

Au Single Metal Atom for Carbon Dioxide Reduction Reaction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
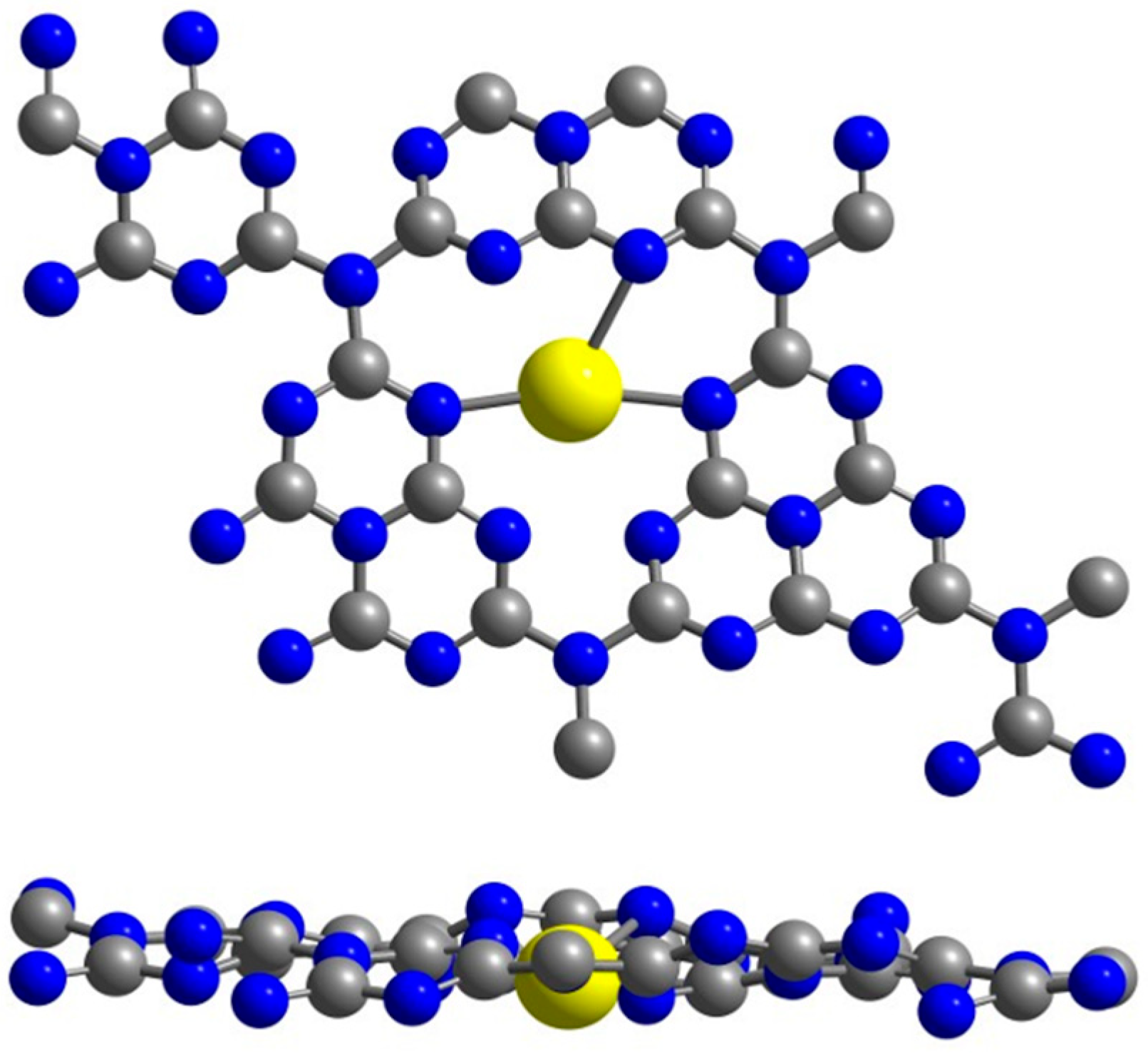
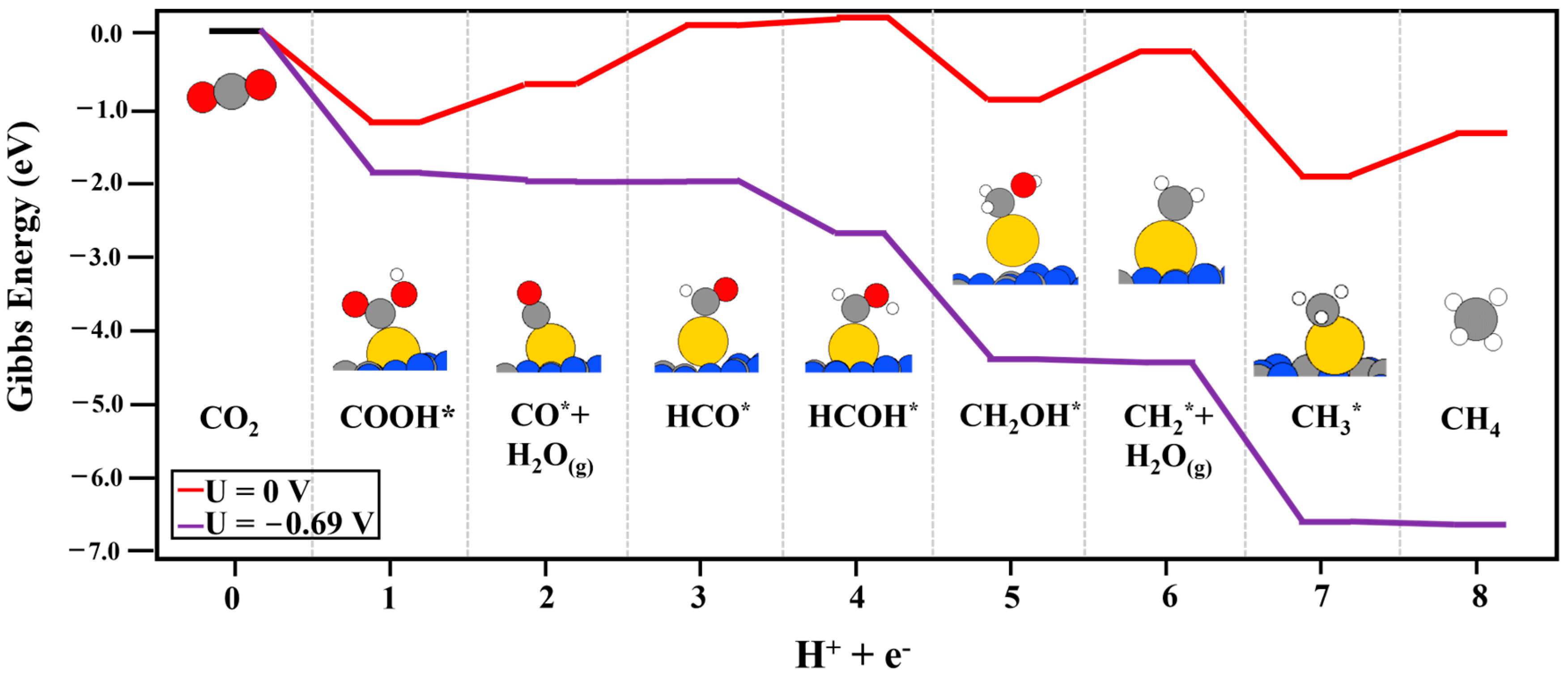
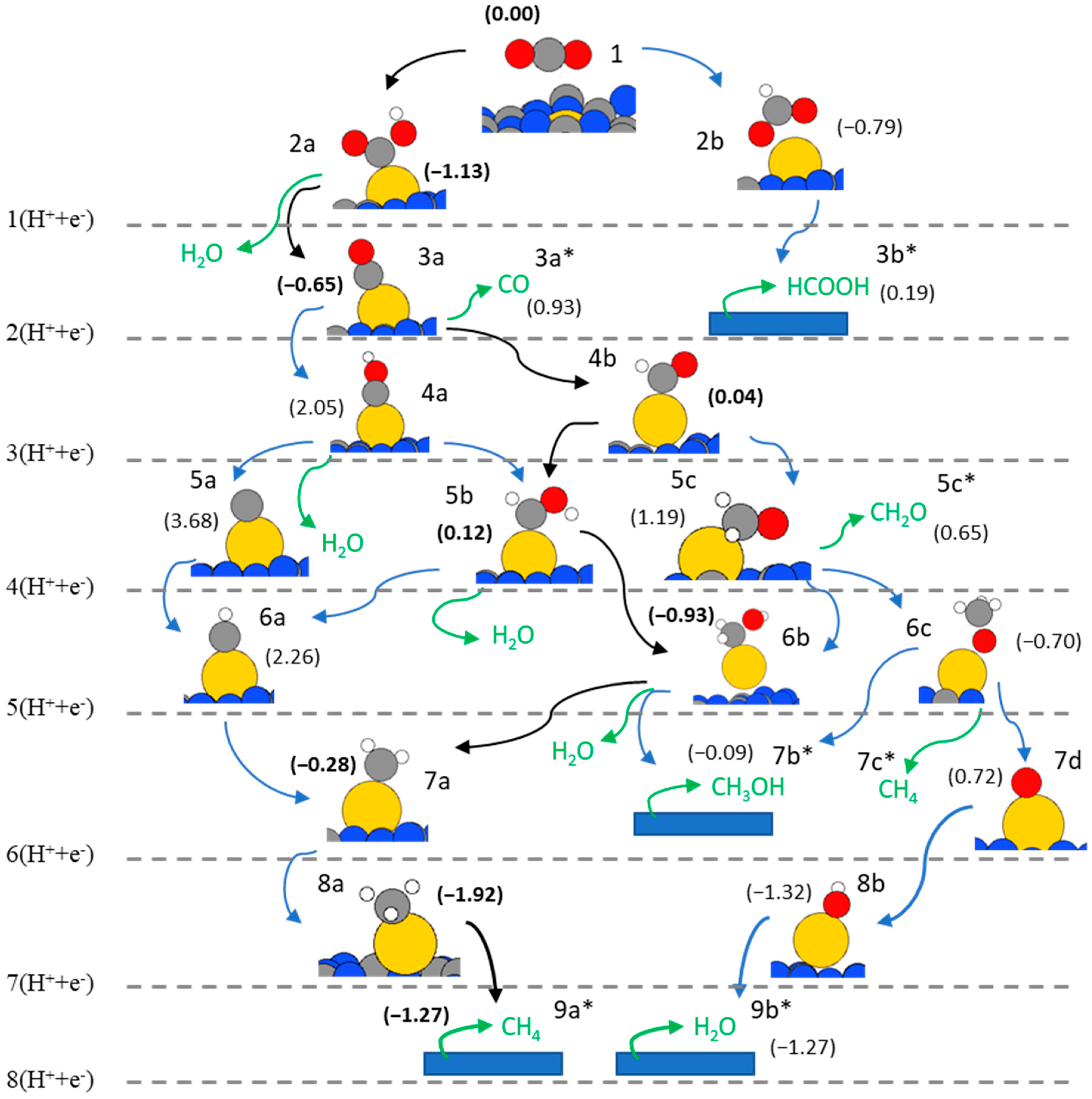
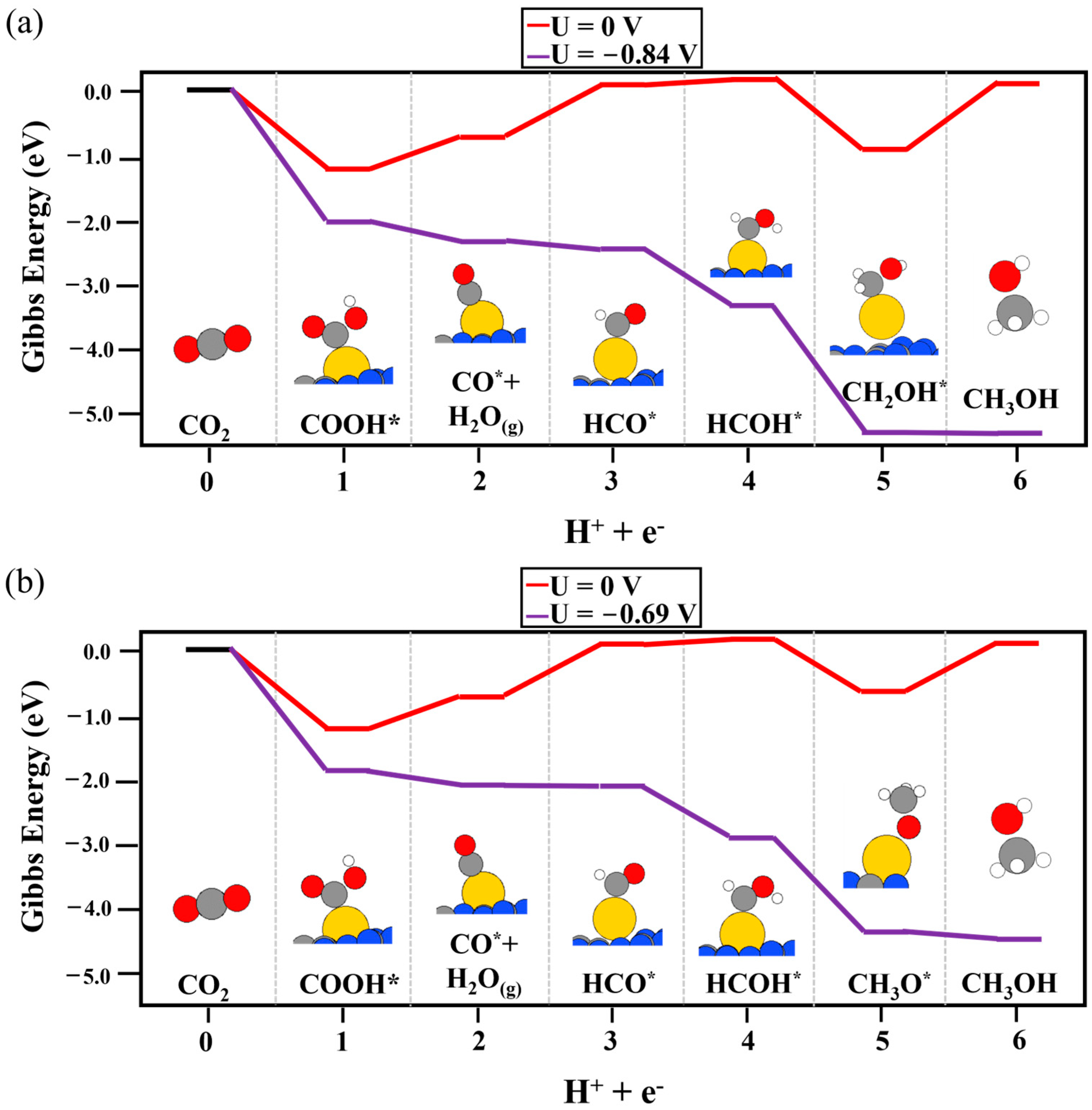
2. Results
3. Conclusions
4. Computational Details and Models
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lim, L. How to make the most of carbon dioxide. Nature 2015, 526, 628–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intergovernmental Panel on Climate Change. Climate Change 2013—The Physical Science Basis, 1st ed.; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Luque-Urrutia, J.A.; Ortiz-García, T.; Solà, M.; Poater, A. Green Energy by Hydrogen Production from Water Splitting, Water Oxidation Catalysis and Acceptorless Dehydrogenative Coupling. Inorganics 2023, 11, 88. [Google Scholar] [CrossRef]
- Poater, J.; Gimferrer, M.; Poater, A. Covalent and Ionic Capacity of MOFs To Sorb Small Gas Molecules. Inorg. Chem. 2018, 57, 6981–6990. [Google Scholar] [CrossRef] [PubMed]
- Rajjak Shaikh, A.; Posada-Pérez, S.; Brotons-Rufes, A.; Pajski, J.J.; Kumar, G.; Mateen, A.; Poater, A.; Solà, M.; Chawla, M.; Cavallo, L. Selective absorption of H2S and CO2 by azole based protic ionic liquids: A combined Density Functional Theory and Molecular Dynamics study. J. Mol. Liq. 2022, 367, 120558. [Google Scholar] [CrossRef]
- Arayachukiat, S.; Yingcharoen, P.; Vummaleti, S.V.C.; Cavallo, L.; Poater, A.; D’Elia, V. Cycloaddition of CO2 to challenging N-tosyl aziridines using a halogen-free niobium complex: Catalytic activity and mechanistic insights. Mol. Catal. 2017, 443, 280–285. [Google Scholar] [CrossRef]
- Vummaleti, S.V.C.; Nolan, S.P.; Cavallo, L.; Talarico, G.; Poater, A. Mechanism of CO2 fixation by Ir–X Bonds (X = OH, OR, N, C). Eur. J. Inorg. Chem. 2015, 2015, 4653–4657. [Google Scholar] [CrossRef]
- Coufourier, S.; Gaignard-Gaillard, Q.; Lohier, J.-F.; Poater, A.; Gaillard, S.; Renaud, J.-L. Hydrogenation of CO2, Hydrogenocarbonate, and Carbonate to Formate in Water using Phosphine Free Bifunctional Iron Complexes. ACS Catal. 2020, 10, 2108–2116. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Holtcamp, M.W. Catalysts for the reactions of epoxides and carbon dioxide. Coord. Chem. Rev. 1996, 27, 155–174. [Google Scholar] [CrossRef]
- Vidal-López, A.; Posada-Pérez, S.; Solà, M.; D’Elia, V.; Poater, A. The Importance of the Bite Angle of Metal(III) Salen Catalysts in the Sequestration of CO2 with Epoxides in Mild Conditions. Green Chem. Eng. 2022, 3, 180–187. [Google Scholar] [CrossRef]
- Aomchad, V.; Del Gobbo, S.; Yingcharoen, P.; Poater, A.; D’Elia, V. Exploring the potential of Group III salen complexes for the conversion of CO2 under ambient conditions. Catal. Today 2021, 375, 324–334. [Google Scholar] [CrossRef]
- Natongchai, W.; Luque-Urrutia, J.A.; Phungpanya, C.; Solà, M.; D’Elia, V.; Poater, A.; Zipse, H. Cycloaddition of CO2 to epoxides by highly nucleophilic 4-aminopyridines: Establishing a relationship between carbon basicity and catalytic performance by experimental and DFT investigations. Org. Chem. Front. 2021, 8, 613–627. [Google Scholar] [CrossRef]
- Sodpiban, O.; Del Gobbo, S.; Barman, S.; Aomchad, V.; Kidkhunthod, P.; Ould-Chikh, S.; Poater, A.; D’Elia, V.; Basset, J.-M. Synthesis of Well-defined Yttrium-based Lewis Acids by Capture of a Reaction Intermediate and Catalytic Application for cycloaddition of CO2 to Epoxides Under Atmospheric Pressure. Catal. Sci. Technol. 2019, 9, 6152–6165. [Google Scholar] [CrossRef]
- Bobadilla, L.F.; Azancot, L.; Luque-Álvarez, L.A.; Torres-Sempere, G.; González-Castaño, M.; Pastor-Pérez, L.; Yu, J.; Ramírez-Reina, T.; Ivanova, S.; Centeno, M.A.; et al. Development of Power-to-X Catalytic Processes for CO2 Valorisation: From the Molecular Level to the Reactor Architecture. Chemistry 2022, 4, 1250–1280. [Google Scholar] [CrossRef]
- Schäppi, R.; Rutz, D.; Dähler, F.; Muroyama, A.; Haueter, P.; Lilliestam, J.; Patt, A.; Furler, P.; Aomchad, V.; Del Gobbo, S.; et al. Drop-in fuels from sunlight and air. Nature 2022, 601, 63–68. [Google Scholar] [CrossRef]
- Preti, D.; Resta, C.; Squarcialupi, S.; Fachinetti, G. Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2011, 50, 12551–12554. [Google Scholar] [CrossRef]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Pomelli, C.S.; Tomasi, J.; Solà, M. Theoretical Study on the Thermodynamics of the Elimination of Formic Acid in the Last Step of the Hydrogenation of CO2 Catalyzed by Rhodium Complexes in the Gas Phase and Supercritical CO2. Organometallics 1998, 17, 3164–3168. [Google Scholar] [CrossRef]
- Italiano, C.; Llorca, J.; Pino, L.; Ferraro, M.; Antonucci, V.; Vita, A. CO and CO2 methanation over Ni catalysts supported on CeO2, Al2O3 and Y2O3 oxides. Appl. Catal. B Environ. 2020, 264, 118494. [Google Scholar] [CrossRef]
- Lam, E.; Noh, G.; Larmier, K.; Safonova, O.V.; Copéret, C. CO2 Hydrogenation on Cu-Catalysts Generated from ZnII Single-Sites: Enhanced CH3OH Selectivity Compared to Cu/ZnO/Al2O3. J. Catal. 2021, 394, 266–272. [Google Scholar] [CrossRef]
- Song, X.; Yang, C.; Li, X.; Wang, Z.; Pei, C.; Zhao, Z.J.; Gong, J. On the Role of Hydroxyl Groups on Cu/Al2O3 in CO2 Hydrogenation. ACS Catal. 2022, 12, 14162–14172. [Google Scholar] [CrossRef]
- Falbo, L.; Visconti, C.G.; Lietti, L.; Szanyi, J. The effect of CO on CO2 methanation over Ru/Al2O3 catalysts: A combined steady-state reactivity and transient DRIFT spectroscopy study. Appl. Catal. B Environ. 2019, 256, 117791. [Google Scholar] [CrossRef]
- Yan, Y.; Wong, R.J.; Ma, Z.; Donat, F.; Xi, S.; Saqline, S.; Fan, Q.; Du, Y.; Borgna, A.; He, Q.; et al. CO2 hydrogenation to methanol on tungsten-doped Cu/CeO2 catalysts. Appl. Catal. B Environ. 2022, 306, 121098. [Google Scholar] [CrossRef]
- Ye, R.-P.; Liao, L.; Reina, T.R.; Liu, J.; Chevella, D.; Jin, Y.; Fan, M.; Liu, J. Engineering Ni/SiO2 catalysts for enhanced CO2 methanation. Fuel 2021, 285, 119151. [Google Scholar] [CrossRef]
- Gonell, F.; Puga, A.V.; Julián-López, B.; García, H.; Corma, A. Copper-doped titania photocatalysts for simultaneous reduction of CO2 and production of H2 from aqueous sulfide. Appl. Catal. B Environ. 2016, 180, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Posada-Pérez, S.; Ramirez, P.J.; Gutierrez, R.A.; Stacchiola, D.J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. The conversion of CO2 to methanol on orthorhombic β-Mo2C and Cu/β-Mo2C catalysts: Mechanism for admetal induced change in the selectivity and activity. Catal. Sci. Technol. 2016, 6, 6766–6777. [Google Scholar] [CrossRef]
- Dixit, M.; Peng, X.; Porosoff, M.D.; Willauer, H.D.; Mpourmpakis, G. Elucidating the role of oxygen coverage in CO2 reduction on Mo2C. Catal. Sci. Technol. 2017, 7, 5521–5529. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Ramírez, P.J.; Evans, J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. Highly active Au/δ-MoC and Cu/δ-MoC catalysts for the conversion of CO2: The metal/C ratio as a key factor defining activity, selectivity, and stability. J. Am. Chem. Soc. 2016, 138, 8269–8278. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.A.; Ramírez, P.J.; Gutierrez, R.A. Highly Active Pt/MoC and Pt/TiC Catalysts for the Low-Temperature Water-Gas Shift Reaction: Effects of the Carbide Metal/Carbon Ratio on the Catalyst Performance. Catal. Today 2017, 289, 47–52. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Solà, M.; Poater, A. Carbon dioxide conversion on supported metal nanoparticles: A brief review. Catalysts 2023, 13, 305. [Google Scholar] [CrossRef]
- Todorova, T.K.; Schreiber, M.W.; Fontcave, M. Mechanistic understanding of CO2 reduction reaction (CO2RR) toward multicarbon products by heterogeneous copper-based catalysts. ACS Catal. 2020, 10, 1754–1768. [Google Scholar] [CrossRef]
- Jiang, T.-W.; Zhou, Y.-W.; Ma, X.-Y.; Qin, X.; Li, H.; Ding, C.; Jiang, B.; Jiang, K.; Cai, W.-B. Spectrometric study of electrochemical CO2 reduction on Pd and Pd-B electrodes. ACS Catal. 2021, 11, 840–848. [Google Scholar] [CrossRef]
- Mistry, H.; Choi, Y.W.; Bagger, A.; Scholten, F.; Bonifacio, C.S.; Sinev, I.; Divins, N.J.; Zegkinoglou, I.; Jeon, H.S.; Kisslinger, K.; et al. Enhanced carbon dioxide electroreduction to carbon monoxide over defect-reach plasma-activated silver catalysts. Angew. Chem. Int. Ed. 2017, 56, 11394–11398. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.L.; Ringe, S.; Tang, M.; Walton, A.; Hahn, C.; Jaramillo, T.F.; Chan, K.; Bell, A.T. Influence of atomic surface structure on the activity of Ag for the electrochemical reduction of CO2 to CO. ACS Catal. 2019, 9, 4006–4014. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, C.W.; Kanan, M.W. Aqueous CO2 reduction at very low overpotential on oxide-derived Au nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. [Google Scholar] [CrossRef]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Roldan-Cuenya, B. Exceptional size-dependent activity enhancement in the electroreduction of CO2 over Au nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef]
- Bonin, J.; Maurin, A.; Robert, M. Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Fe and Co metal based complexes. Recent advances. Coord. Chem. Rev. 2017, 334, 184–198. [Google Scholar] [CrossRef]
- Rendón-Calle, A.; Builes, S.; Calle-Vallejo, F. How symmetry factors cause potential- and facet-dependent pathway shifts during CO2 reduction to CH4 on Cu electrodes. Curr. Opin. Electrochem. 2018, 9, 158–165. [Google Scholar] [CrossRef]
- Diaz-Morales, O.; Ledezma-Yanez, I.; Koper, M.T.M.; Calle-Vallejo, F. Guidelines for the Rational Design of Ni-Based Double Hydroxide Electrocatalysts for the Oxygen Evolution Reaction. ACS Catal. 2015, 9, 5380–5387. [Google Scholar] [CrossRef]
- Rendón-Calle, A.; Low, Q.H.; Hong, S.H.L.; Builes, S.; Yao, B.S.; Calle-Vallejo, F. A brief review of the computational modeling of CO2 electroreduction on Cu electrodes. Appl. Catal. 2021, 285, 119776. [Google Scholar] [CrossRef]
- Reske, R.; Mistry, H.; Behafarid, F.; Roldan-Cuenya, B.; Strasser, P. Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef]
- Woldu, A.R.; Huang, Z.; Zhao, P.; Hu, L.; Astruc, D. Electrochemical CO2 reduction (CO2RR) to multi-carbon products over copper-based catalysts. Coord. Chem. Rev. 2022, 454, 214340. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Norskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Nie, X.; Luo, W.; Janik, M.J.; Asthagiri, A. Reaction mechanisms of CO2 electrochemical reduction on Cu(1 1 1) determined with density functional theory. J. Catal. 2014, 312, 108–122. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Goodpaster, J.D.; Bell, A.T.; Head-Gordon, M. Identification of Possible Pathways for C–C Bond Formation during Electrochemical Reduction of CO2: New Theoretical Insights from an Improved Electrochemical Model. J. Phys. Chem. Lett. 2016, 7, 1471–1477. [Google Scholar] [CrossRef]
- Datye, A.K.; Guo, H. Single atom catalysis poised to transition from an academic curiosity to an industrially relevant technology. Nat. Commun. 2021, 12, 895. [Google Scholar] [CrossRef]
- Liu, L.; Corma, A. Metal catalysts for heterogeneous catalysis: From single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [Green Version]
- Basset, J.M.; Baudouin, A.; Bayard, F.; Candy, J.-P.; Copéret, C.; De Mallmann, A.; Godard, G.; Kuntz, E.; Lefebvre, F.; Lucas, C.; et al. Modern Surface Organometallic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 23–73. [Google Scholar]
- Rivera-Cárcamo, C.; Serp, P. Single atom catalysts on carbon-based materials. ChemCatChem 2018, 10, 5058–5091. [Google Scholar] [CrossRef]
- Xu, Y.-T.; Xie, M.-Y.; Zhong, H.; Cao, Y. In situ clustering of single-atom copper precatalysts in a metal-organic framework for efficient electrocatalytic nitrate-to-ammonia reduction. ACS Catal. 2022, 12, 8698–8706. [Google Scholar] [CrossRef]
- Jurado, L.; Esvan, J.; Luque-Álvarez, L.A.; Bobadilla, L.F.; Odriozola, J.A.; Posada-Pérez, S.; Poater, A.; Comas-Vives, A.; Axet, M.R. Highly dispersed Rh single atoms over graphitic carbon nitride as a robust catalyst for the hydroformylation reaction. Catal. Sci. Technol. 2023, 13, 1425–1436. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q.; Zhang, L.c.; Wu, Y.; Chen, H.; Li, T.; Xu, M.; Bao, S.-J. A gel-limiting strategy for large-scale fabrication of Fe-N-C single-atom ORR catalysts. J. Mater. Chem. A 2021, 9, 7137–7142. [Google Scholar] [CrossRef]
- Ying, Y.; Fan, K.; Luo, X.; Qiao, J.; Huang, H. Unravelling the origin of bifunctional OER/ORR activity for single-atom catalysts supported on C2N by DFT and machine learning. J. Mater. Chem. A 2021, 9, 16860–16867. [Google Scholar] [CrossRef]
- Zeng, L.; Dai, C.; Liu, B.; Xue, C. Oxygen-assisted stabilization of single-atom Au during photocatalytic hydrogen evolution. J. Mater. Chem. A 2019, 7, 24217–24221. [Google Scholar] [CrossRef]
- Li, M.; Wang, H.; Luo, W.; Sherrell, P.C.; Chen, J.; Yang, J. Heterogeneous single-atom catalysts for electrochemical CO2 reduction reaction. Adv. Mater. 2020, 32, e2001848. [Google Scholar] [CrossRef]
- Azofra, L.M.; MacFarlane, D.R.; Sun, C. A DFT study of planar vs. corrugated graphene-like carbon nitride (g-C3N4) and its role in the catalytic performance of CO2 conversion. Phys. Chem. Chem. Phys. 2016, 18, 18507–18514. [Google Scholar] [CrossRef]
- Gao, G.; Jiao, Y.; Waclawik, E.; Du, A. Single Atom (Pd/Pt) Supported on Graphitic Carbon Nitride as an Efficient Photocatalyst for Visible-Light Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2016, 138, 6292–6297. [Google Scholar] [CrossRef] [Green Version]
- Cometto, C.; Ugolotti, A.; Grazietti, E.; Moretto, A.; Bottaro, G.; Armelao, L.; Di Valentin, C.; Calvillo, L.; Granozzi, G. Copper single-atoms embedded in 2D graphitic carbon nitride for the CO2 reduction. npj 2D Mater. Appl. 2021, 5, 63. [Google Scholar] [CrossRef]
- Peng, Y.; Lu, B.; Chen, S. Carbon-Supported Single Atom Catalysts for Electrochemical Energy Conversion and Storage. Adv. Mater. 2018, 30, 1801995. [Google Scholar] [CrossRef]
- Li, J.; Yan, O.; Li, K.; You, J.; Wang, H.; Cui, W.; Cen, W.; Chu, Y.; Dong, F. Cu supported on polymeric carbon nitride for selective CO2 reduction into CH4: A combined kinetic and thermodynamics investigation. J. Mater. Chem. A 2019, 7, 17014–17021. [Google Scholar] [CrossRef]
- Liu, H.; Huang, Q.; An, W.; Wang, Y.; Men, Y.; Liu, S. Dual-atom active sites embedded in two-dimensional C2N for efficient CO2 electroreduction: A computational study. J. Energy Chem. 2021, 61, 507–516. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Skorodumova, N.V.; Mentus, S.V.; Pašti, I.A. Surface Pourbaix plots of M@N4-graphene single-atom electrocatalysts from density functional theory thermodynamic modeling. Electrochim. Acta 2022, 412, 140155. [Google Scholar] [CrossRef]
- Yu, J.; Chen, C.; Zhang, Q.; Lin, J.; Yang, X.; Gu, L.; Zhang, H.; Liu, Z.; Wang, Y.; Zhang, S.; et al. Au Atoms Anchored on Amorphous C3N4 for Single-Site Raman Enhancement. J. Am. Chem. Soc. 2022, 144, 21908–21915. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Vidal-López, A.; Solà, M.; Poater, A. 2D carbon nitride as a support with single Cu, Ag, and Au atoms for carbon dioxide reduction reaction. Phys. Chem. Chem. Phys. 2023, 25, 8574–8582. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, L.; Pei, Y. Single metal atom catalysts supported on g-C3N4 for formic acid dehydrogenation: A combining density functional theory and machine learning study. J. Phys. Chem. C 2021, 125, 22513–22521. [Google Scholar] [CrossRef]
- Cui, X.; An, W.; Liu, X.; Wang, H.; Men, Y.; Wang, J. C2N-graphene supported single-atom catalysts for CO2 electrochemical reduction reaction: Mechanistic insight and catalysts screening. Nanoscale 2018, 10, 15262–15272. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Wisesa, P.; McGill, K.; Mueller, T. Efficient generation of generalized Monkhorst-Pack grids through the use of informatics. Phys. Rev. B 2016, 93, 155109. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; An, W.; Wang, Z.; Wang, Y.; Men, Y.; Du, Y. Electrochemical CO2 reduction reaction on M@Cu(211) bimetallic single-atom surface alloys: Mechanism, kinetics, and catalyst screening. ACS Sustain. Chem. Eng. 2020, 8, 210–222. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidal-López, A.; Posada-Pérez, S.; Solà, M.; Poater, A. Au Single Metal Atom for Carbon Dioxide Reduction Reaction. Chemistry 2023, 5, 1395-1406. https://doi.org/10.3390/chemistry5020095
Vidal-López A, Posada-Pérez S, Solà M, Poater A. Au Single Metal Atom for Carbon Dioxide Reduction Reaction. Chemistry. 2023; 5(2):1395-1406. https://doi.org/10.3390/chemistry5020095
Chicago/Turabian StyleVidal-López, Anna, Sergio Posada-Pérez, Miquel Solà, and Albert Poater. 2023. "Au Single Metal Atom for Carbon Dioxide Reduction Reaction" Chemistry 5, no. 2: 1395-1406. https://doi.org/10.3390/chemistry5020095