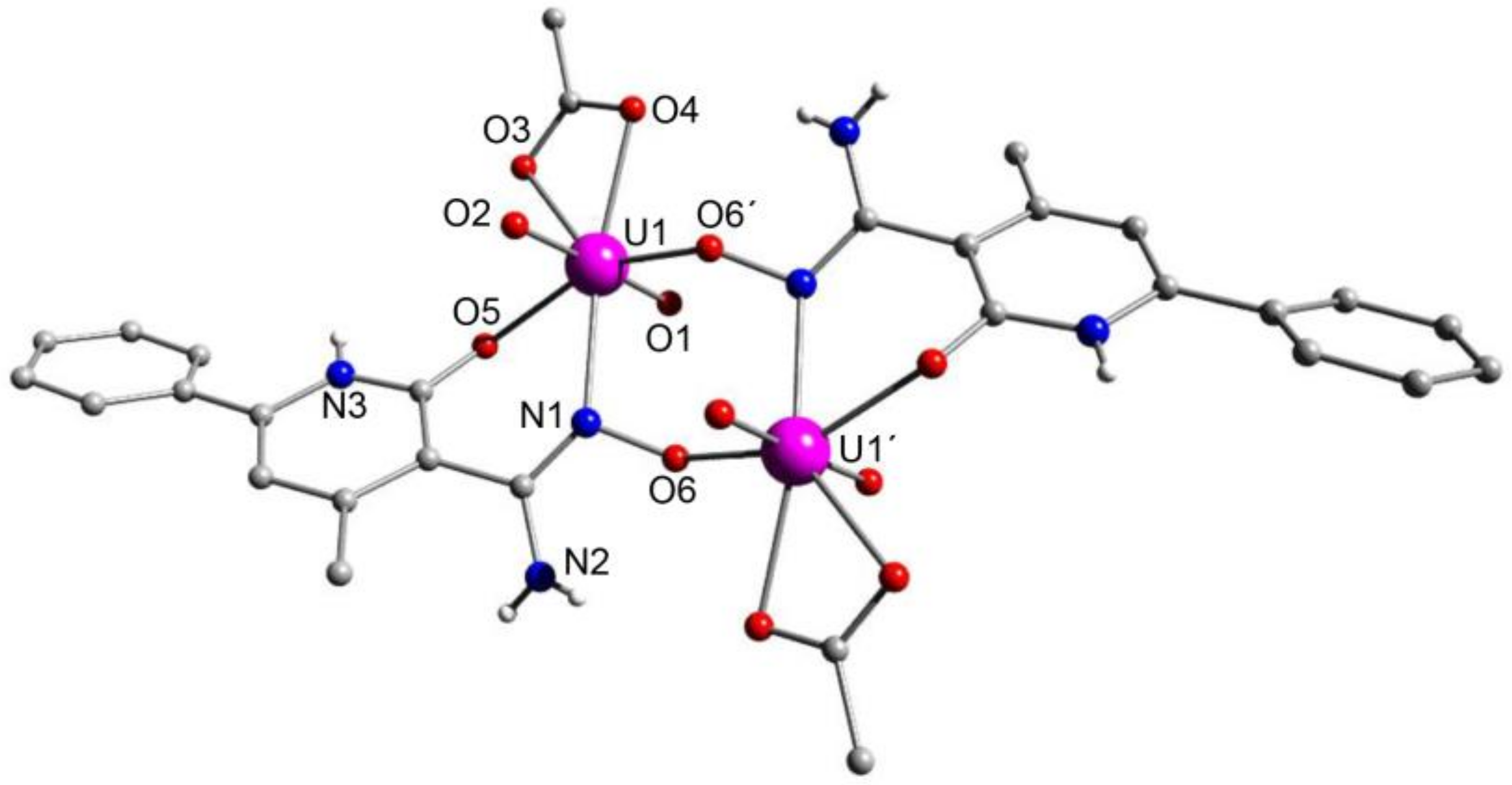
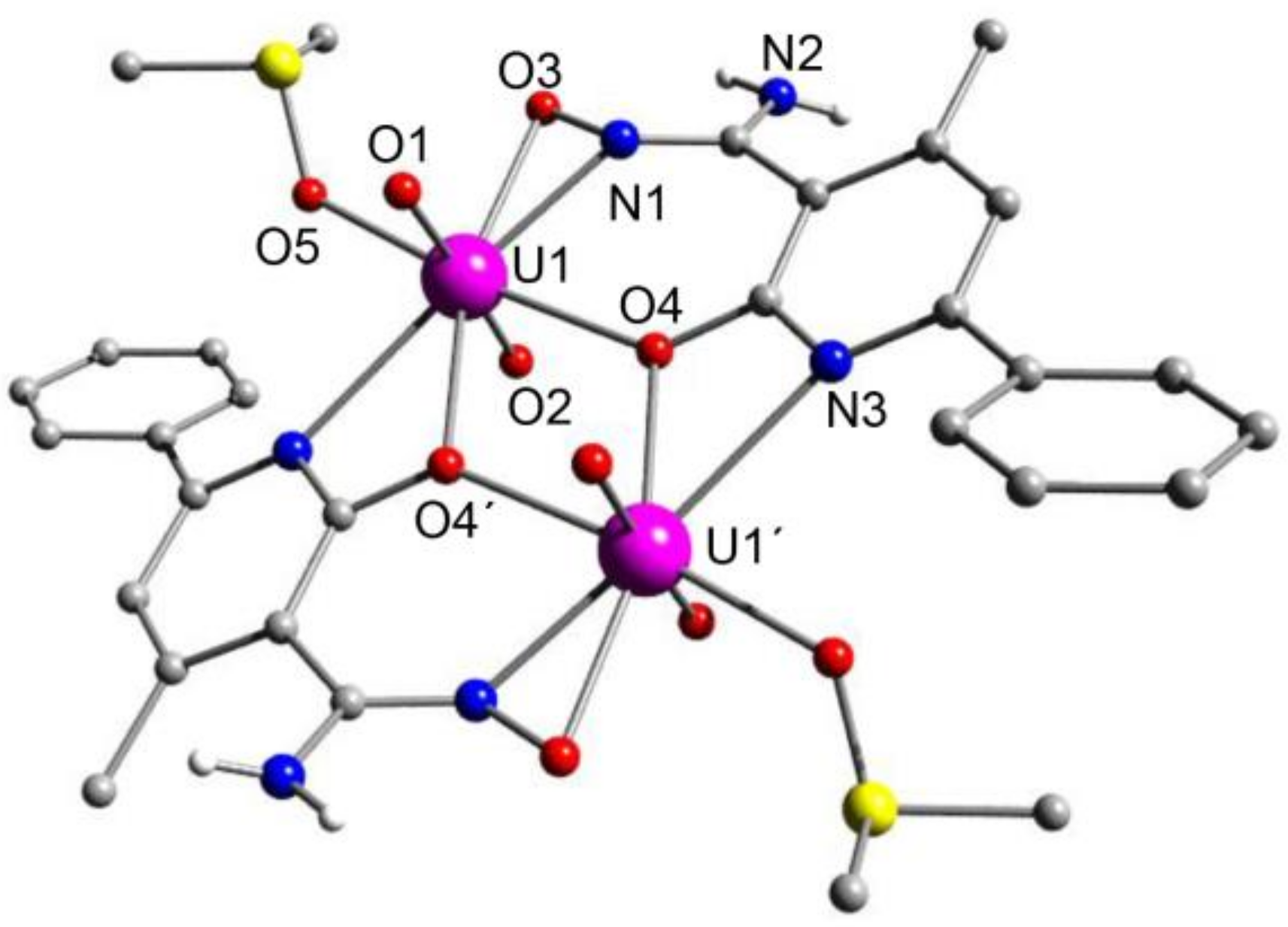
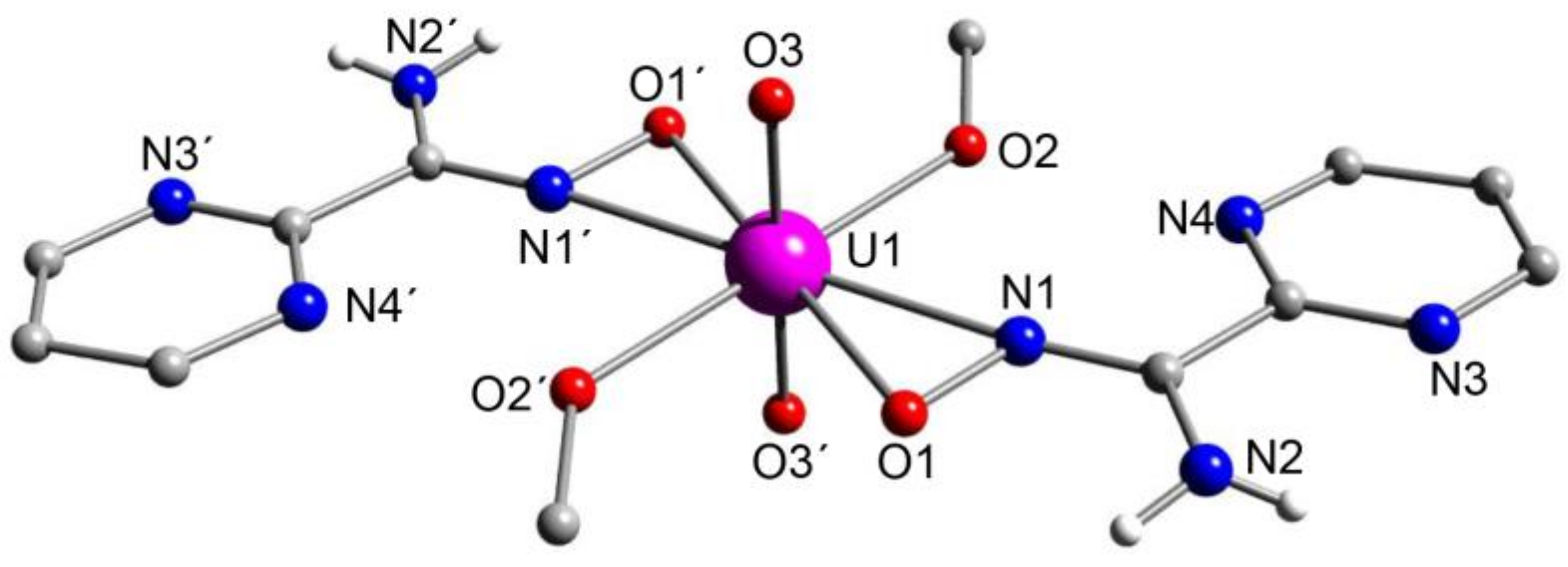
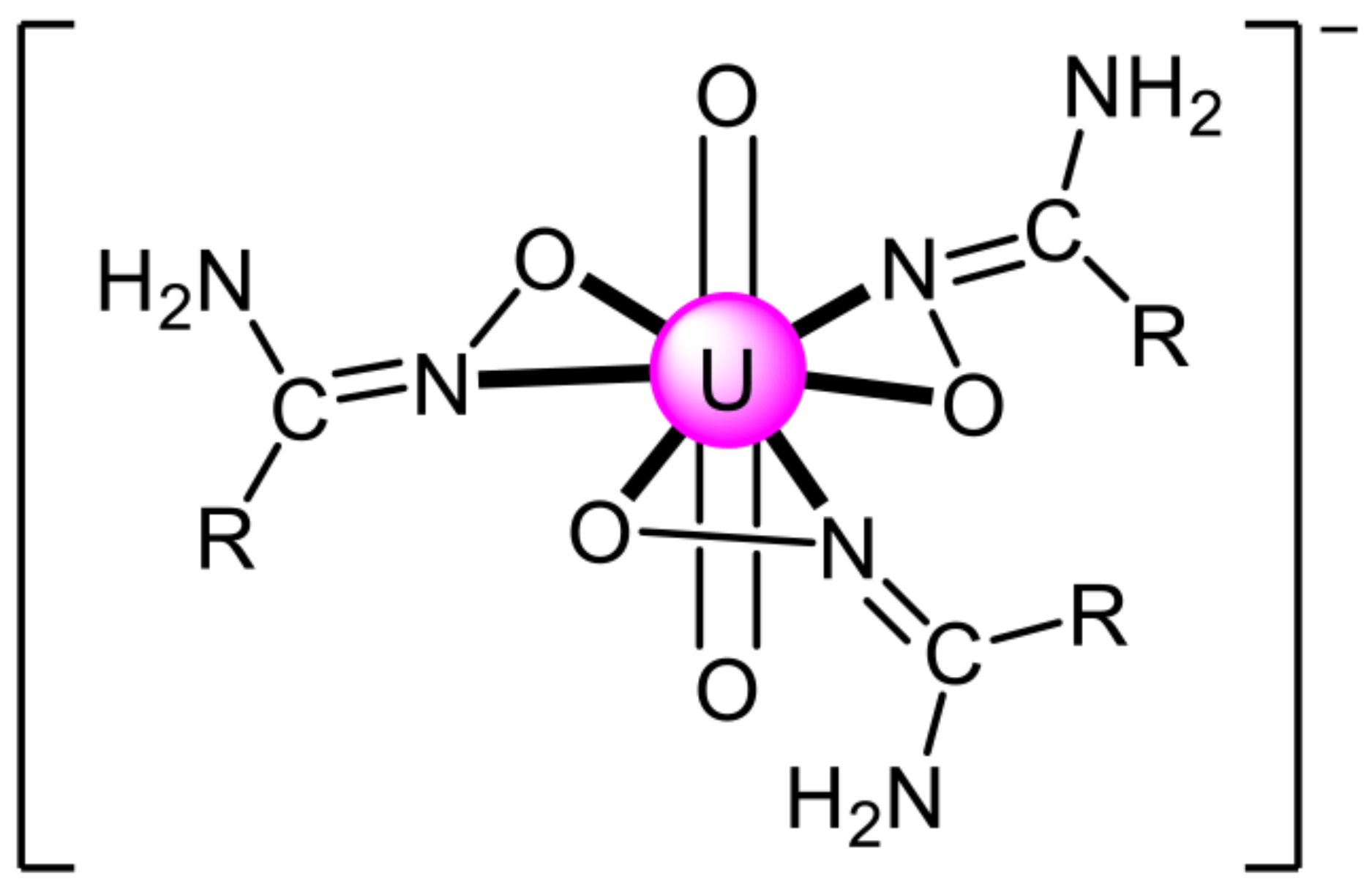
5.8. Uranyl Complexes with New Amidoxime-Based Ligands
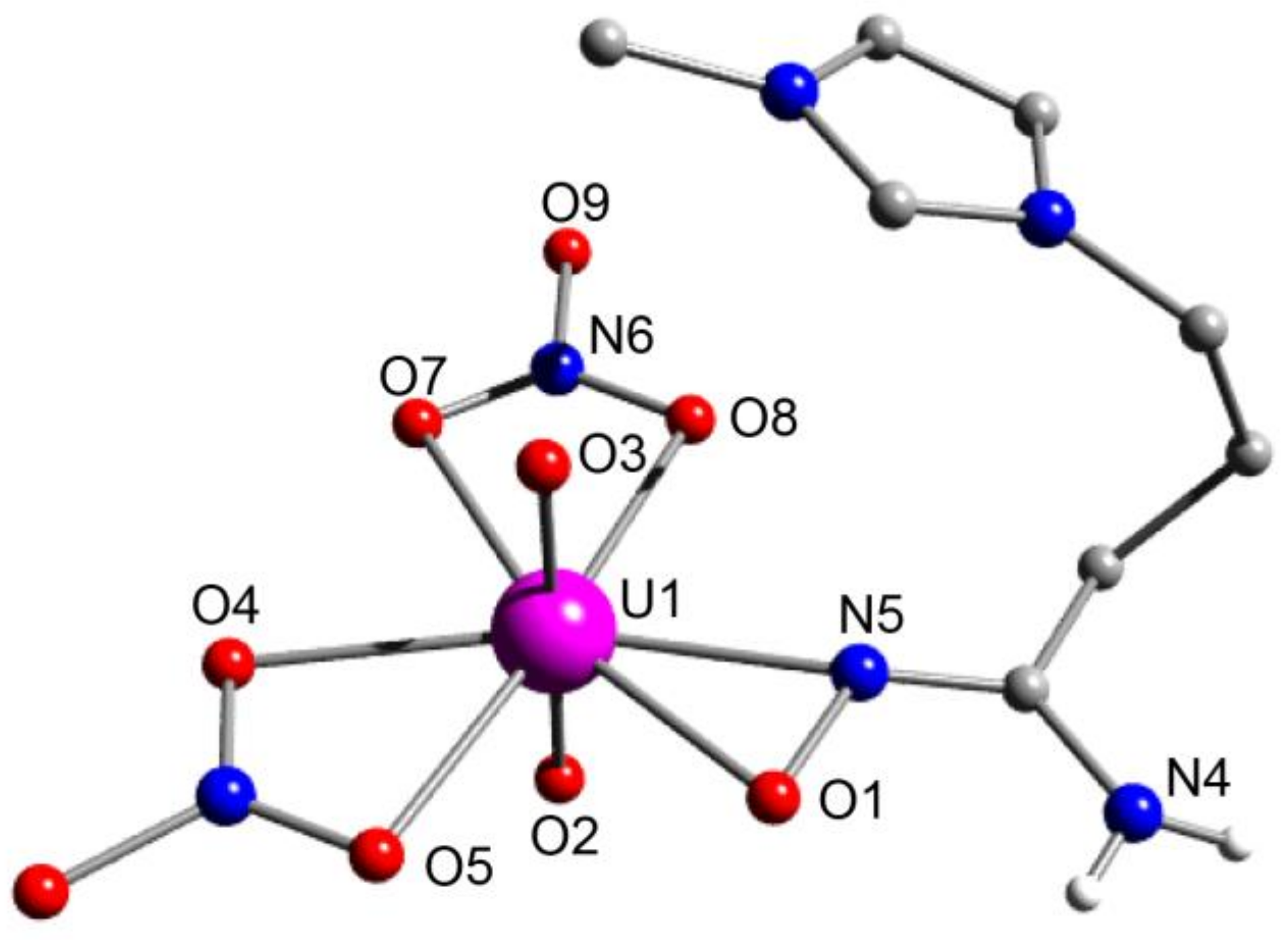
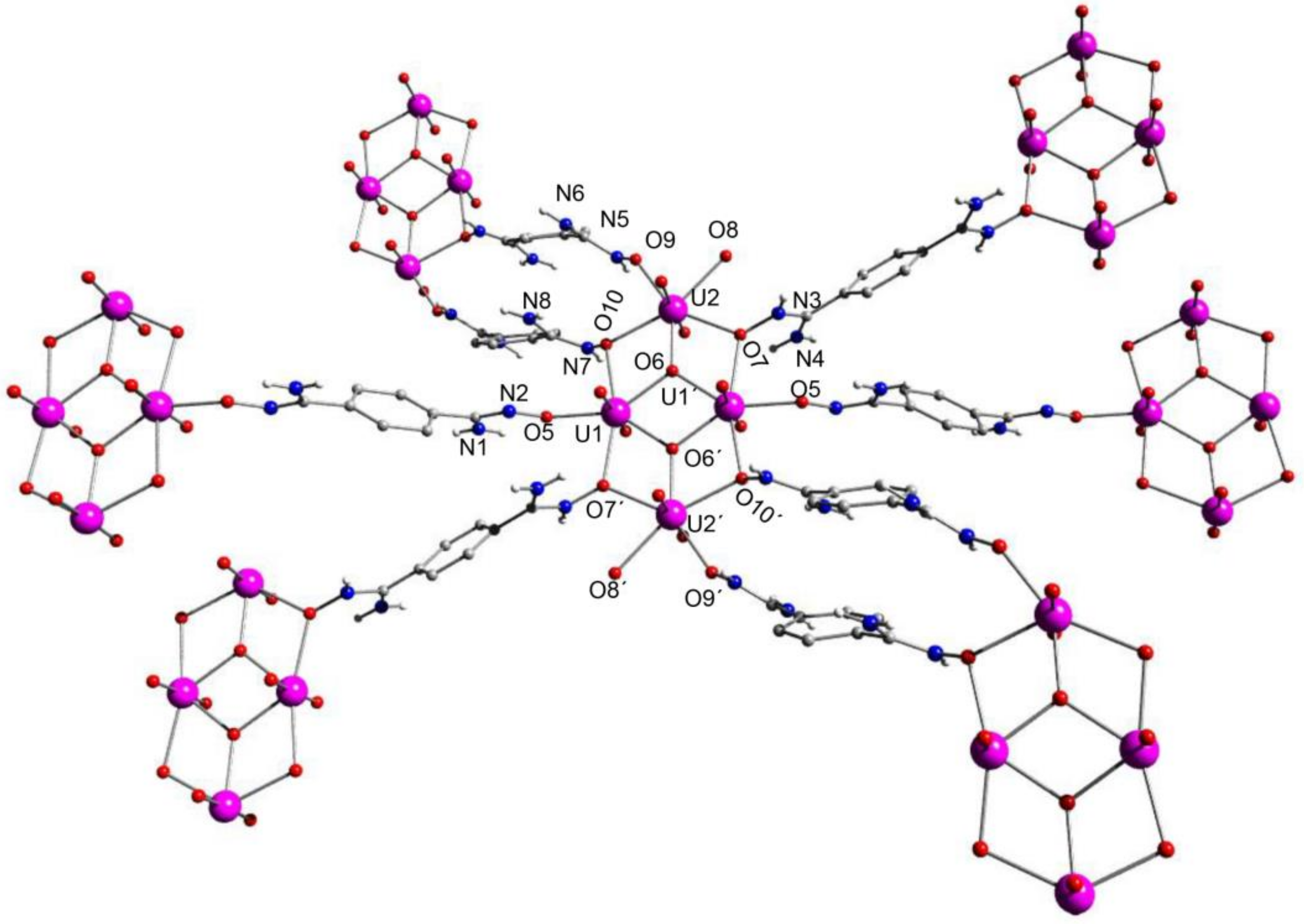
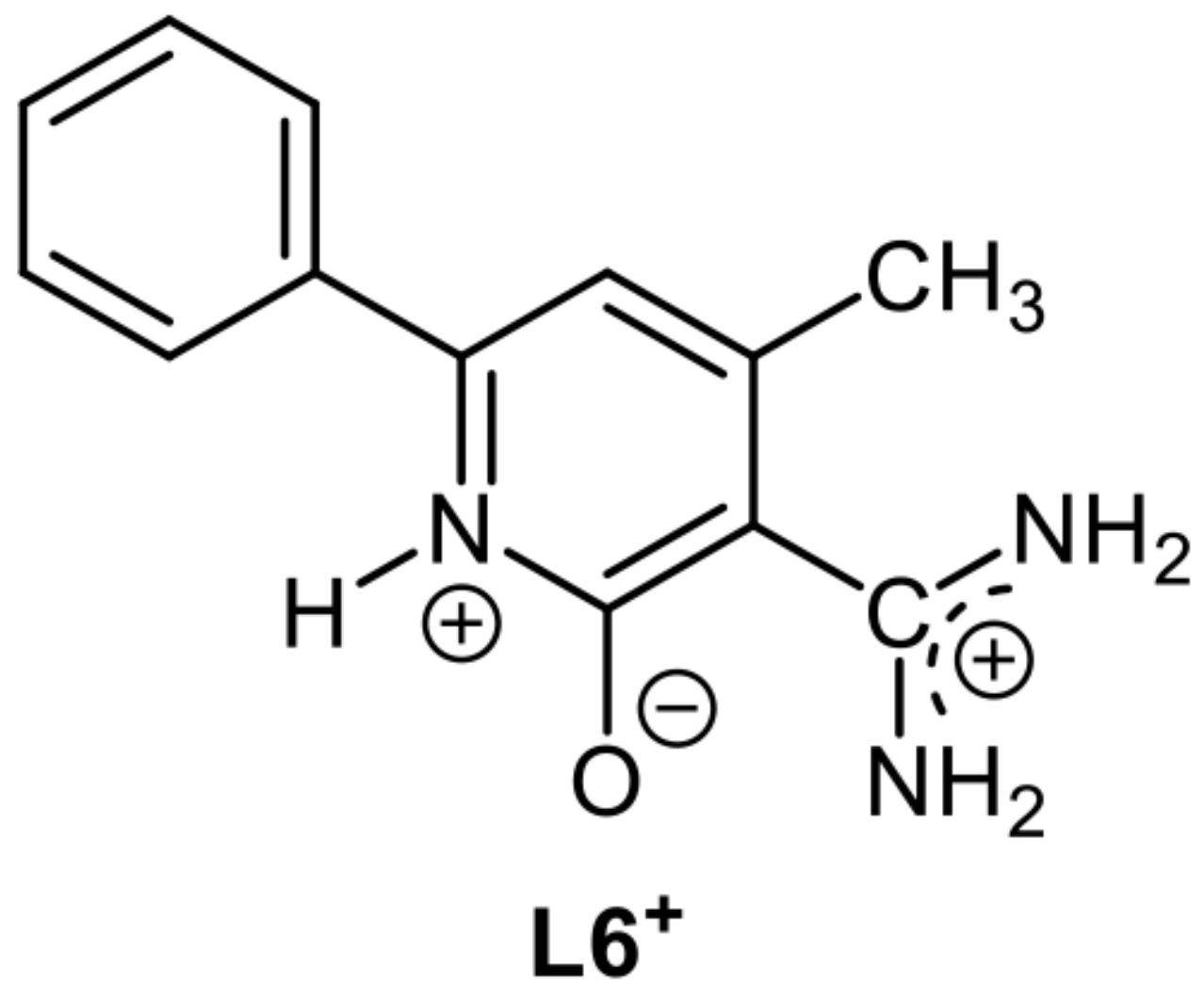
5.9. An overlooked Motif to Uranyl Binding on Poly(Amidoxime) Adsorbents
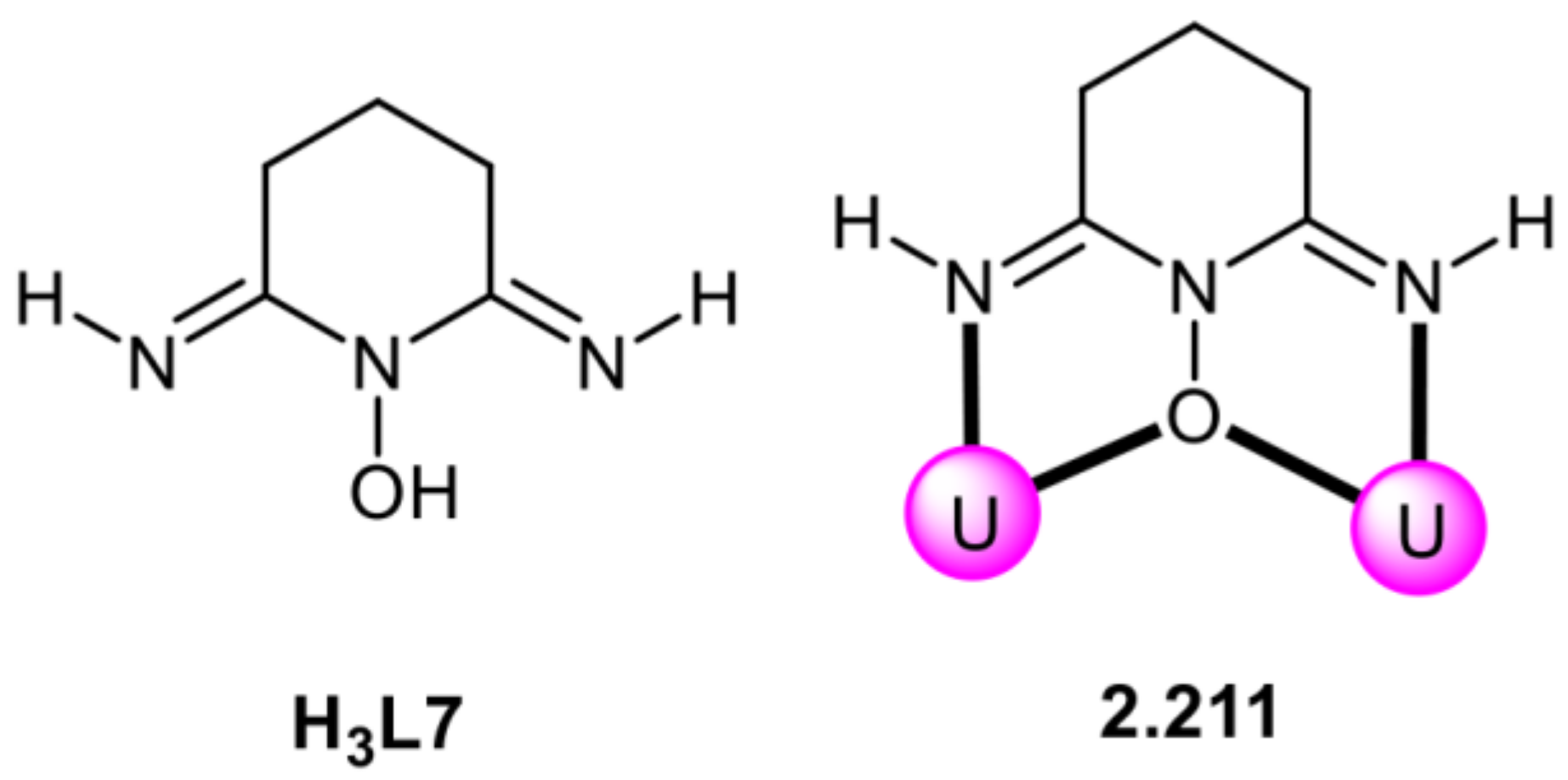
6. EXAFS Studies
7. Concluding Remarks and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liddle, S.T. The Renaissance of Non-Aqueous Uranium Chemistry. Angew. Chem. Int. Ed. 2015, 54, 8604–8641. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Bokach, N.A.; Kukushkin, V.Y. Coordination chemistry and metal-involving reactions of amidoximes: Relevance to the chemistry of oximes and oxime ligands. Coord. Chem. Rev. 2016, 313, 62–93. [Google Scholar] [CrossRef] [Green Version]
- Astheimer, L.; Schenk, H.J.; Witte, E.G.; Schwochau, K. Development of Sorbers for the Recovery of Uranium from Seawater. Part 2. The Accumulation of Uranium from Seawater by Resins Containing Amidoxime and Imidoxime Functional Groups. Sep. Sci. Technol. 1983, 18, 307–339. [Google Scholar] [CrossRef]
- Yue, Y.; Mayes, R.T.; Kim, J.; Fulvio, P.F.; Sun, X.-G.; Tsouris, C.; Chen, J.; Brown, S.; Dai, S. Seawater Uranium Sorbents: Preparation from a Mesoporous Copolymer Initiator by Atom-Transfer Radical Polymerization. Angew. Chem. Int. Ed. 2013, 52, 13458–13462. [Google Scholar] [CrossRef]
- Das, S.; Oyola, Y.; Mayes, R.T.; Janke, C.J.; Kuo, L.-J.; Gill, G.; Wood, J.R.; Dai, S. Extracting Uranium from Seawater: Promising AF Series Adsorbents. Ind. Eng. Chem. Res. 2016, 55, 4110–4117. [Google Scholar] [CrossRef]
- Ladshaw, A.P.; Wiechert, A.I.; Das, S.; Yiacoumi, S.; Tsouris, C. Amidoxime Polymers for Uranium Adsorption: Influence of Comonomers and Temperature. Materials 2017, 10, 1268. [Google Scholar] [CrossRef] [Green Version]
- Dungan, K.; Butler, G.; Livens, F.R.; Warren, L.M. Uranium from seawater-Infinite resource or improbable aspiration? Prog. Nucl. Energy 2017, 99, 81–85. [Google Scholar] [CrossRef]
- Parker, B.F.; Zhang, Z.; Rao, L.; Arnold, J. An overview and recent progress in the chemistry of uranium extraction from seawater. Dalton Trans. 2018, 47, 639–644. [Google Scholar] [CrossRef]
- Tang, N.; Liang, J.; Niu, C.; Wang, H.; Luo, Y.; Xing, W.; Ye, S.; Liang, C.; Guo, H.; Guo, J.; et al. Amidoxime-based materials for uranium recovery and removal. J. Mater. Chem. A 2020, 8, 7588–7625. [Google Scholar] [CrossRef]
- Qin, Z.; Ren, Y.; Shi, S.; Yang, C.; Yu, J.; Wang, S.; Jia, J.; Yu, H.; Wang, X. The enhanced uranyl-amidoxime binding by the electron-donating substituents. RSC Adv. 2017, 7, 18639–18642. [Google Scholar] [CrossRef] [Green Version]
- Endrizzi, F.; Melchior, A.; Tolazzi, M.; Rao, L. Complexation of uranium(VI) with glutarimidoxioxime: Thermodynamic and computational studies. Dalton Trans. 2015, 44, 13835–13844. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Tian, G.; Xu, C.; Rao, L.; Vukovic, S.; Kang, S.O.; Hay, B.P. Quantifying the binding strength of U(VI) with phthalimidedioxime in comparison with glutarimidedioxime. Dalton Trans. 2014, 43, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Abney, C.W.; Liu, S.; Lin, W. Tuning Amidoximate to Enhance Uranyl Binding: A Density Functional Theory Study. J. Phys. Chem. A 2013, 117, 11558–11565. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Pei, S.; Chen, B.; Ye, L.; Yu, H.; Hu, S. Density functional theory investigations on the binding modes of amidoximes with uranyl ions. Dalton Tans. 2016, 45, 3120–3129. [Google Scholar] [CrossRef]
- Vukovich, S.; Hay, B.P. De Novo Structure-Based Design of Bis-amidoxime Uranophiles. Inorg. Chem. 2013, 52, 7805–7810. [Google Scholar] [CrossRef]
- Ladshaw, A.P.; Ivanov, A.S.; Das, S.; Bryantsev, V.S.; Tsouris, C.; Yiacoumi, S. First-Principles Integrated Adsorption Modeling for Selective Capture of Uranium from Seawater by Polyamidoxime Sorbent Materials. ACS Appl. Mater. Interfaces 2018, 10, 12580–12593. [Google Scholar] [CrossRef]
- Priest, C.; Li, B.; Jiang, D.-E. Uranyl-Glutardiamidoxime Binding from First Principles Molecular Dynamics, Classical Molecular Dynamics, and Free-Energy Simulations. Inorg. Chem. 2017, 56, 9497–9504. [Google Scholar] [CrossRef] [PubMed]
- Tsantis, S.T.; Tzimopoulos, D.I.; Holynska, M.; Perlepes, S.P. Oligonuclear Actinoid Complexes with Schiff Bases as Ligands-Older Achievements and Recent Progress. Int. J. Mol. Sci. 2020, 21, 555. [Google Scholar] [CrossRef] [Green Version]
- Tsantis, S.T.; Lada, Z.G.; Tzimopoulos, D.I.; Bekiari, V.; Psycharis, V.; Raptopoulou, C.P.; Perlepes, S.P. Two different coordination modes of the Schiff base derived from ortho-vanillin and 2-(2-aminomethyl)pyridine in a mononuclear uranyl complex. Heliyon 2022, 8, e09705. [Google Scholar] [CrossRef]
- Tsantis, S.T.; Bekiari, V.; Raptopoulou, C.P.; Tzimopoulos, D.I.; Psycharis, V.; Perlepes, S.P. Dioxidouranium(VI) complexes with Schiff bases possessing an ONO donor set: Synthetic, structural and spectroscopic studies. Polyhedron 2018, 152, 172–178. [Google Scholar] [CrossRef]
- Tsantis, S.T.; Danelli, P.; Tzimopoulos, D.I.; Raptopoulou, C.P.; Psycharis, V.; Perlepes, S.P. Pentanuclear Thorium(IV) Coordination Cluster from the Use of Di(2-pyridyl) Ketone. Inorg. Chem. 2021, 60, 11888–11892. [Google Scholar] [CrossRef]
- Tsantis, S.T.; Lagou-Rekka, A.; Konidaris, K.F.; Raptopoulou, C.P.; Bekiari, V.; Psycharis, V.; Perlepes, S.P. Tetranuclear oxido-bridged thorium(IV) clusters obtained using tridentate Schiff bases. Dalton Trans. 2019, 48, 15668–15678. [Google Scholar] [CrossRef]
- Tsantis, S.T.; Zagoraiou, E.; Savvidou, A.; Raptopoulou, C.P.; Psycharis, V.; Szyrwiel, L.; Holynska, M.; Perlepes, S.P. Binding of oxime group to uranyl ion. Dalton Trans. 2016, 45, 9307–9319. [Google Scholar] [CrossRef]
- Tsantis, S.T. Chemistry of Uranyl Complexes and Its Relevance to Topical Technological Aspects. Ph.D. Thesis, University of Patras, Patras, Greece, 2020. [Google Scholar]
- Iliopoulou, M. Efforts to Structurally Model the Uranyl/Vanadium Competition in Seawater Extraction of Uranium Using Amidoxime Adsorbents. Master’s Thesis, University of Patras, Patras, Greece, 2023. [Google Scholar]
- Coxall, R.A.; Harris, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; Winpenny, R.E.P. Inter-ligand reactions: In situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2000, 2349–2356. [Google Scholar] [CrossRef]
- Abney, C.W.; Mayes, R.T.; Saito, T.; Dai, S. Materials for the Recovery of Uranium from Seawater. Chem. Rev. 2017, 117, 13935–14013. [Google Scholar] [CrossRef]
- Burns, C.J.; Neu, M.P.; Boukhalfa, H.; Gutowski, K.E.; Bridges, N.J.; Rogers, R.D. The Actinides. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 3, pp. 189–330. [Google Scholar]
- Boreen, M.A.; Arnold, J. The synthesis and versatile reducing power of low-valent uranium complexes. Dalton Trans. 2020, 49, 15124–15138. [Google Scholar] [CrossRef] [PubMed]
- Barluzzi, L.; Giblin, S.R.; Mansikkamaki, A.; Layfield, R.A. Identification of Oxidation State +1 in a Molecular Uranium Complex. J. Am. Chem. Soc. 2022, 144, 18229–18233. [Google Scholar] [CrossRef]
- Bart, S. Bonding with actinides. Nat. Chem. 2017, 9, 832. [Google Scholar]
- Kindra, D.R.; Evans, W.J. Magnetic susceptibility of uranium complexes. Chem. Rev. 2014, 114, 8865–8882. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, A.; Anderson, N.H.; Tatebe, C.J.; Stanley, D.A.; Zeller, M.; Bart, S.C. Insight into geometric preferences in uranium(VI) mixed tris(imido) systems. Chem. Commun. 2020, 56, 11138–11141. [Google Scholar] [CrossRef]
- Andreychuk, N.R.; Vidjayacoumar, B.; Price, J.S.; Kervazo, S.; Peeples, C.A.; Emslie, D.J.H.; Vallet, V.; Gomes, A.S.P.; Real, F.; Schreckenbach, G.; et al. Uranium(IV) alkyl cations: Synthesis, structures, comparison with thorium(IV) analogues, and the influence of arene-coordination on thermal stability and ethylene polymerization activity. Chem. Sci. 2022, 13, 13748–13763. [Google Scholar] [CrossRef] [PubMed]
- Knecht, S.; Jensen, H.J.A.; Saue, T. Relativistic quantum chemical calculations show that the uranium molecule U2 has a quadruple bond. Nat. Chem. 2018, 11, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Barluzzi, L.; Falcone, M.; Mazzanti, M. Small molecule activation by multimetallic uranium complexes supported by siloxide ligands. Chem. Commun. 2019, 55, 13031–13047. [Google Scholar] [CrossRef]
- Kaltsoyannis, N. Does Covalency Increase or Decrease across the Actinide Series? Implications for Minor Actinide Partitioning. Inorg. Chem. 2013, 52, 3407–3413. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.J.; Harwood, L.M.; Laventine, D.M.; Lewis, F.W. Use of Soft Heterocyclic N-Donor Ligands to Separate Actinides and Lanthanides. Inorg. Chem. 2013, 52, 3414–3428. [Google Scholar] [CrossRef] [PubMed]
- King, D.M.; Cleaves, P.A.; Wooles, A.J.; Gardner, B.M.; Chilton, N.F.; Tuna, F.; Lewis, W.; McInnes, E.J.L.; Liddle, S.T. Molecular and electronic structure of terminal and alkali metal-capped uranium(V) nitride complexes. Nat. Commun. 2016, 7, 13773. [Google Scholar] [CrossRef] [Green Version]
- Meihaus, K.R.; Long, J.R. Actinide-based single-molecule magnets. Dalton Trans. 2015, 44, 2517–2528. [Google Scholar] [CrossRef]
- Mills, D.P.; Moro, F.; McMaster, J.; van Slageren, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. A delocalized arene-bridged diuranium single-molecule magnet. Nat. Chem. 2011, 3, 454–460. [Google Scholar] [CrossRef]
- Dey, S.; Velmurugan, G.; Rajaraman, G. How important is the coordinating atom in controlling magnetic anisotropy in uranium(III) single-ion magnets? A theoretical perspective. Dalton Trans. 2019, 48, 8976–8988. [Google Scholar] [CrossRef]
- Fortier, S.; Hayton, T.W. Oxo ligand functionalization in the uranyl ion (UO22+). Coord. Chem. Rev. 2010, 254, 197–214. [Google Scholar] [CrossRef]
- Cowie, B.E.; Purkis, J.M.; Austin, J.; Love, J.B.; Arnold, P.L. Thermal and Photochemical Reduction and Functionalization Chemistry of the Uranyl Dication, [UVIO2]2+. Chem. Rev. 2019, 119, 10595–10637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, J.; Chen, F.-Y.; Gao, C.-Y.; Tian, H.-R.; Pan, Q.-J.; Sun, Z.-M. Porous anionic uranyl-organic networks for highly efficient Cs+ adsorption and investigation of the mechanism. Inorg. Chem. 2018, 57, 4419–4426. [Google Scholar] [CrossRef] [PubMed]
- Rudkevich, D.M.; Verboom, W.; Brzozka, A.; Palys, M.J.; Stauthamer, W.P.R.V.; van Hummel, G.J.; Franken, S.M.; Harkema, S.; Engbersen, J.F.J.; Reinhoudt, D.N. Functionalized UO2 salenes: Neutral receptor for anions. J. Am. Chem. Soc. 1994, 116, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Odoh, S.O.; Bondarevsky, G.D.; Karpus, J.; Cui, Q.; He, C.; Spezia, R.; Gagliardi, L. UO22+ uptake by proteins: Understanding the binding features of the super uranyl binding protein and design of a protein with higher affinity. J. Am. Chem. Soc. 2014, 136, 17484–17494. [Google Scholar] [CrossRef]
- Xu, M.; Eckard, P.; Burns, P.C. Organic Functionalization of Uranyl Peroxide Clusters to Impact Solubility. Inorg. Chem. 2020, 59, 9881–9888. [Google Scholar] [CrossRef] [PubMed]
- Gutowski, K.E.; Cocalia, V.A.; Griffin, S.T.; Bridges, N.J.; Dixon, D.A.; Rogers, R.D. Interactions of 1-methylimidazole with UO2(CH3CO2)2 and UO2(NO3)2: Structural, spectroscopic, and theoretical evidence for imidazole binding to the uranyl ion. J. Am. Chem. Soc. 2007, 129, 526–536. [Google Scholar] [CrossRef]
- Tsipis, A.C. cis- and trans-Ligand Effects of the Inverse trans-Influence in [UVI(O)(L)Cl4]0/− (L=Unidentate Ligand) Complexes. Inorg. Chem. 2020, 59, 8946–8959. [Google Scholar] [CrossRef]
- Harrowfield, J.; Thuery, P. Uranyl Ion Complexes of Polycarboxylates: Steps towards Isolated Photoactive Cavities. Chemistry 2020, 2, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Costisor, O.; Linert, W. 4f and 5f metal ion directed Schiff condensation. Rev. Inorg. Chem. 2004, 24, 61–95. [Google Scholar] [CrossRef]
- Van Axel Castelli, V.; Cort Dalla, A.; Mandolini, L. Supramolecular catalysis of 1,4-thiol addition by salophen-uranyl complexes. J. Am. Chem. Soc. 1998, 120, 12688–12689. [Google Scholar] [CrossRef]
- Hawkins, C.A.; Bustillos, C.G.; Copping, R.; Scott, B.L.; May, I.; Nilsson, M. Challenging conventional f-element separation chemistry-reversing uranyl(VI)/lanthanide(III) solvent extraction selectivity. Chem. Commun. 2014, 50, 8670–8673. [Google Scholar] [CrossRef] [Green Version]
- Weng, Z.; Zhang, Z.-h.; Olds, T.; Sterniczuk, M.; Burns, P.C. Copper(I) and Copper(II) Uranyl Heterometallic Hybrid Materials. Inorg. Chem. 2014, 53, 7993–7998. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Y.; Silver, M.A.; Liu, W.; Duan, T.; Yin, X.; Chen, L.; Diwu, J.; Chai, Z.; Wang, S. Tunable 4f/5f Bimodal Emission in Europium-Incorporated Uranyl Coordination Polymers. Inorg. Chem. 2018, 57, 575–582. [Google Scholar] [CrossRef]
- Behera, N.; Sethi, S. Unprecedented Catalytic Behavior of Uranyl(VI) Compounds in Chemical Reactions. Eur. J. Inorg. Chem. 2021, 2021, 95–111. [Google Scholar] [CrossRef]
- Sahyoun, T.; Arrault, A.; Schneider, R. Amidoximes and Oximes: Synthesis, Structure, and Their Key Role as NO Donors. Molecules 2019, 24, 2470. [Google Scholar] [CrossRef] [Green Version]
- Efthymiou, C.G.; Cunha-Silva, L.; Perlepes, S.P.; Brechin, E.K.; Inglis, R.; Evangelisti, M.; Papatriantafyllopoulou, C. In search of molecules displaying ferromagnetic exchange: Multiple-decker Ni12 and Ni16 complexes from the use of pyridine-2-amidoxime. Dalton Trans. 2016, 45, 17409–17419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
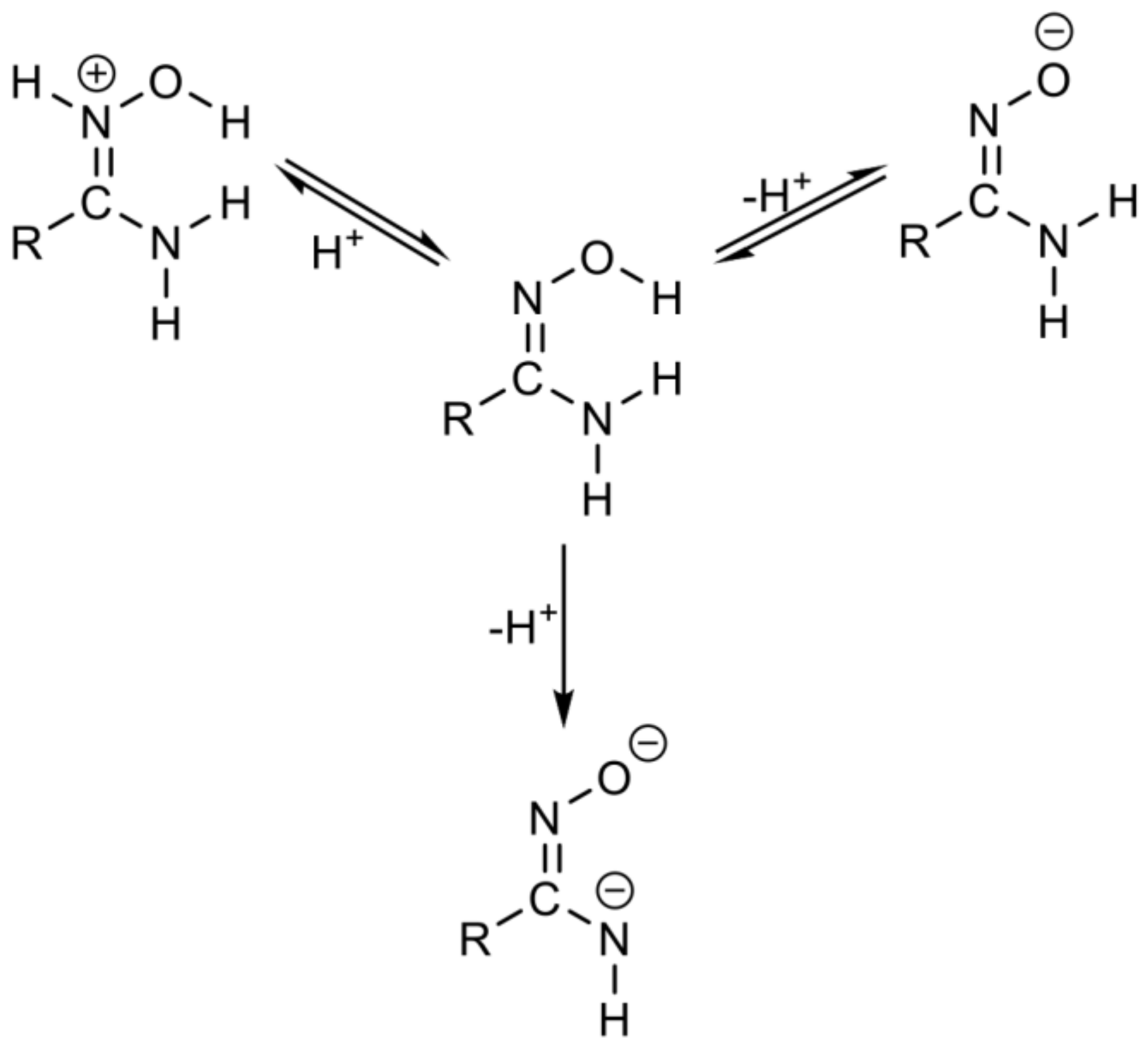
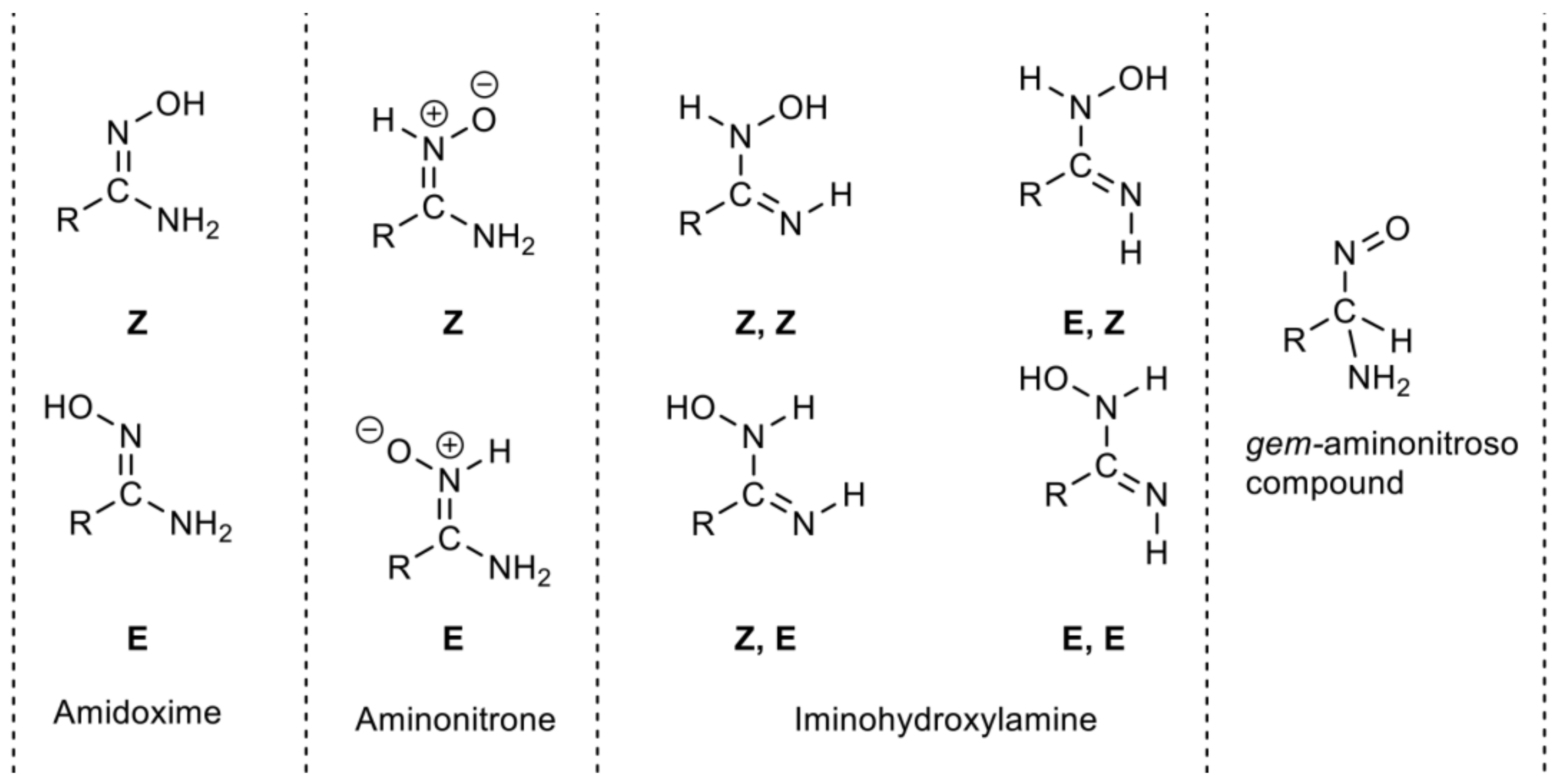
- Novikov, A.S.; Bolotin, D.S. Tautomerism of amidoximes and other oximes species. J. Phys. Org. Chem. 2018, 31, e3772. [Google Scholar] [CrossRef]
- Tavakol, H.; Arshadi, S. Theoretical investigation of tautomerism in N-hydroxy amidines. J. Mol. Model. 2009, 15, 807–816. [Google Scholar] [CrossRef] [PubMed]
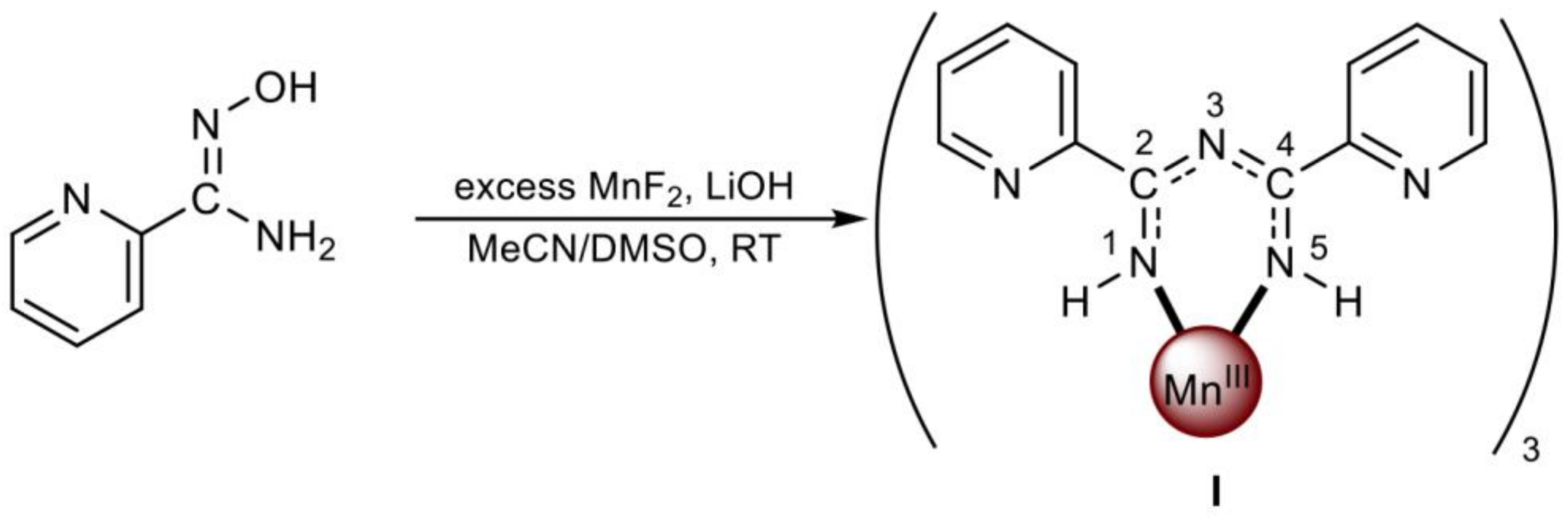
- Duros, V.; Sartzi, H.; Teat, S.J.; Sanakis, Y.; Roubeau, O.; Perlepes, S.P. Tris{2,4-bis(2-pyridyl)-1,3,5-triazapentanedienato}manganese(III), a complex derived from a unique metal ion-assisted transformation of pyridine-2-amidoxime. Inorg. Chem. Commun. 2014, 50, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Tsouris, C. Fuel from seawater. Nat. Energy 2017, 2, 17022. [Google Scholar] [CrossRef]
- Das, S.; Oyola, Y.; Mayes, R.T.; Janke, C.J.; Kuo, L.-J.; Gill, G.; Wood, J.R.; Dai, S. Extracting Uranium from Seawater: Promising AI Series Adsorbents. Ind. Eng. Chem. Res. 2016, 55, 4103–4109. [Google Scholar] [CrossRef]
- Gill, G.A.; Kuo, L.-J.; Janke, C.J.; Park, J.; Jeters, R.T.; Bonheyo, G.T.; Pan, H.-B.; Wai, C.; Khangaonkar, T.; Bianucci, L.; et al. The Uranium from Seawater Program at the Pacific Northwest National Laboratory: Overview of Marine Testing, Adsorbent Characterization, Adsorbent Durability, Adsorbent Toxicity, and Deployment Studies. Ind. Eng. Chem. Res. 2016, 55, 4264–4277. [Google Scholar] [CrossRef]
- Davies, R.V.; Kennedy, J.; McIlroy, R.W.; Spence, R.; Hill, K.M. Extraction of Uranium from Sea Water. Nature 1964, 203, 1110–1115. [Google Scholar] [CrossRef]
- Schenk, H.J.; Astheimer, L.; Witte, E.G.; Schwochau, K. Development of Sorbers for the Recovery of Uranium from Seawater. 1. Assessment of Key Parameters and Screening Studies of Sorber Materials. Sep. Sci. Technol. 1982, 17, 1293–1308. [Google Scholar] [CrossRef]
- Yue, Y.; Mayes, R.T.; Gill, G.; Kuo, L.-J.; Wood, J.; Binder, A.; Brown, S.; Dai, S. Macroporous monoliths for trace metal extraction from seawater. RSC Adv. 2015, 5, 50005–50010. [Google Scholar] [CrossRef]
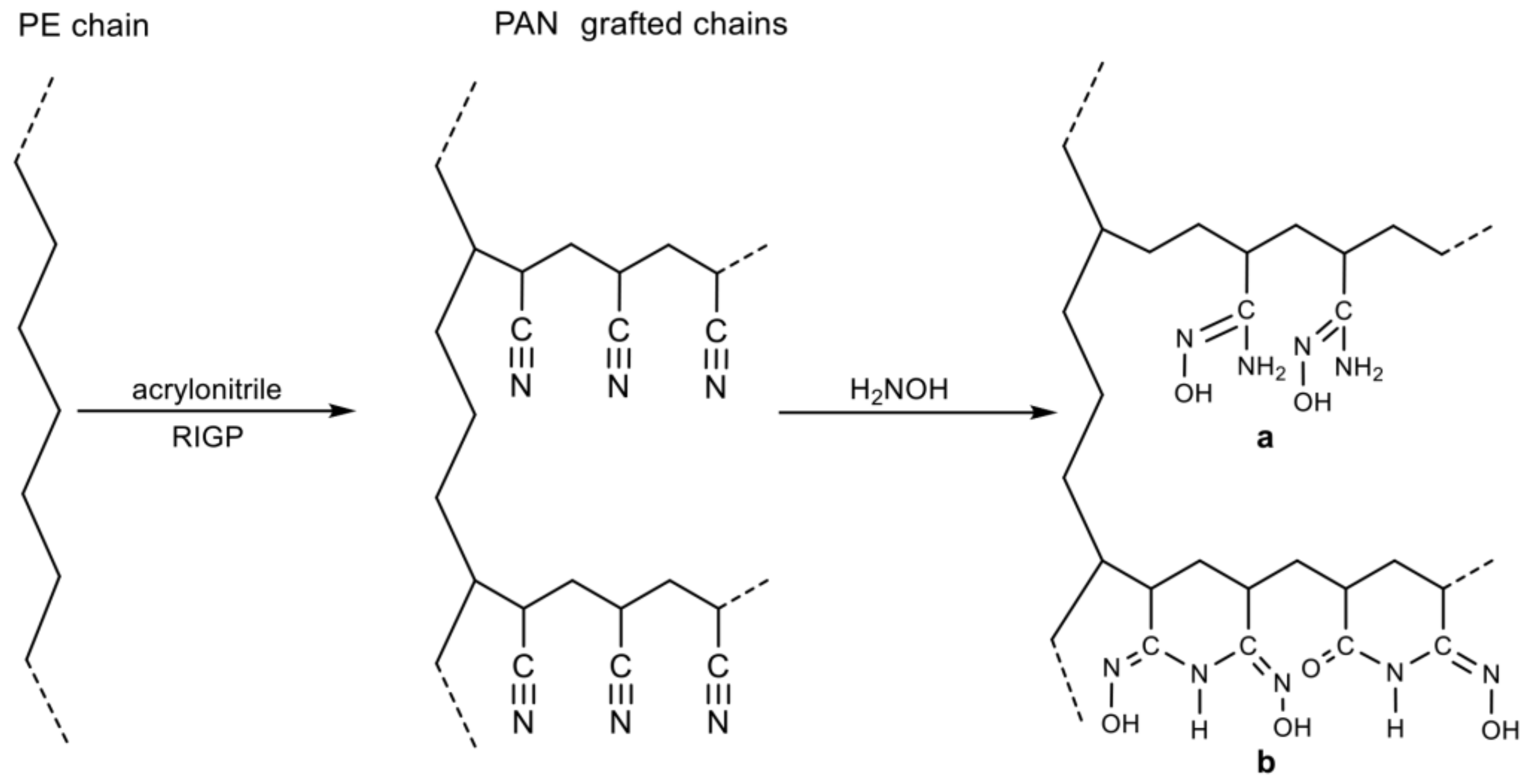
- Hu, J.; Ma, H.; Xing, Z.; Liu, X.; Xu, L.; Li, R.; Lin, C.; Wang, M.; Li, J.; Wu, G. Preparation of Amidoximated Ultrahigh Molecular Weight Polyethylene Fiber by Radiation Grafting and Uranium Adsorption Test. Ind. Eng. Chem. Res. 2016, 55, 4118–4124. [Google Scholar] [CrossRef]
- Mehio, N.; Lashely, M.A.; Nugent, J.W.; Tucker, L.; Correia, B.; Do-Thanh, C.-L.; Dai, S.; Hancock, R.D.; Bryantsev, V.S. Acidity of the Amidoxime Functional Group in Aqueous Solution: A Combined Experimental and Computational Study. J. Phys. Chem. B 2015, 119, 3567–3576. [Google Scholar] [CrossRef]
- Lashley, M.; Mehio, N.; Nugent, J.W.; Holguin, E.; Do-Thanh, C.-L.; Bryantsev, V.S.; Dai, S.; Hancock, R.D. Amidoximes as ligand functionalities for braided polymeric materials for the recovery of uranium from seawater. Polyhedron 2016, 109, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Aguila, B.; Sun, Q.; Cassady, H.; Abney, C.W.; Li, B.; Ma, S. Design Strategies to Enhance Amidoxime Chelators for Uranium Recovery. ACS Appl. Mater. Interfaces 2019, 11, 30919–30926. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, Y.; Liu, X.; Li, Y.; Wang, J.; Chen, Z.; Yang, H.; Hu, B.; Chen, C.; Tang, Z.; et al. Functional nanomaterials for selective uranium recovery from seawater: Material design, extraction properties and mechanisms. Coord. Chem. Rev. 2023, 483, 215097. [Google Scholar] [CrossRef]
- Liu, C.; Hsu, P.-C.; Xie, J.; Zhao, J.; Wu, T.; Wang, H.; Liu, W.; Zhang, J.; Chu, S.; Cui, L. A half-wave rectified alternating current electrochemical method for uranium extraction from seawater. Nat. Energy 2017, 2, 17007. [Google Scholar] [CrossRef]
- Witte, E.G.; Schowochau, K.S.; Henkel, G.; Krebs, B. Uranyl Complexes of Acetamidoxime and Benzamidoxime. Preparation, Characterization, and Crystal Structure. Inorg. Chim. Acta 1984, 94, 323–331. [Google Scholar] [CrossRef]
- Kravchuk, D.V.; Diaz, A.B.; Carolan, M.E.; Mpundu, E.A.; Cwiertny, D.M.; Forbes, T.Z. Uranyl Speciation on the Surface of the Amidoximated Polyacrylonitrile Mats. Inorg. Chem. 2020, 59, 8134–8145. [Google Scholar] [CrossRef]
- Vukovic, S.; Watson, L.A.; Kang, S.O.; Custelcean, R.; Hay, B.P. How Amidoximate Binds the Uranyl Cation. Inorg. Chem. 2012, 51, 3855–3859. [Google Scholar] [CrossRef] [PubMed]
- Barber, P.S.; Kelley, S.P.; Rogers, R.D. Highly selective extraction of the uranyl ion with hydrophobic amidoxime-functionalized ionic liquids via η2 coordination. RSC Adv. 2012, 2, 8526–8530. [Google Scholar] [CrossRef]
- Tian, G.; Teat, S.J.; Zhang, Z.; Rao, L. Sequestering uranium from seawater: Binding strength and modes of uranyl complexes with glutarimidedioxime. Dalton Trans. 2012, 41, 11579–11586. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, K.J.; Do-Thanh, C.-L.; Penchoff, D.A.; Cramer, S.A.; Murdock, C.R.; Lu, Z.; Harrison, R.J.; Camden, J.P.; Jenkins, D.M. The synthesis and spectroscopic characterization of an aromatic uranium amidoxime complex. Inorg. Chim. Acta 2014, 421, 374–379. [Google Scholar] [CrossRef]
- Kelley, S.P.; Barber, P.S.; Mullins, P.H.K.; Rogers, R.D. Structural clues to UO22+/VO2+ competition in seawater using amidoxime-based extractants. Chem. Commun. 2014, 50, 12504–12507. [Google Scholar] [CrossRef]
- Sun, Q.; Aguila, B.; Perman, J.; Ivanov, A.S.; Bryantsev, V.S.; Earl, L.D.; Abney, C.W.; Wojtas, L.; Ma, S. Bio-inspired nano-traps for uranium extraction from seawater and recovery from nuclear waste. Nat. Commun. 2018, 9, 1644. [Google Scholar] [CrossRef] [Green Version]
- Mishra, M.K.; Patil, V.; Kelley, S.P.; Rogers, R.D. A Uranyl Metal Organic Framework Arising from the Coordination of a Partially Hydrolyzed Tetrauranyl Mode with the Tautomerically Diverse 1,4-(diamidoximyl)benzene Ligand. Crystal Growth Des. 2019, 19, 5466–5470. [Google Scholar] [CrossRef]
- Decato, D.A.; Berryman, O.B. Structural and computational characterization of a bridging zwitterionic-amidoxime uranyl complex. Org. Chem. Front. 2019, 6, 1038–1043. [Google Scholar] [CrossRef]
- Parker, B.F.; Hohloch, S.; Pankhurst, J.R.; Zhang, Z.; Love, J.B.; Arnold, J.; Rao, L. Interactions of vanadium ligands: Redox activity. Dalton Trans. 2018, 47, 5695–5702. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Teat, S.J.; Rao, L. Thermodynamic studies of U(VI) complexation with glutardiamidoxime for sequestration of uranium from seawater. Dalton Trans. 2013, 42, 5690–5696. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.S.; Leggett, C.J.; Parker, B.F.; Zhang, Z.; Arnold, J.; Dai, S.; Abney, C.W.; Bryantsev, V.S.; Rao, L. Origin of the unusually strong and selective binding of vanadium by polyamidoximes in seawater. Nat. Commun. 2017, 8, 1560. [Google Scholar] [CrossRef] [Green Version]
- Mehio, N.; Ivanov, A.S.; Ladshaw, A.P.; Dai, S.; Bryanstev, V.S. Theoretical Study of Oxovanadium(IV) complexation with formamidoximate: Implications for the Design of Uranyl-Selective Adsorbents. Ind. Eng. Chem. Res. 2016, 55, 4231–4240. [Google Scholar] [CrossRef]
- Mehio, N.; Johnson, J.C.; Dai, S.; Bryantsev, V.S. Theoretical study of the coordination behavior of formate and formamidoximate with dioxovanadium(V) cation: Implications for selectivity towards uranyl. Phys. Chem. Chem. Phys. 2015, 17, 31715–31726. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.F.; Zhang, Z.; Leggett, C.J.; Arnold, J.; Rao, L. Kinetics of complexation of V(V), U(VI) and Fe(III) with glutaroimide-dioxime: Studies by stopped-flow and conventional adsorption spectroscopy. Dalton Trans. 2017, 46, 11084–11096. [Google Scholar] [CrossRef]
- Ivanov, A.S.; Bryantsev, V.S. Assessing ligand selectivity for uranium over vanadium ions to aid in the discovery of superior adsorbents for extraction of UO22+ from seawater. Dalton Trans. 2016, 45, 10744–10751. [Google Scholar] [CrossRef]
- Kennedy, Z.C.; Cardenas, A.J.P.; Corbey, J.F.; Warner, M.G. 2,6-Diiminopiperidin-1-ol: An overlooked motif relevant to uranyl and transition metal binding on poly(amidoxime) adsorbents. Chem. Commun. 2016, 52, 8802–8805. [Google Scholar] [CrossRef]
- Abney, C.W.; Mayes, R.T.; Piechowicz, M.; Lin, Z.; Bryantsev, V.S.; Veith, G.M.; Dai, S.; Lin, W. XAFS investigation of polyamidoxime-bound uranyl contests the paradigm from small molecule studies. Energy Environ. Sci. 2016, 9, 448–453. [Google Scholar] [CrossRef]
- Zhang, L.; Qie, M.; Su, J.; Zhang, S.; Zhou, J.; Li, J.; Wang, Y.; Yang, S.; Wang, S.; Li, J.; et al. Tris-amidoximate uranyl via η2 binding mode coordinated in aqueous solution shown by X-ray absorption spectroscopy and density functional theory methods. J. Synchrotron Rad. 2018, 25, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Endrizzi, F.; Rao, L. Chemical Speciation of Uranium(VI) in Marine Environments: Complexation of Calcium and Magnesium Ions with [(UO2)(CO3)3]4− and the Effect on the Extraction of Uranium from Seawater. Chem. Eur. J. 2014, 20, 14499–14506. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Priest, C.; Jiang, D.-E. Displacement of carbonates in Ca2UO2(CO3)3 by amidoxime-based ligands from free-energy simulations. Dalton Trans. 2018, 47, 1604–1613. [Google Scholar] [CrossRef]
- Priest, C.; Li, B.; Jiang, D.-E. Understanding the Binding of a Bifunctional Amidoximate-Carboxylate Ligand with Uranyl in Seawater. J. Phys. Chem. B 2018, 122, 12060–12066. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Z.; Lan, J.-H.; Wu, Q.-Y.; Luo, Q.; Zhao, Y.-L.; Wang, X.-K.; Chai, Z.-F.; Shi, W.-Q. Theoretical Insights on the Interaction of Uranium with Amidoxime and Carboxyl Groups. Inorg. Chem. 2014, 53, 9466–9476. [Google Scholar] [CrossRef]




































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
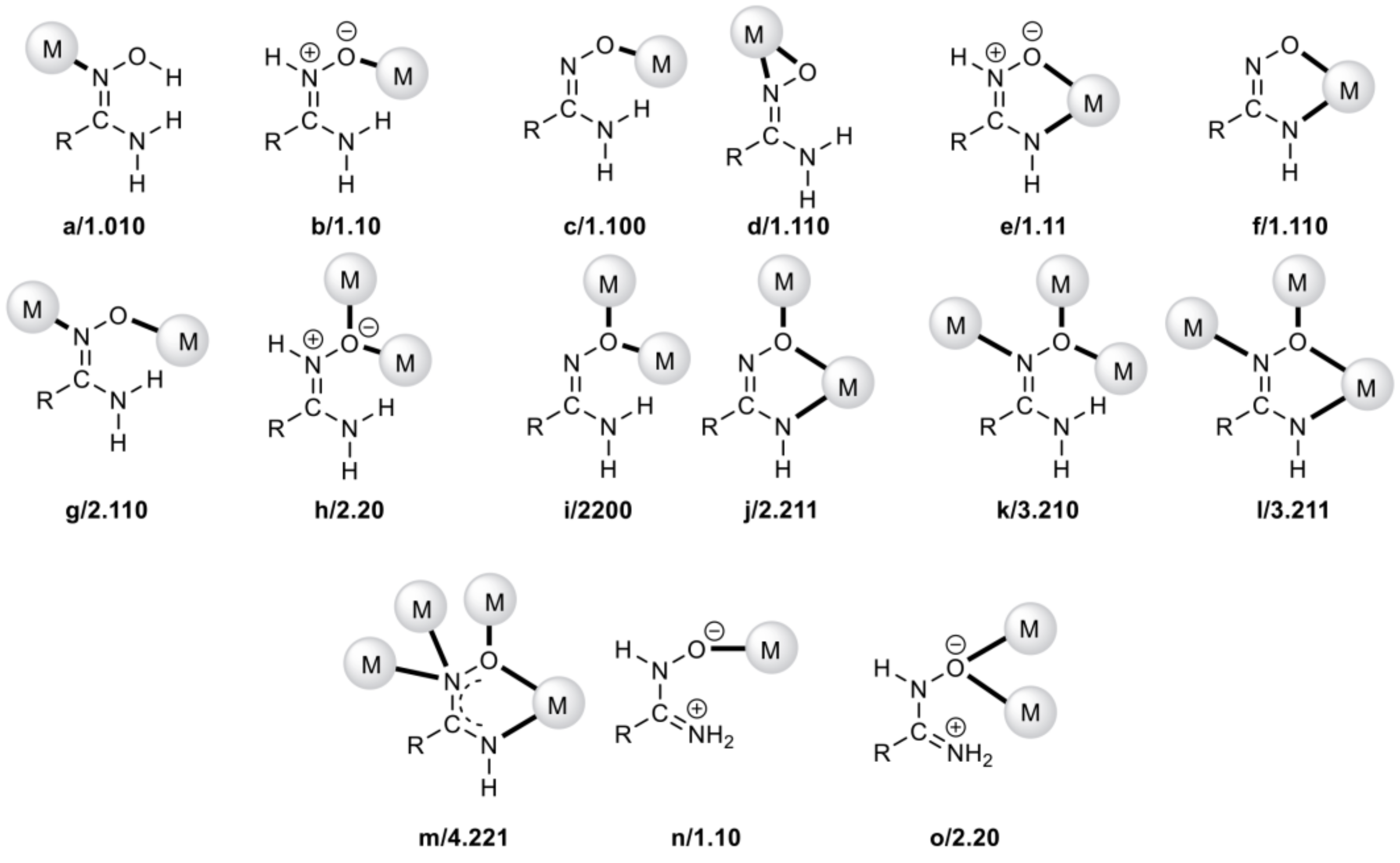
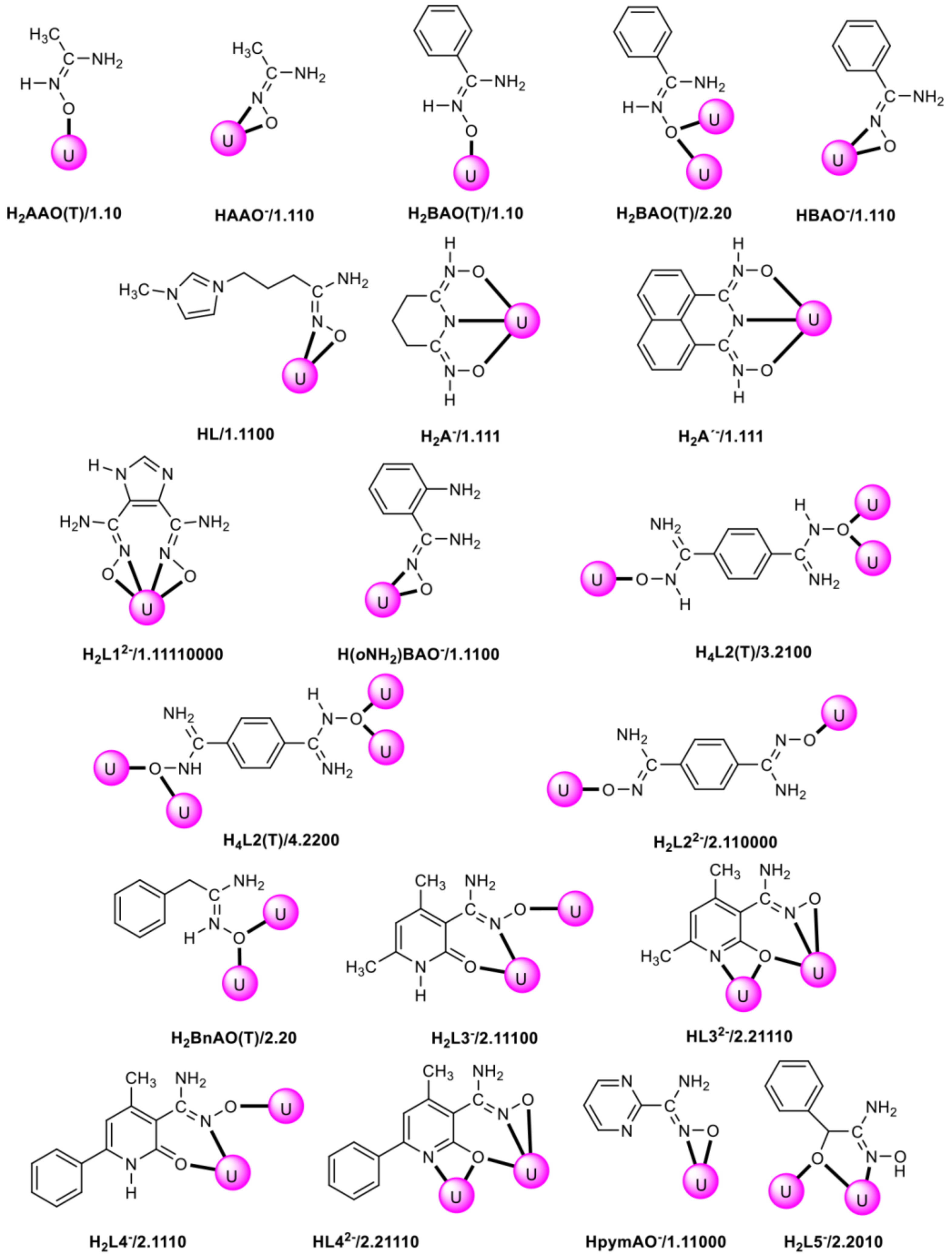
| Complex Number | Formula 1,2 | Coordination Mode 3,4 | Refs. |
|---|---|---|---|
| 1 | [UO2{H2AAO(T)}4(NO3)2 | 1.10 | [75] |
| 2 | [UO2(NO3)2{H2AAO(T)}](H2O) | 1.10 | [76] |
| 3 | [UO2(NO3)2{H2AAO(T)}2] | 1.10 | [76] |
| 4 | [UO2(HAAO)2(MeOH)2] | 1.110 | [77] |
| 5 | [UO2{H2BAO(T)}]4(NO3)2 | 1.10 | [75] |
| 6 | [(UO2)4(O)2{H2BAO(T)}8(H2O)2](NO3)4 | 1.10, 2.20 | [76] |
| 7 | [(UO2)4(O)2{H2BAO(T)}10Na(NO3)2](NO3)3 | 1.10, 2.20 | [76] |
| 8 | [UO2(HBAO)2(MeOH)2] | 1.110 | [76,77] |
| 9 | [(UO2)3(NO3)3(HBAO)3] | 1.110 | [76] |
| 10 | [UO2(NO3)2(HL)] | 1.1100 | [78] |
| 11 | [UO2(H2A)2] | 1.111 | [79] |
| 12 | [UO2(H2A’)(NO3)(ΜeOH)] | 1.111 | [80] |
| 13 | [UO2(H2L1)(H2O)2] | 1.11110000 | [81] |
| 14 | [UO2{oNH2)BAO}2(MeOH)2] | 1.1100 | [82] |
| 15 | {[(UO2)4(O)2{H4L2(T)}3(H2L2)(H2O)2]Cl2}n | 3.2110, 4.2200, 2.110000 | [83] |
| 16 | [(UO2)2(NO3)4{H2BnAO(T)}2] | 2.20 | [84] |
| 17 | [(UO2)2(NO3)2(H2L3)2] | 2.11100 | [24] |
| 18 | [(UO2)2HL3)2(DMSO)2] | 2.21110 | [24] |
| 19 | [(UO2)2(O2CMe)2(H2L4)2] | 2.11100 | [24,25] |
| 20 | [(UO2)2(HL4)2(DMSO)2] | 2.21110 | [24,25] |
| 21 | [UO2(HpymAO)2(MeOH)2] | 1.11000 | [24] |
| 22 | (Et3NH)2[(UO2)3(O)(O2CMe)3(H2L5)3] | 2.2010 | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsantis, S.T.; Iliopoulou, M.; Tzimopoulos, D.I.; Perlepes, S.P. Synthetic and Structural Chemistry of Uranyl-Amidoxime Complexes: Technological Implications. Chemistry 2023, 5, 1419-1453. https://doi.org/10.3390/chemistry5020097
Tsantis ST, Iliopoulou M, Tzimopoulos DI, Perlepes SP. Synthetic and Structural Chemistry of Uranyl-Amidoxime Complexes: Technological Implications. Chemistry. 2023; 5(2):1419-1453. https://doi.org/10.3390/chemistry5020097
Chicago/Turabian StyleTsantis, Sokratis T., Maria Iliopoulou, Demetrios I. Tzimopoulos, and Spyros P. Perlepes. 2023. "Synthetic and Structural Chemistry of Uranyl-Amidoxime Complexes: Technological Implications" Chemistry 5, no. 2: 1419-1453. https://doi.org/10.3390/chemistry5020097