
The Miracle of Vitamin B12 Biochemistry
Department of Chemistry, Columbia University, New York, NY 10027, USA
Reactions 2024, 5(1), 20-76; https://doi.org/10.3390/reactions5010002
Submission received: 30 October 2023
/
Revised: 13 December 2023
/
Accepted: 29 December 2023
/
Published: 5 January 2024
(This article belongs to the Special Issue Feature Papers in Reactions in 2023)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:For decades, the comparison of experimental data with theoretical results in studying the biochemistry of vitamin B12 has been very confusing. While the methylcobalamin cofactor-dependent Methionine Synthase process can undergo unlimited turnovers, and some of the adenosylcobalamin-dependent processes run with close-to-unity equilibrium constants (e.g., with close-to-zero energy barriers), the DFT and QM/MM based on density functional theory, the most used and appreciated methods for calculating the electronic structure of molecules, have been showing a much shorter than experimental-determined Co-N distances in the vitamin B12 cofactors of Co+2 and the inadequate large energetic barriers of their enzymology bioprocesses. The confusion was even larger since some in vitro experimental data showed large barriers to the vitamin B12 cofactor reactions (which in fact play a destructive role in the Methionine Synthase process and which barriers were caused mostly by the influence of the solvents in which the reaction took place). It reached the point where solid contributions to the study of the biochemical processes of vitamin B12 were almost officially questioning the correctness of the experimental determination of the Co-N chemical bond distances in the cobalt(II) cofactors of vitamin B12. Unexpectedly, all the theoretical biochemistry of the vitamin B12 cofactors began to agree with all in vivo experimental data only when they were treated with the MCSCF method, the method that considers the orbital mixing, or in other words, the Pseudo-Jahn–Teller Effect. MCSCF data establish unknown mechanistic details of the methyl radical and hydrogen transfers, the origin of the electronic transfers between bioreagents, and the nature and the relationship between the bioreactions. The Pseudo-Jahn–Teller Effect, e.g., orbital mixing, governs vitamin B12 chemistry in general and provides insight into particular details of vitamin B12-dependent reactions in the human body. It turns out that the DFT or QM/MM based on DFT method theoretical data are incongruent with the experimental data due to their limitations, e.g., the unaccounted-for effects of orbital mixing.
1. Introduction
A brief history of the research development of vitamin B12 and the most general information about the biochemistry and structure of its cofactors are covered here. During the second half of the 19th century, the study of what later became known as vitamin B12 began with descriptions of cases of pernicious anemia, neuropathy, and megaloblasts as a myelopathy effect in bone marrow. Thomas Addison, William Oster, William Gardner, Paul Ehrlich, and Ludwig Lichtheim [1] were significant individuals involved [1]. These conditions were later determined to be associated with vitamin B12 deficiency. In the early 20th century, effective treatment of pernicious anemia was established by using quantities of liver concentrate (later determined to contain vitamin B12) along with an intrinsic factor contained in gastric juice, with contributions by George Whipple, Edwin Cohn, William Castle, William P. Murphy, and George Minot [2,3,4,5]. Whipple, Murphy, and Minot shared the 1934 Nobel Prize in Physiology or Medicine for their work [5]. Afterward, Alexander R. Todd, Mary Shaw Shorb, and Karl Folkers collaborated to develop an assay to isolate the “anti-pernicious anemia factor” in the liver, naming it vitamin B12. Todd later significantly determined the structure of vitamin B12, for which he received the 1957 Nobel Prize in Chemistry [6,7]. In 1964, Dorothy Hodgkin was awarded the Nobel Prize in Chemistry for using crystallographic techniques to determine the vitamin’s complete structure [7]. Another Nobel laureate, Robert Burns Woodward (in collaboration with Albert Eschenmoser) completed a formal synthesis consisting of 72 stages of cyanocobalamin, a nonactive provitamin form of vitamin B12 in 1972 [8,9].
Vitamin B12 or cobalamin, is the largest and most complex of the 13 vitamins required by humans through consumption to maintain proper physiological health and was extensively studied [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159]. Humans are known to directly use two vitamers of B12 as cofactors in several vital enzymatic processes as well as provitamins that are convertible to active forms; however, numerous other pseudovitamins are not convertible to biologically active forms of B12 for human processes. Numerous bacteria and archaea synthesize vitamin B12. Humans obtain it by eating animal products that require it and thus have accumulated stores of it in their various tissues (especially the liver); plants do not require vitamin B12 for any analogous enzymatic process, so unfermented plants are not good sources of the vitamin. It is a water-soluble vitamin that has a complex and multifaceted absorption pathway requiring numerous proteases, binding proteins, and transport proteins. Vitamin B12 is known to be important for human DNA synthesis, fatty acid metabolism, amino acid metabolism, the nervous system, and the circulatory system. In humans, adenosylcobalamin is known to be a cofactor in a number of enzymatic processes. In particular, it is well known to be a cofactor in Methylmalonyl Coenzyme A Mutase bioprocesses in mitochondria that break down odd-chain fatty acids, certain amino acids, and cholesterol for use in the tricarboxylic acid cycle. The other B12 vitamer that humans are known to use is methylcobalamin. It is a known cofactor for Methionine Synthase that regenerates methionine in the S-adenosylmethionine (SAMe) cycle, allowing for the methylation of nucleic acids, histones, phospholipids, and proteins. In a healthy body, up to 5 mg of vitamin B12 is stored primarily in the liver but also in numerous other tissues. Since the liver can store at least several years’ worth of vitamin B12, individuals are not likely to be deficient unless they primarily have malabsorption conditions or have plant-based diets. Governmental consumption recommendations are consistently in the range of a low to medium amount in micrograms per day (depending on age, sex, weight, and pregnancy status) without a clear indication of any harm from excessive amounts. Vitamin B12 often fortifies food staples and is included in common multivitamin products in its chemically stable cyanocobalamin provitamin form. Since known syntheses consist of many steps and are very low-yielding, industrial production of vitamin B12 relies on fermentation processes involving specific microorganisms.
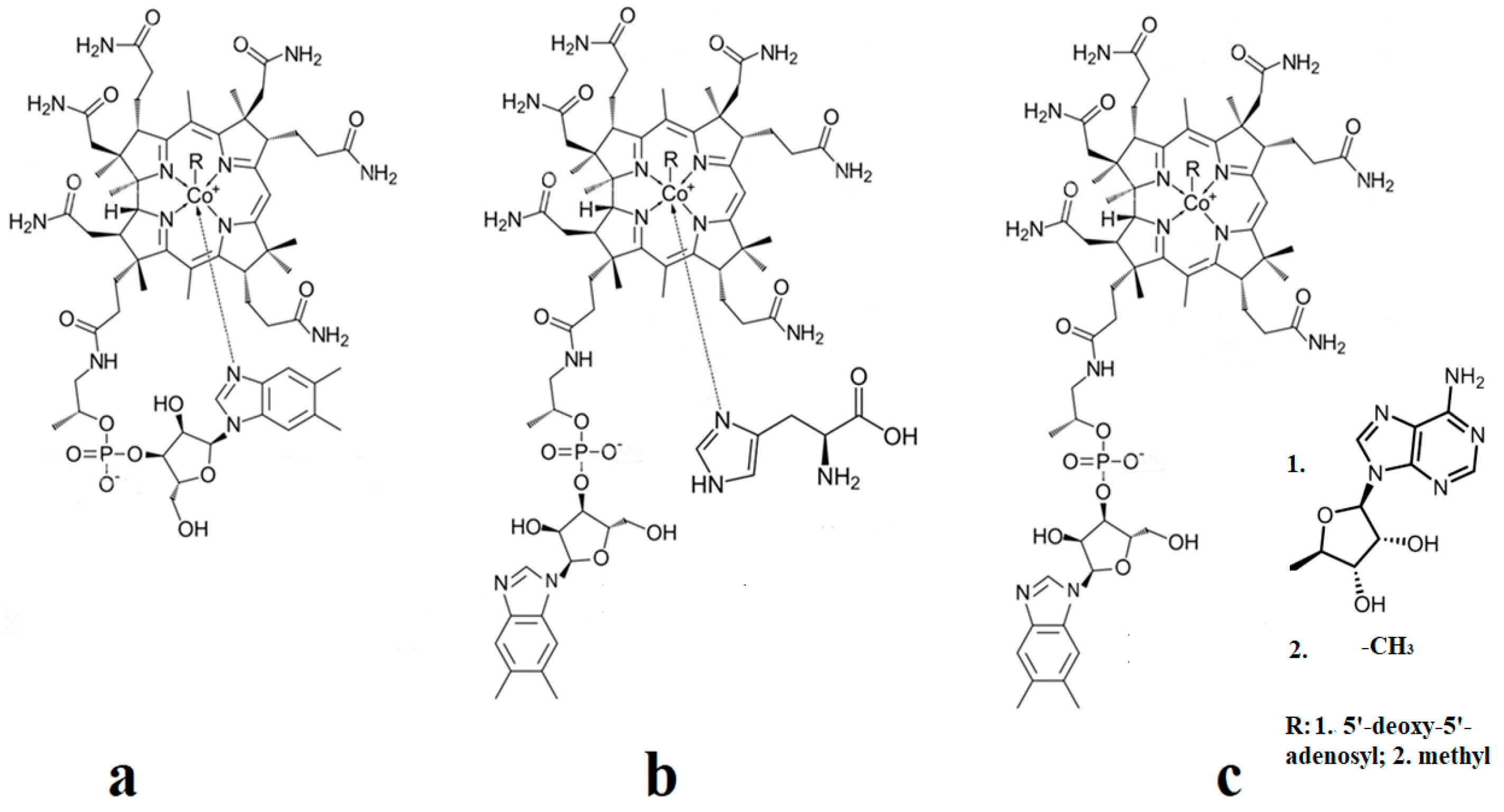
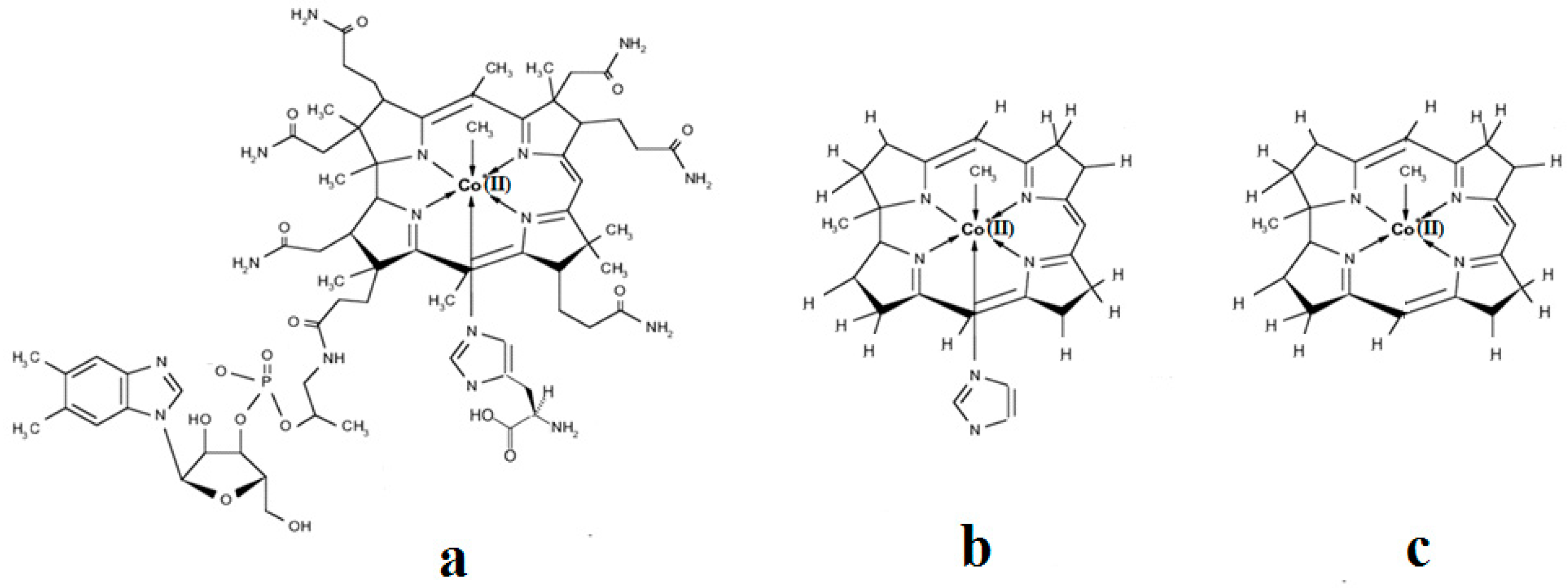
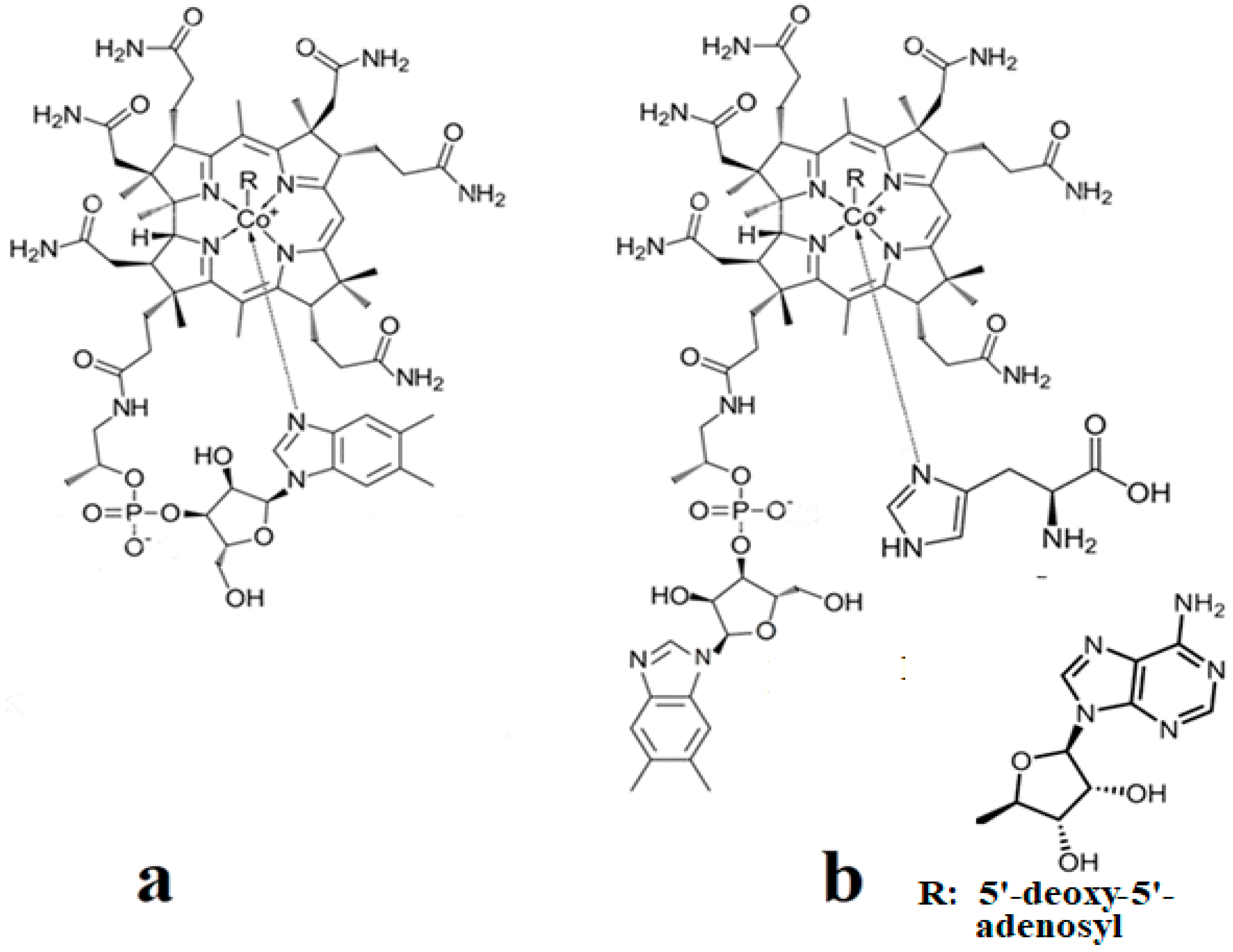
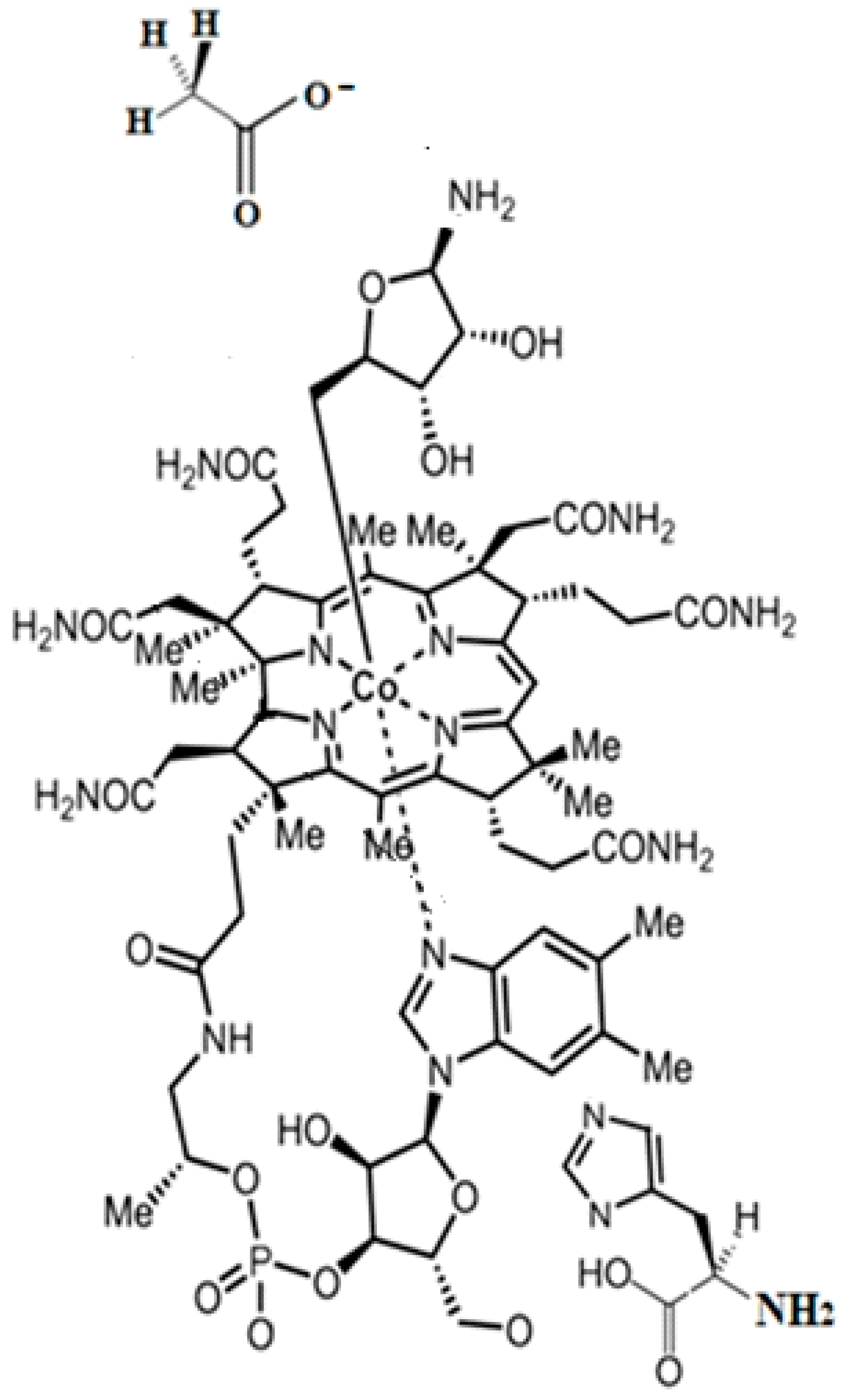
The methylcobalamin and adenosylcobalamin cofactors are mammals’ only known active vitamers of vitamin B12. All active and inactive forms of vitamin B12 have one cobalt ion as the coordinating center and one corrin ring as the equatorial ligand (Figure 1).
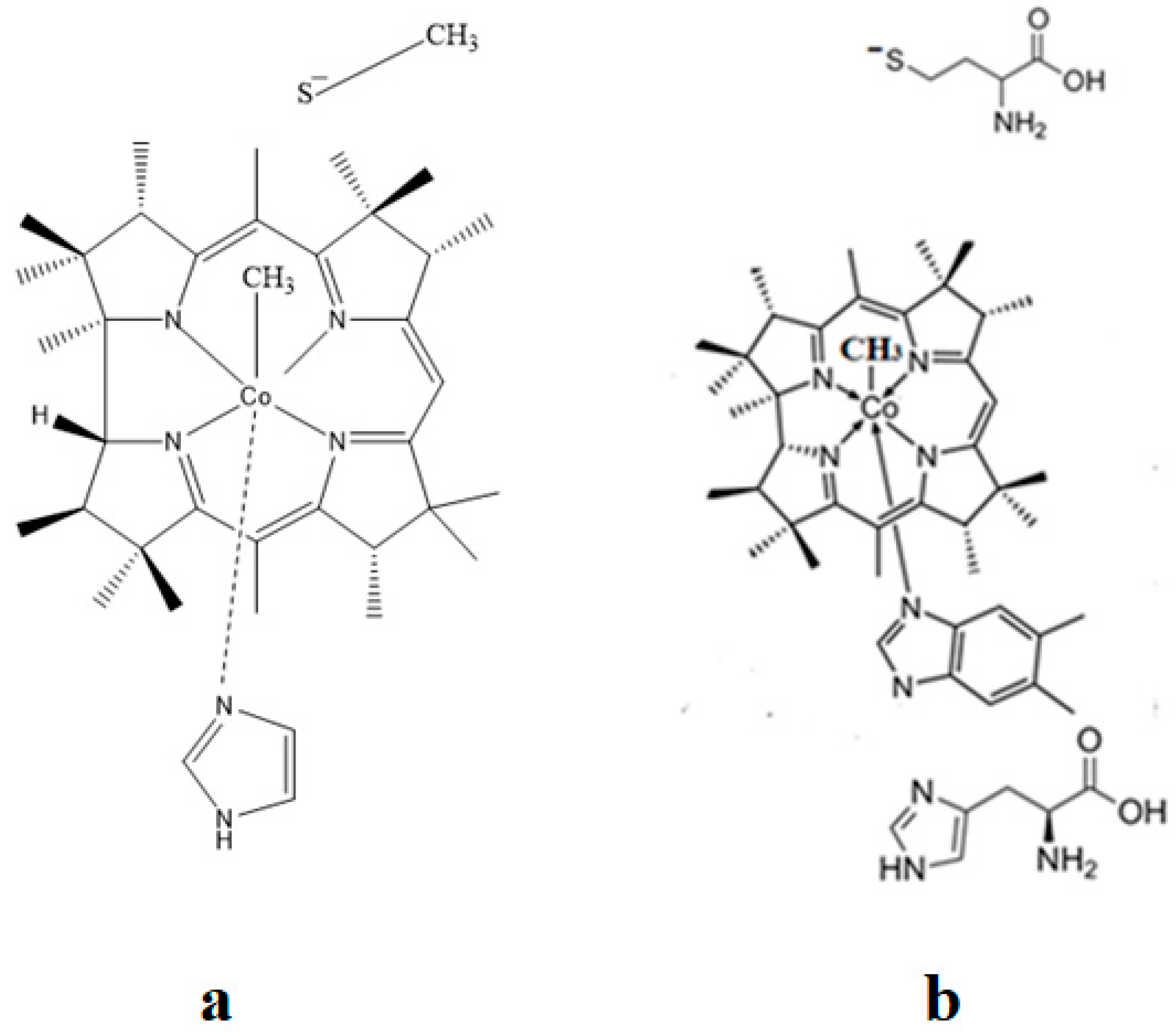
The corrin structure has one lateral chain with dimethylbenzimidazole at the end that can coordinate with the cobalt center as one of two axial ligands [10,11]. The oxidation state of the cobalt ion is equal to +3 and is offset by two negative charges of the corrin ring and one negative charge of a phosphate ion (PO4−) on the lateral chain so that the total charge of vitamin B12 is equal to the charge of the second axial ligand R [12,13,14,15,16]. The vitamin B12 isomer with the dimethylbenzimidazole ligand coordinated with the cobalt ion is referred to as the base-on form, while the isomer with the dimethylbenzimidazole group not coordinated with the cobalt ion is referred to as the base-off form [16,17,18]. In living matter, the dimethylbenzimidazole ligand is replaced in some of the vitamin B12-dependent processes involving Methionine Synthase, Glutamate Mutase, and Methylmalonyl-CoA Mutase with a histidine molecule [11,14,17,18].
A general description of the electronic and geometric structures of the active vitamin B12 forms in solutions has been elucidated by the Schrauzer group [19]. The Schrauzer group, which also addressed the issue of breaking the Co-C bond in the cyanocobalamin provitamin under the influence of thiols and dithiols in biochemical reactions, was one of the first groups of researchers to address the mechanism of bioactivity of a relevant form of vitamin B12 [20,21]. The Schrauzer group also demonstrated the similarity of many vitamin B12 bioreactions with reactions in which vitamin B12 is replaced with cobalt dioxymine, discovering that the central atom of the vitamin B12 forms can be reduced to the +1 oxidation state, becoming very reactive in biochemical reactions [21]. The –CH3 group transfer from methylcobalamin to thiols was elucidated some time afterward [22,23]. Various inorganic chemistry properties, including the cis and trans influences in the vitamin B12 forms, were analyzed by Pratt [23]. Banerjee and colleagues furthered the understanding of the mechanism involving the methylcobalamin cofactor with the Methionine Synthase process. Banerjee proposed that after a preliminary reduction to cob(II)alamin, the S-adenosyl-L-methionine (AdoMet) compound participates in the Methionine Synthase process by reducing the cobalt from a +2 oxidation state to that of +1 [24,25,26]. Banerjee and colleagues also demonstrated that the cobalamin with the central cobalt in the +1 oxidation state transfers the methyl radical from the 5-methyltetrahydrofolate molecule [27,28]. Unlike methylcobalamin, which acts as a cofactor in only one known bioprocess involving Methionine Synthase, adenosylcobalamin [29,30,31] is known to be a cofactor in a whole series of biological processes [32,33,34] in which the structures of several biological substrates change in unusual ways [32,33,34,35,36]. Matthews has studied various methyl transfer reactions with the participation of the methylcobalamin cofactor [37]. The known biochemical processes involving the adenosylcobalamin cofactor do not require the preliminary reduction of the central cobalt oxidation state from +3 to +2 but function in a simultaneous manner. An intermediate product of these processes is thought to be the 5′-deoxy-5′-adenosyl radical, which has not been detected so far in any experiment. A transfer of hydrogen from the substrates along with the breaking of the Co-C bond has also been observed in the adenosylcobalamin cofactor-dependent bioprocesses, which is believed to be confirmed by the impossibility of experimental determination of the 5′-deoxy-5′-adenosyl radical [34,35].
The catalytic mechanisms of the methylcobalamin cofactor-dependent Methionine Synthase process and the various adenosylcobalamin cofactor-dependent processes were partially inferred [38,39,40] and partially demonstrated [41,42,43]; however, many details of these mechanisms and the factors that influence them were unknown [44,45] for many years after initial work to outline many of the general features. Because of this, the international community continued to study these mechanisms both experimentally and theoretically. A major contribution to these studies belongs to the group led by Matthews, which was largely dedicated to determining several aspects of the Methionine Synthase process [46,47,48]. Pratt and Banerjee made continued consistent contributions to the study of the processes involving vitamin B12 cofactors [50,51,52]. The mechanisms of adenosylcobalamin cofactor-dependent processes are more complex [53,54,55,56] and include the transfer of hydrogen from substrates [57,58,59,60,61] as well as breaking the Co-C bond process [62,63,64,65,66]. In addition, the number of bioprocesses [55,66,67,68] requiring the adenosylcobalamin cofactor is much higher (12 bioprocesses attributed at present) [65,69,70,71,72,73,74], so both the number of researchers involved in their study and the number of publications [75,76,77,78,79,80] on these processes are larger [81,82,83,84,85]. Thermodynamics and kinetics [85,86,87] were also a part of the general adenosylcobalamin cofactor-dependent studies. The Co-C cleavage reaction rate constants in the methylcobalamin cofactor and the dependence of the parameters of this reaction on the nature of the solvent were determined by the Savéant and Birke groups [88,89,90]. Interestingly, the catalytic properties of vitamin B12 cofactors [91,92,93,94,95] have been successfully used to catalyze reactions [96,97,98] to obtain certain organic compounds [99]. Theoretical methods [100,101,102,103,104] have been used to study the properties and action of vitamin B12 cofactors in enzymatic processes [105,106,107,108,109,110]. In particular, spectroscopic processes, the trans effect, substrate implication in vitamin B12 bioreactions, and the processes of breaking the Co-C and Co-N axial bonds have been studied [102,111,112,113,114], mainly by using the DFT method [115,116,117,118,119] or by using the QM/MM method, in which the QM part is treated as the DFT method [120,121,122,123,124] by using the models [125,126,127,128,129] of the vitamin B12 cofactors [130,131,132,133]. The authors claimed that the DFT method [134,135,136,137,138] was suitable for studies of vitamin B12 bioprocesses [139,140]. The MCSCF method has been used sporadically [124]. More recently, the CASSCF method was used to determine the role of vitamin B12 cofactors in enzymatic processes [141,142,143,144].
Vitamin B12 bioactivity is necessary for human DNA synthesis [145], red blood cell maturation and maintenance, mammalian cellular metabolism, and a healthy nervous system [146]. Vitamin B12 deficiency leads to weak muscles, numbness or a tingling feeling in the extremities, difficulty walking, nausea, mood changes, depression, memory loss, disorientation, dementia, decreased appetite, weight loss, irritability, lack of energy, skin deterioration, and death in severe cases [147]. Alzheimer’s disease is also connected to vitamin B12 deficiency [148]. Knowledge of the mechanisms of vitamin B12-dependent processes is necessary for the correct application and further development of vitamin B12 treatments for patients with inherent genetic diseases, acquired malabsorption diseases, and diet-related deficiencies [149,150,151,152]. This review is dedicated to the study of the mechanisms involving vitamin B12 cofactors in humans.
2. Experimental Mechanistic Evidence
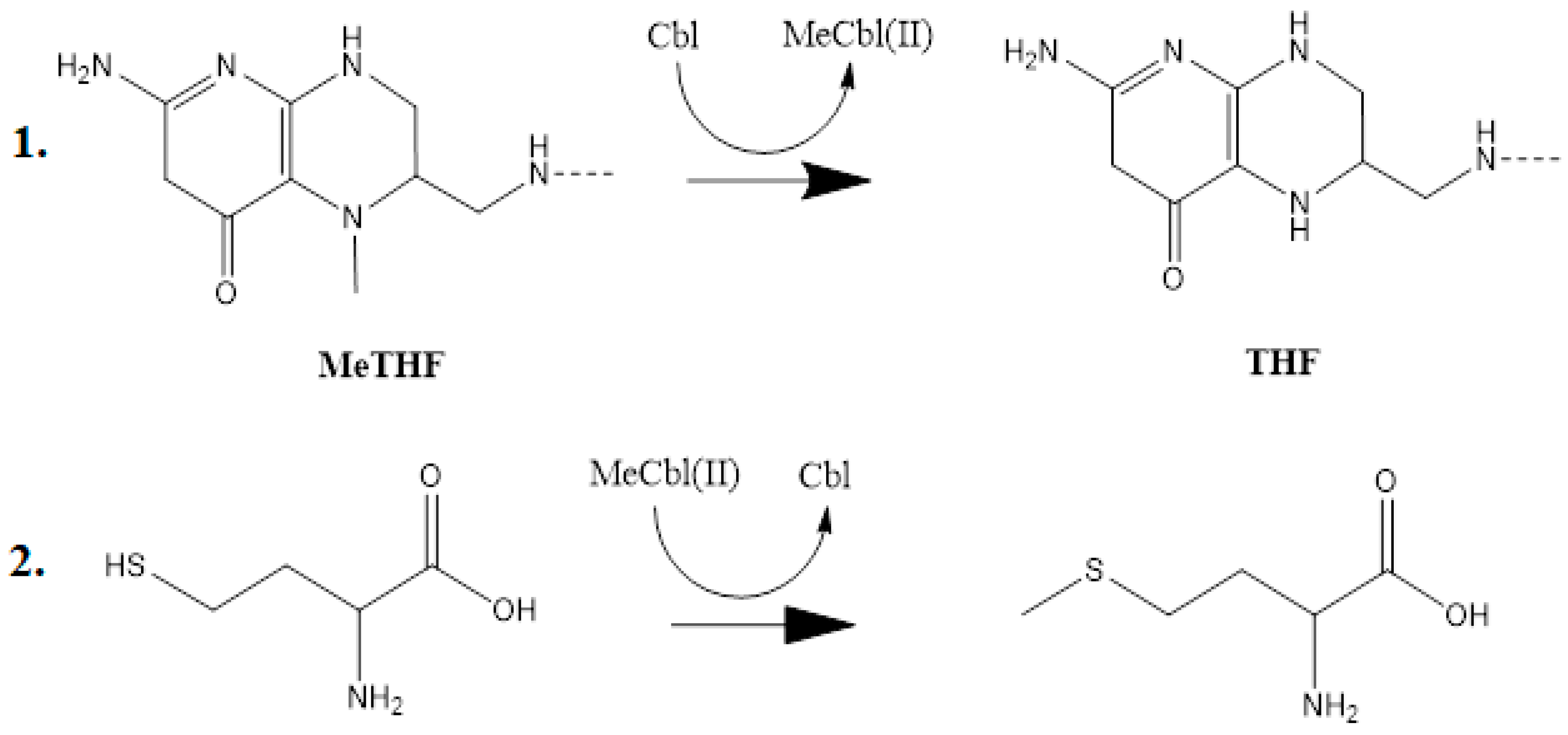
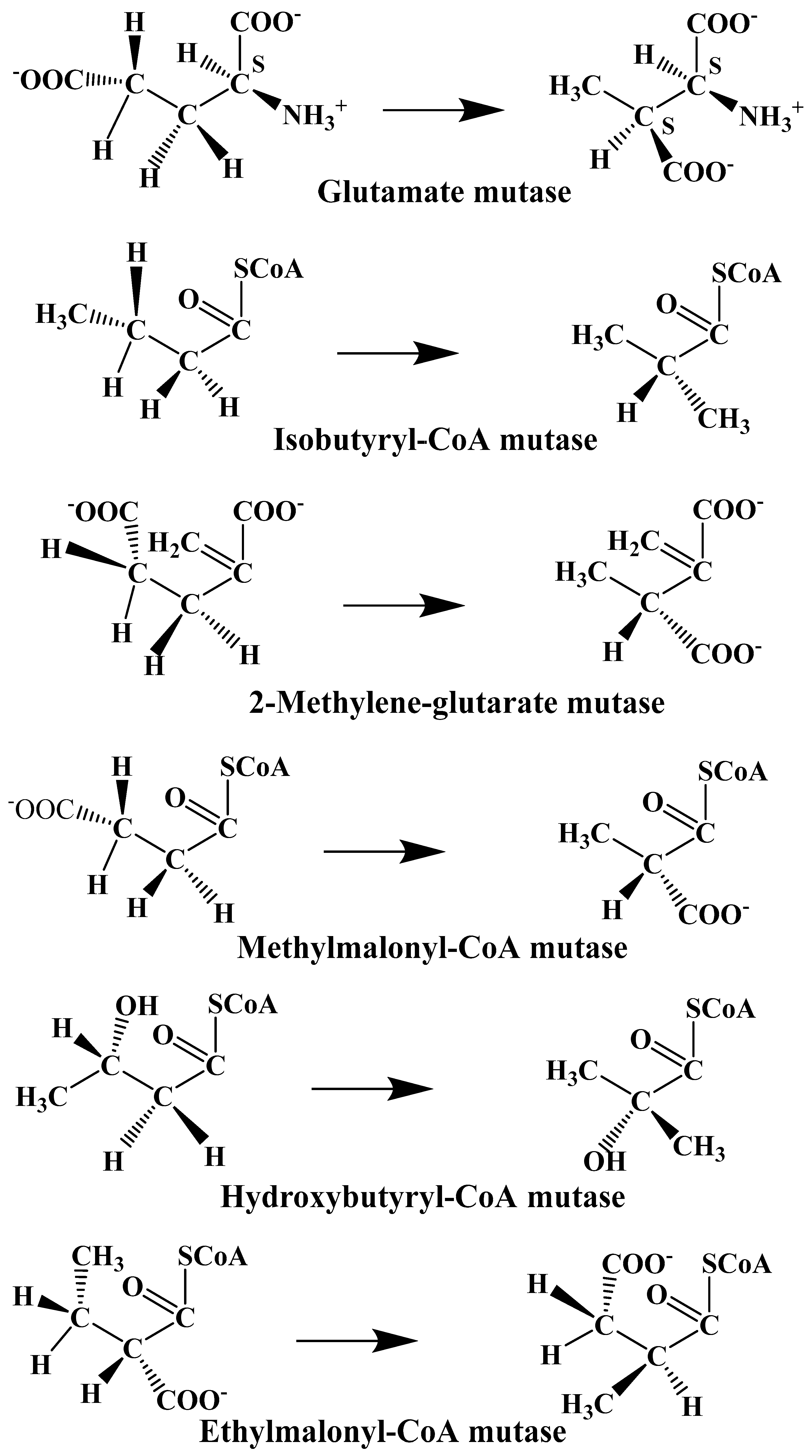
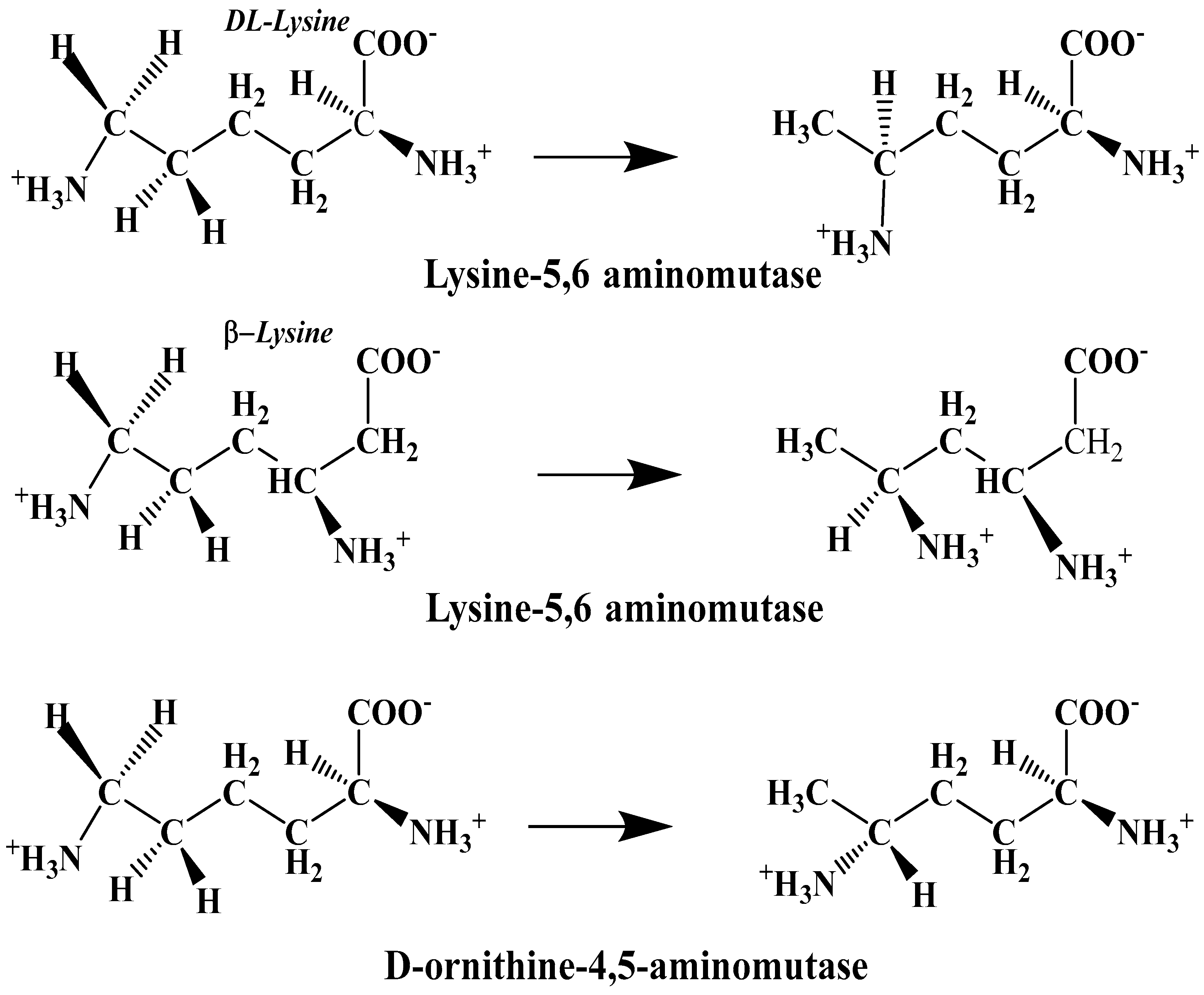
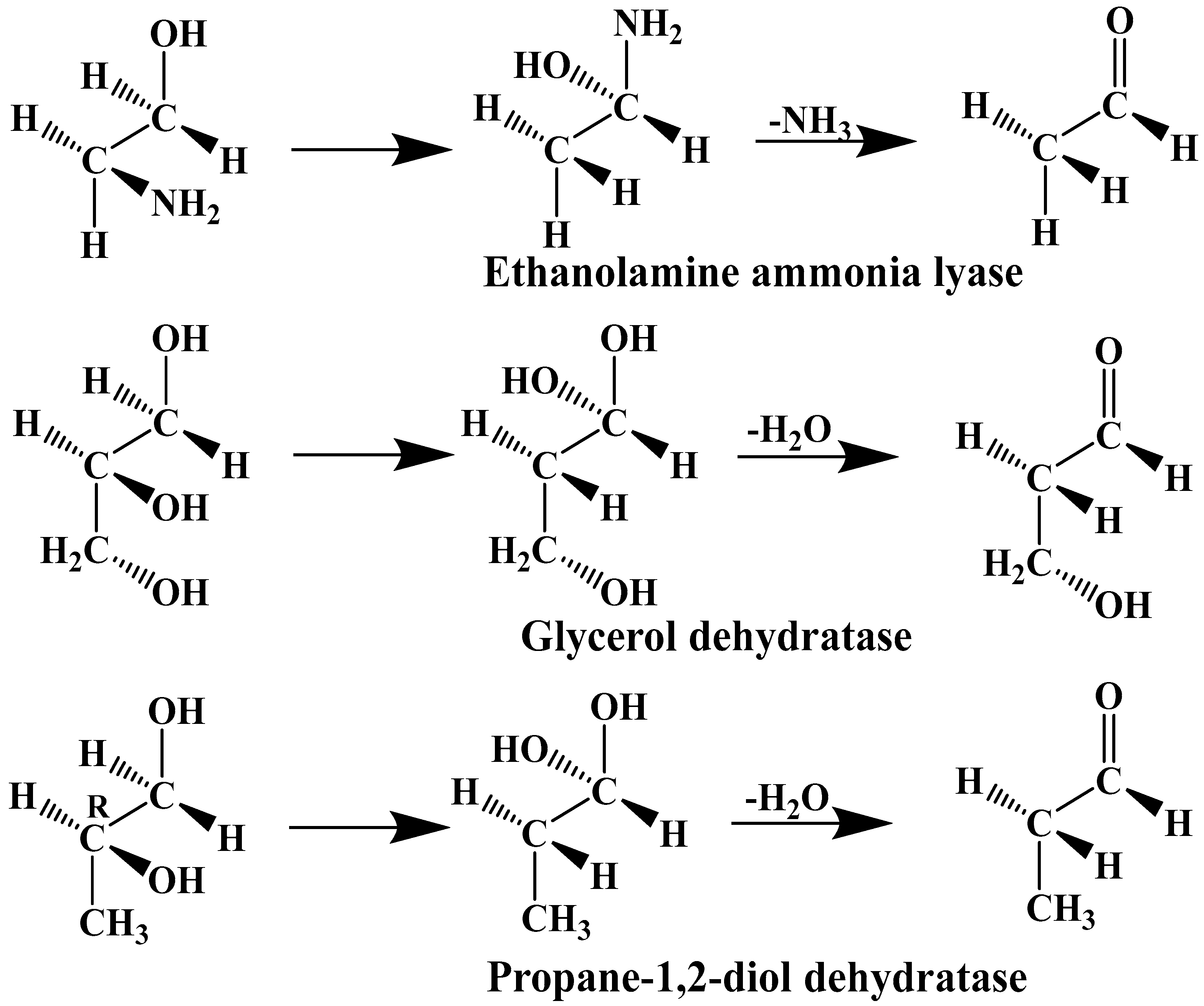
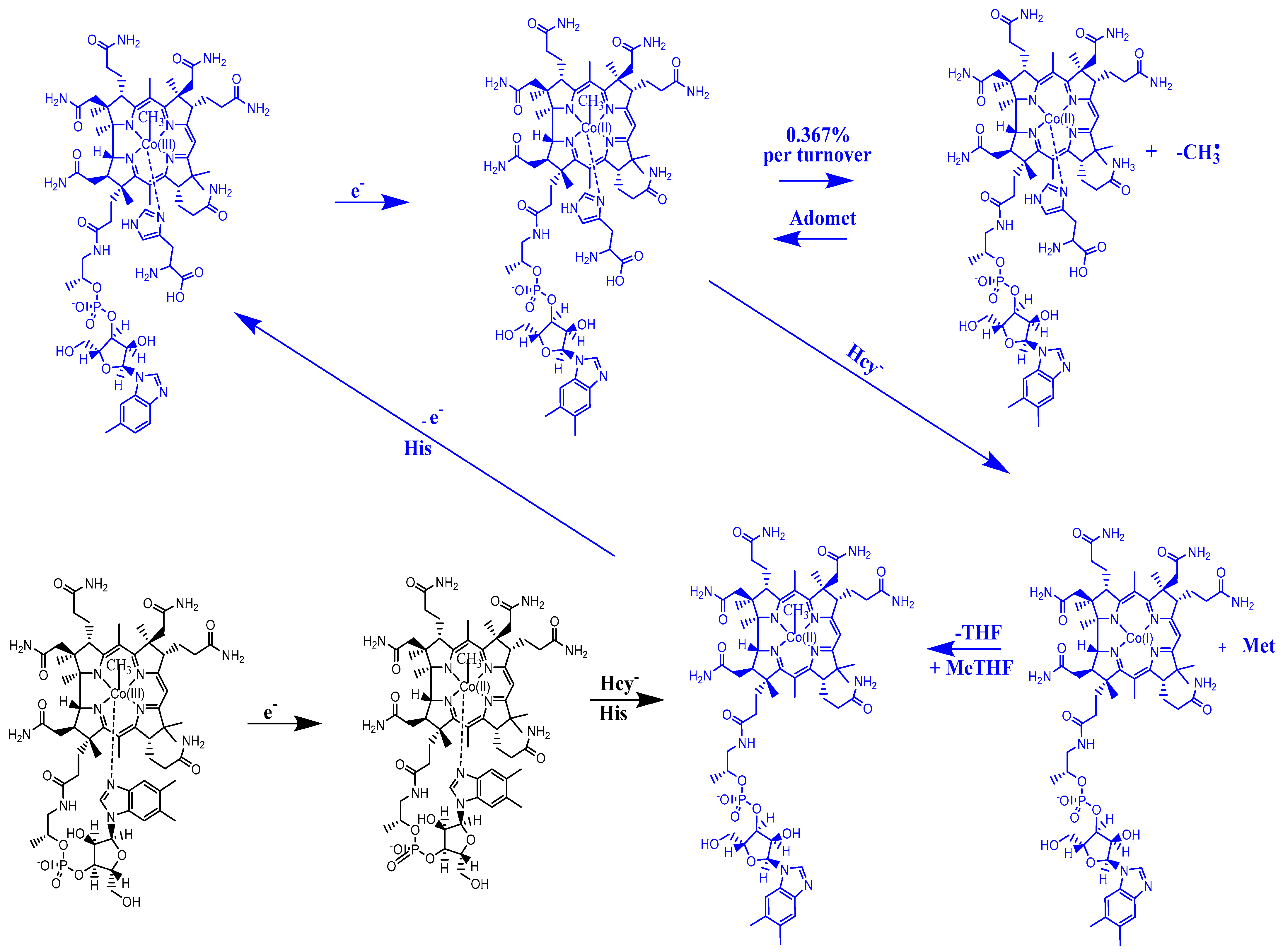
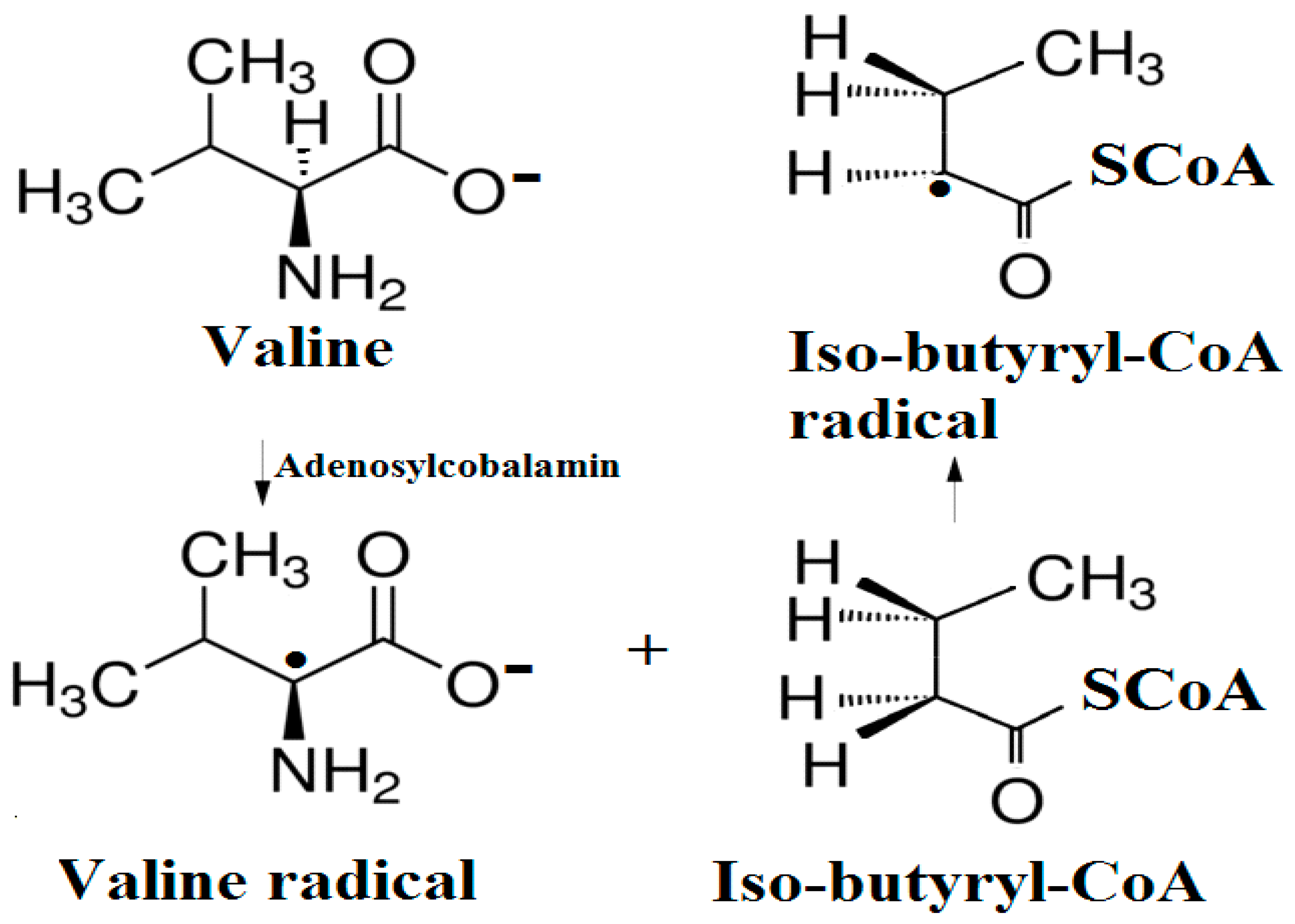
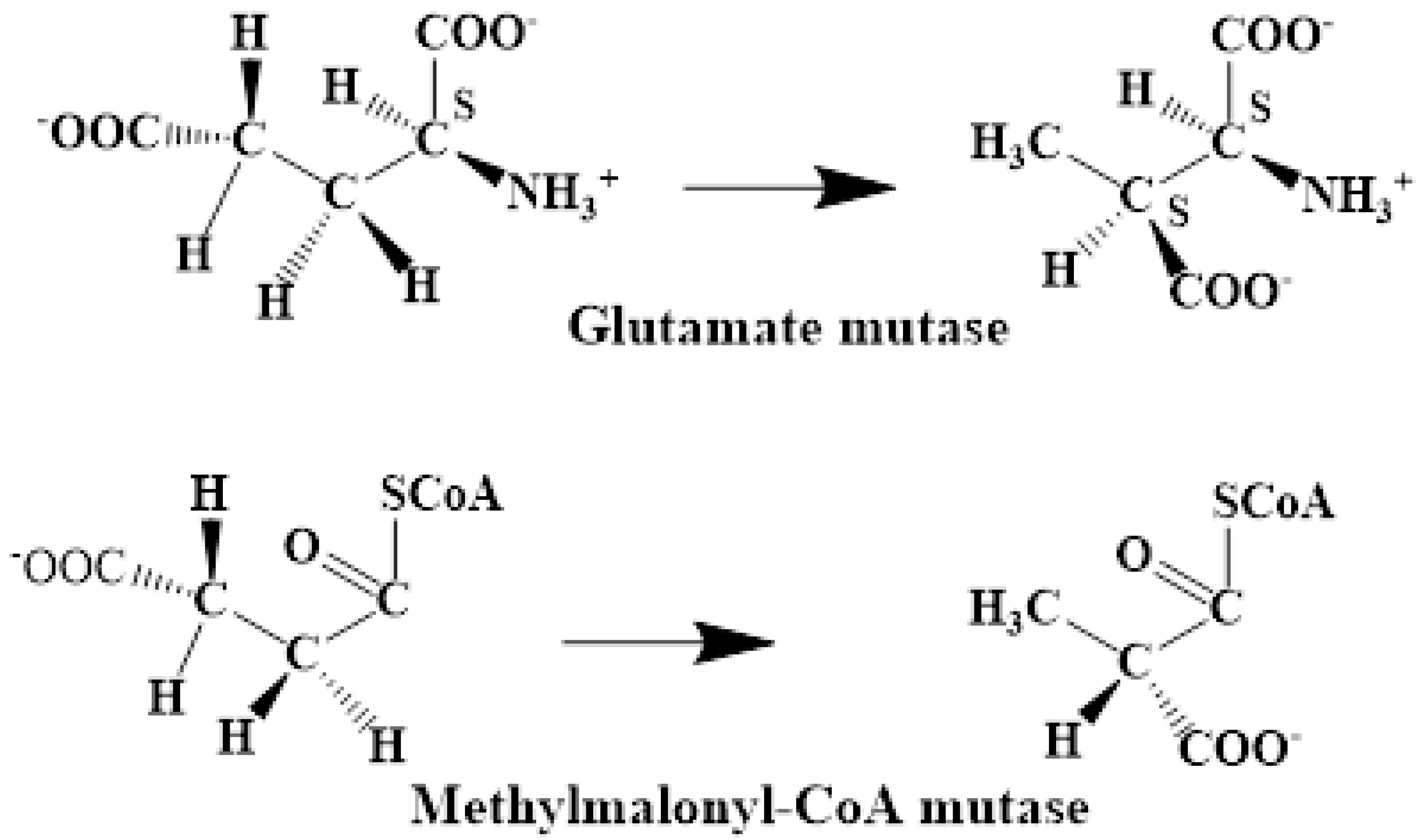
The experimental data regarding all the biochemical processes of the methylcobalamin and adenosylcobalamin cofactors are presented here. The methylcobalamin cofactor plays a key role in the synthesis of methionine from homocysteine via transferring the methyl radical from the 5-methyltetrahydrofolate. Formally, the process takes place in two stages. In the first stage (Figure 2), the methyl radical is transferred from the 5-methyltetrahydrofolate molecule to the two-electron-reduced cobalamin cofactor. In the second stage, the same methyl radical is transferred to homocysteine, thus forming the methionine molecule. The 5′-deoxy-5′-adenosyl-ligand-radical is one of the most active radicals in all known enzymatic processes within mammals [153]. One of the biochemical sources of the 5′-deoxy-5′-adenosyl radical is the adenosylcobalamin cofactor [153]. Unlike methylcobalamin, which is considered a cofactor in the Methionine Synthase process, the adenosylcobalamin cofactor transforms a whole series of substrates from their common structures into unusual isomers [65]. These isomers further participate in biochemical processes, some releasing small molecules. Depending on the type of isomeric transformation and whether small molecules are removed, these transformations have been divided into skeleton mutase, amino-mutase, and eliminase bioprocesses (Figure 3, Figure 4 and Figure 5). Skeleton mutases are named as such due to the unique modification of the carcass of their carbon atoms. Six adenosylcobalamin-dependent skeleton mutase bioprocesses are currently known: Glutamate Mutase, isobutyryl-CoA mutase, 2-methylene glutarate mutase, Methylmalonyl-CoA Mutase, hydroxybutyryl-CoA mutase, and ethyl malonyl-CoA mutase (Figure 3). Aminomutases are named as such due to the position change of the amine group in the newly formed isomers. Three adenosylcobalamin-dependent aminomutase bioprocesses are currently known: lysine-5,6 amino mutase with the participation of D, L-lysine substrate, lysine-5,6 amino mutase with the participation of the B-lysine substrate, and D-ornithine-4,5 amino mutase (Figure 4). Eliminase processes are named as such due to the elimination of small molecules by the newly formed isomers. Presently, three adenosylcobalamin-dependent eliminase bioprocesses are known: ethanolamine ammonia-lyase, glycerol dehydratase, and propane 1, 2-diol dehydratase (Figure 5).
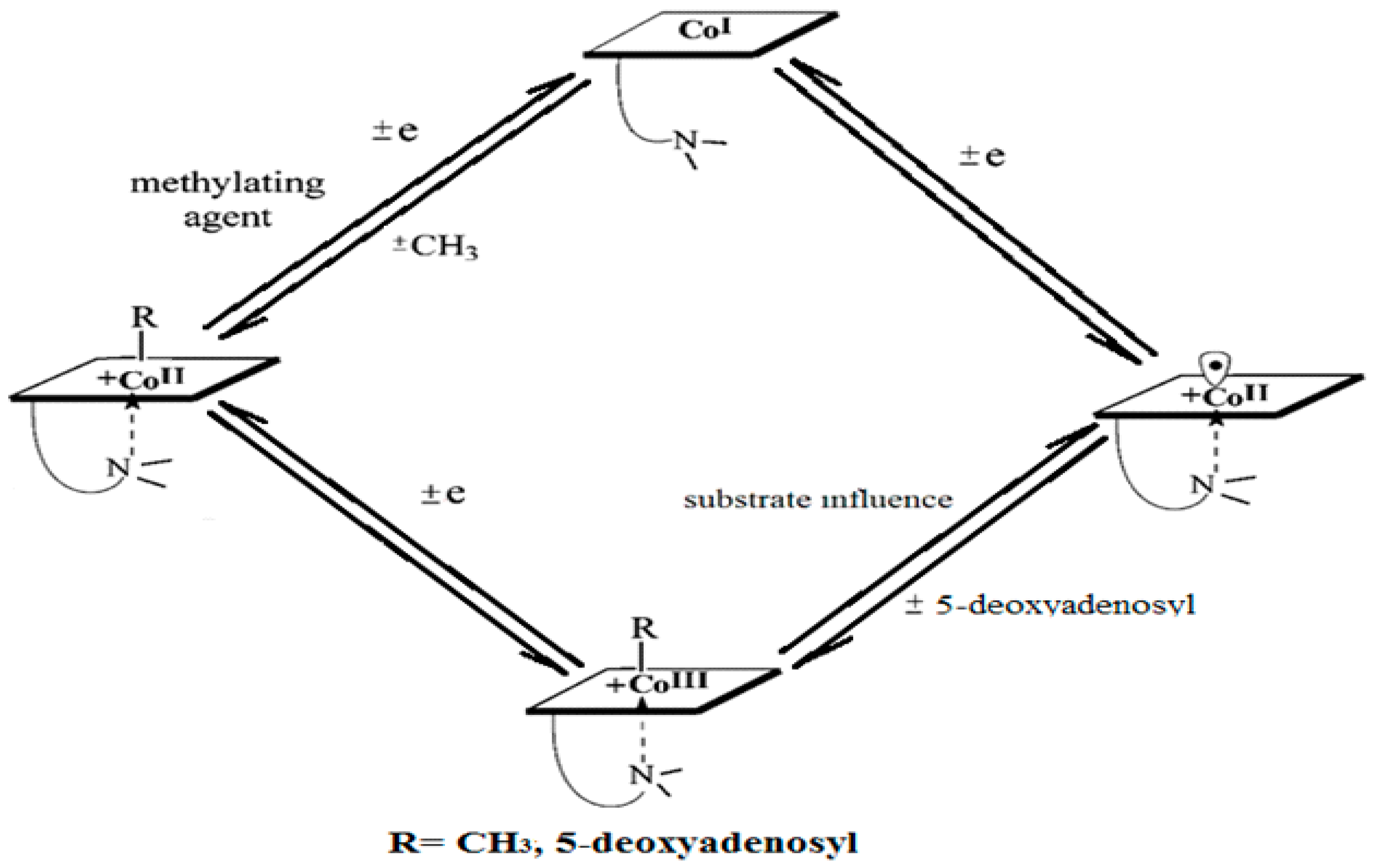
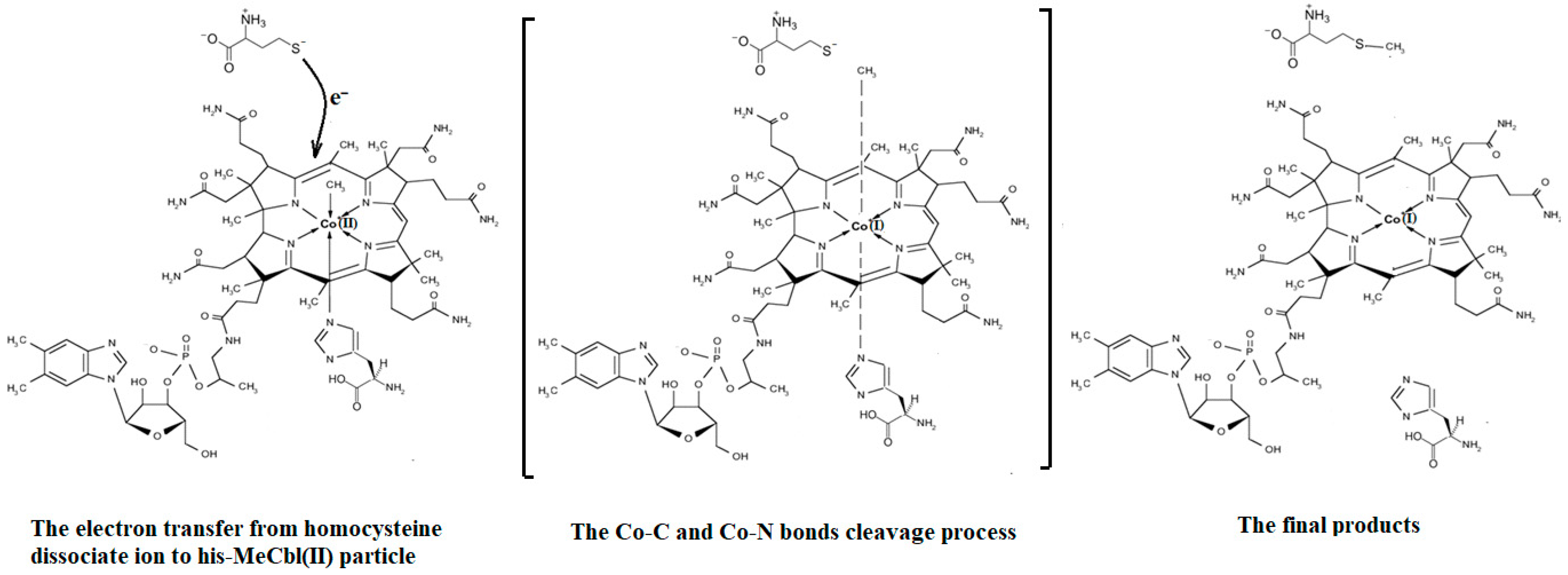
A common feature of the methylcobalamin and adenosylcobalamin cofactor-dependent bioprocesses is the cycling in the oxidation state of the central cobalt atom from +3 to +1 during their turnovers. In the Methionine Synthase process, the methylcobalamin cofactor is reduced first with one electron. Then, methylcobalamin is demethylated and is reduced with another electron so that the central cobalt ion has a +1 oxidation state. Following the turnover of the Methionine Synthase process in the reverse direction, the methylcobalamin cofactor with +1 charge of the central atom is methylated by a methylating agent, usually a 5-methyltetrahydrofolate system, which is also oxidized with one or two electrons. This concludes the turnover of the Methionine Synthase process (Figure 6).
Unlike in methylcobalamin, the Co-C bond in the adenosylcobalamin is broken in the +3 oxidation state of the central cobalt ion with the participation of the substrates of the adenosylcobalamin cofactor-dependent bioprocesses. This assessment of adenosylcobalamin cofactor-dependent catalysis is not drawn from the concrete experimental data but is deduced from the context of in vivo processes. Several other differences exist between the methylcobalamin-dependent Methionine Synthase process and the adenosylcobalamin cofactor-dependent bioprocesses. The breaking of the Co-C bond in the methylcobalamin cofactor is thought to be heterolytic in nature, while that of the adenosylcobalamin cofactor is thought to be homolytic in nature. Additionally, there is no experimental evidence of substrate involvement in the methylcobalamin cofactor Co-C bond cleavage during the Methionine Synthase process, while substrate involvement in Co-C cleavage in the adenosylcobalamin cofactor is experimentally demonstrated. Furthermore, the Co-C bond cleavage of the methylcobalamin cofactor leads to the +1 oxidation state of the central cobalt ion in the Methionine Synthase process, while the breaking of the Co-C bond in adenosylcobalamin-dependent processes gives rise to the cofactor species with a +2 oxidation state on the central cobalt ion and to the 5′-deoxy-5′-adenosyl radical.
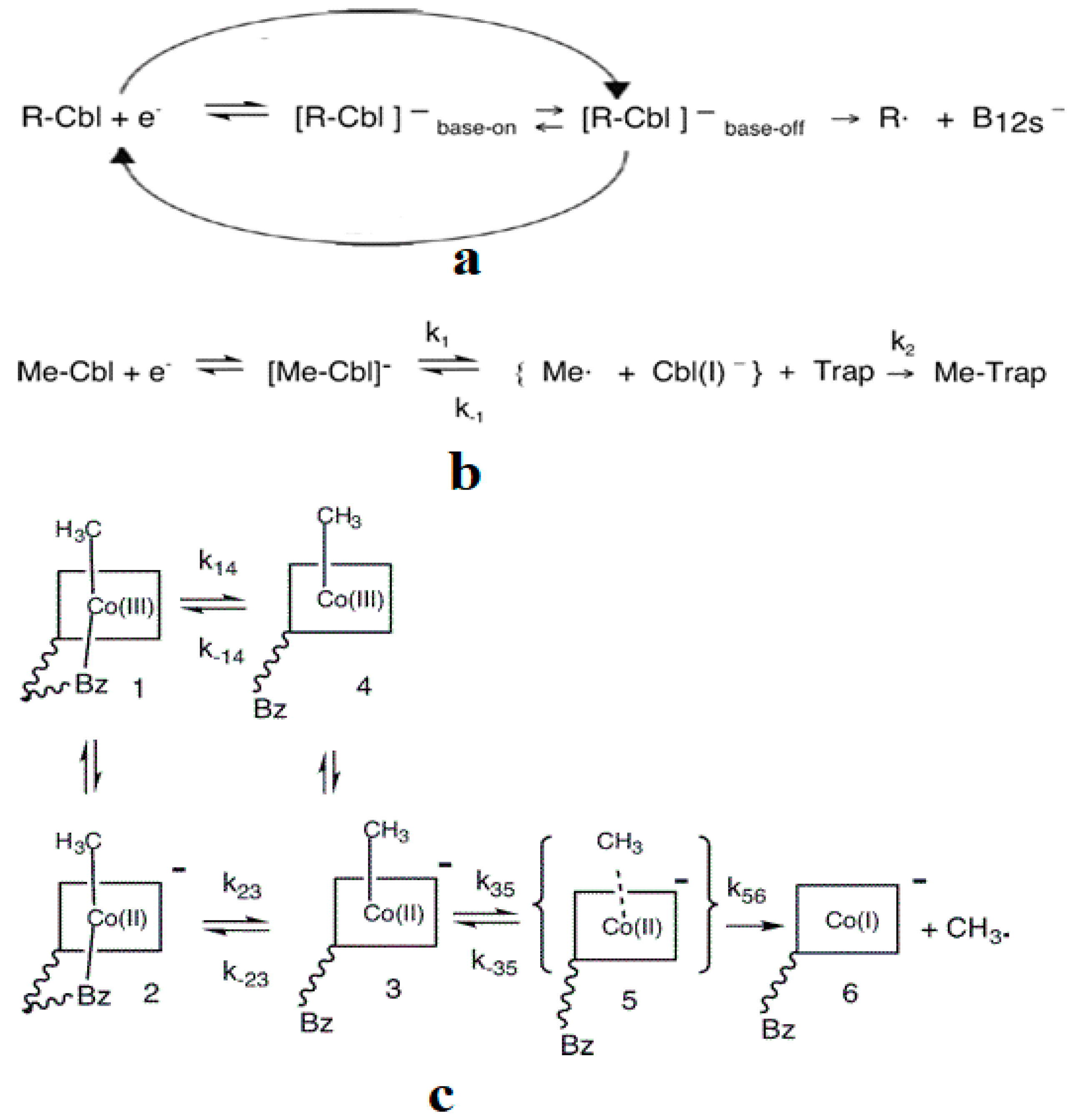
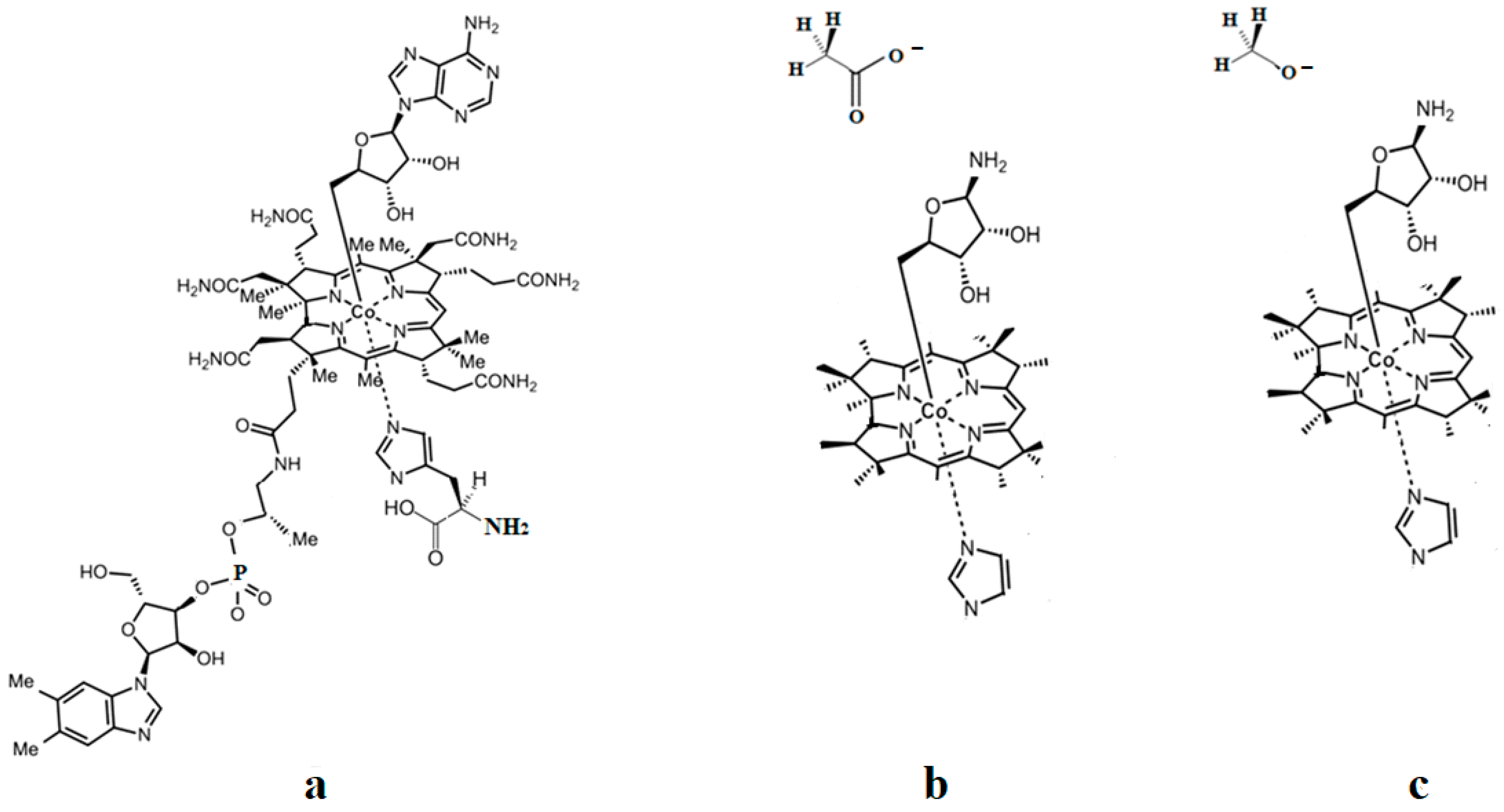
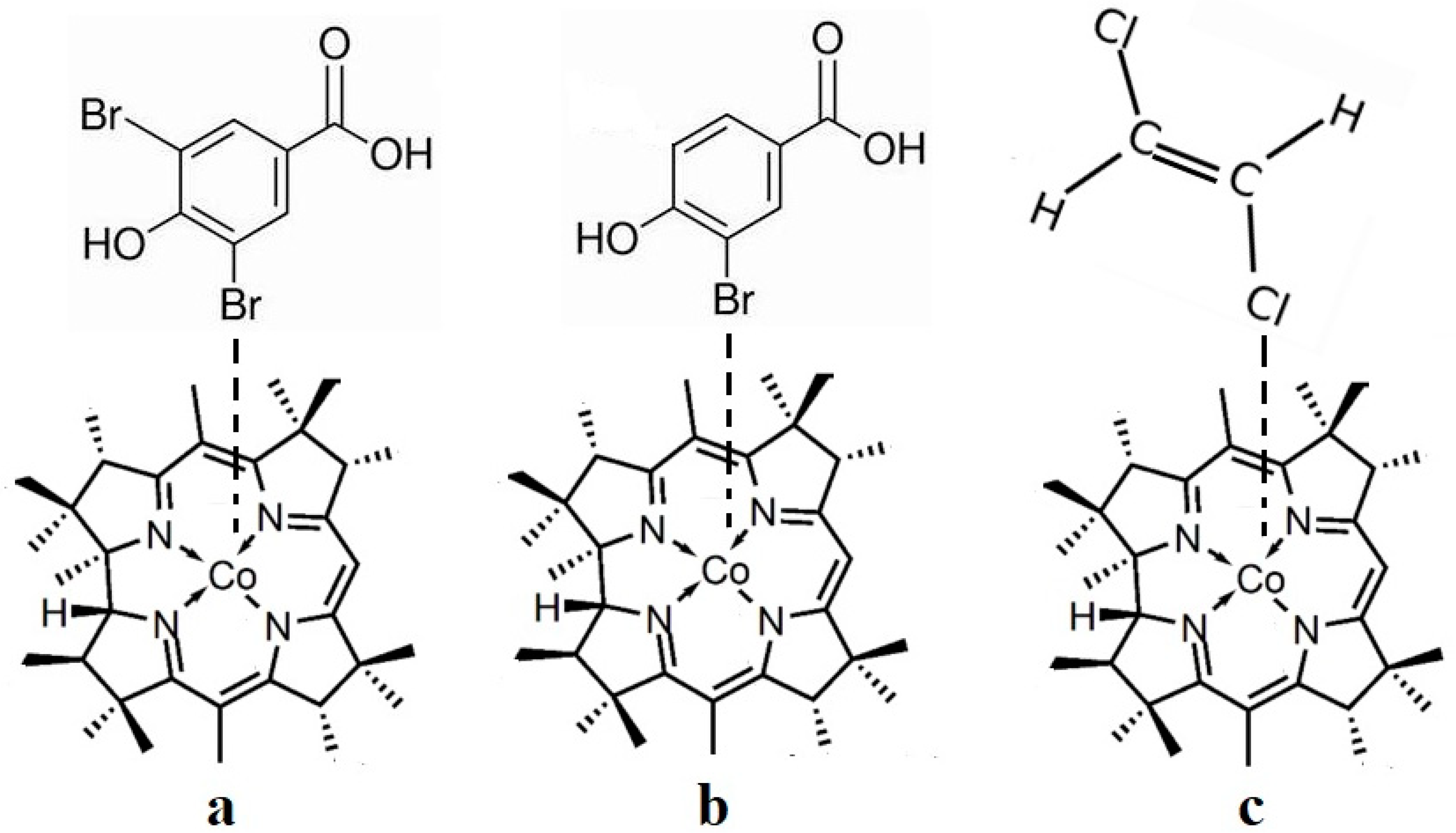
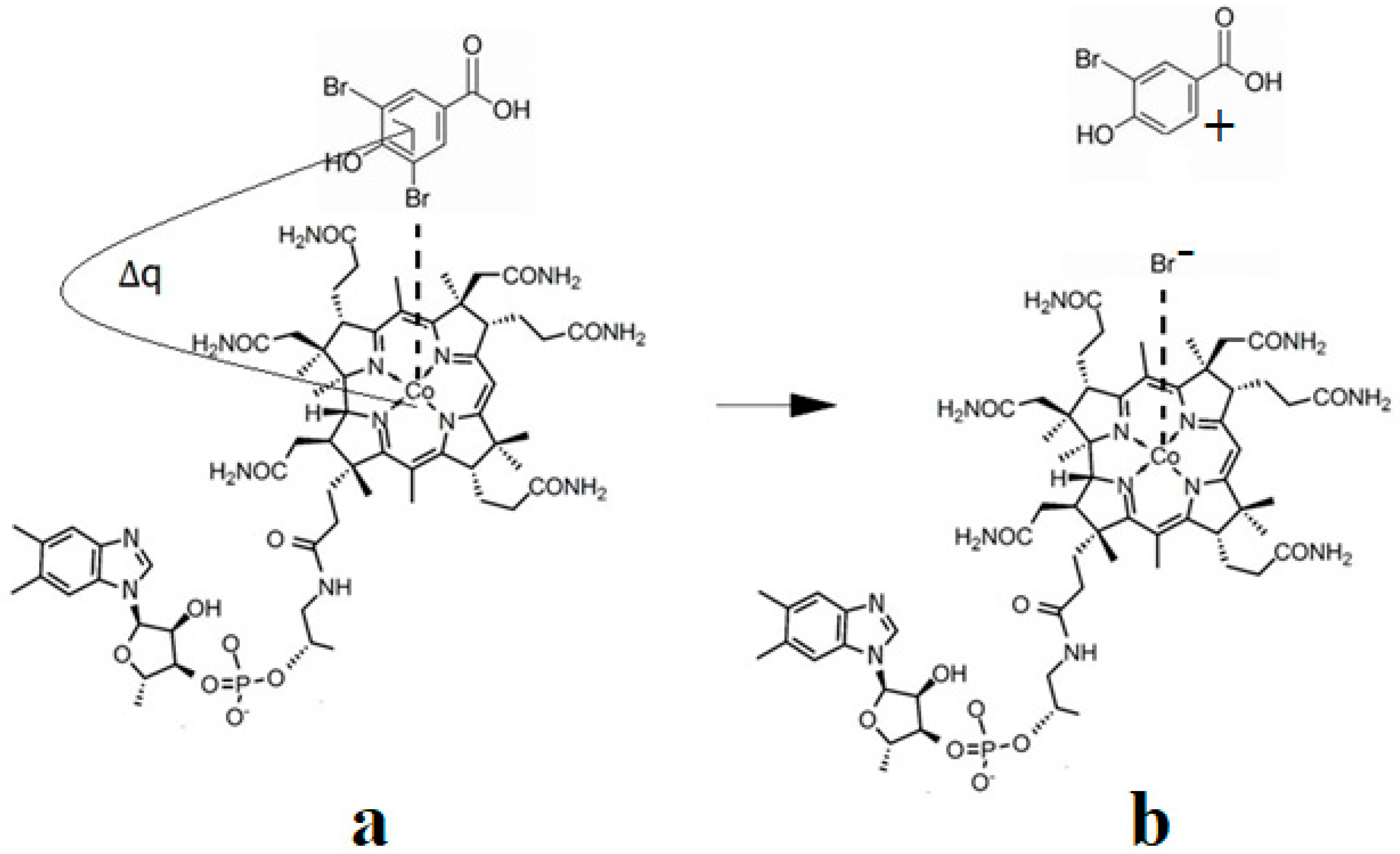
As described above, all the enzymatic processes involving vitamin B12 cofactors have the central cobalt atom cyclically changing its oxidation state between +3 and +1. This served as a starting point for studying the methylcobalamin cofactor mechanism in its dependent biochemical processes by using electrochemical methods such as cyclic voltammetry [88,89,90,154]. As for the adenosylcobalamin cofactor-dependent biochemical processes, cyclic voltammetry was not used due to its more complex processes, which include hydrogen transfer from the substrate to the 5′-deoxy-5′-adenosyl ligand. Lexa and Savéant [88] studied the Co-C and Co-N axial bond cleavage of methylcobalamin under the influence of the central atom oxidation state reduction using cyclic voltammetry. It should be noted that the complete studies were performed only for the cobinamide molecule, promoting the idea that the active particle in the Methionine Synthase process is the base-off version of the methylcobalamin cofactor. Nevertheless, the authors presented a presumed mechanism of the electrolytic process for the Co-C and Co-N axial bond cleavages in the methylcobalamin cofactor molecule (Figure 7a).
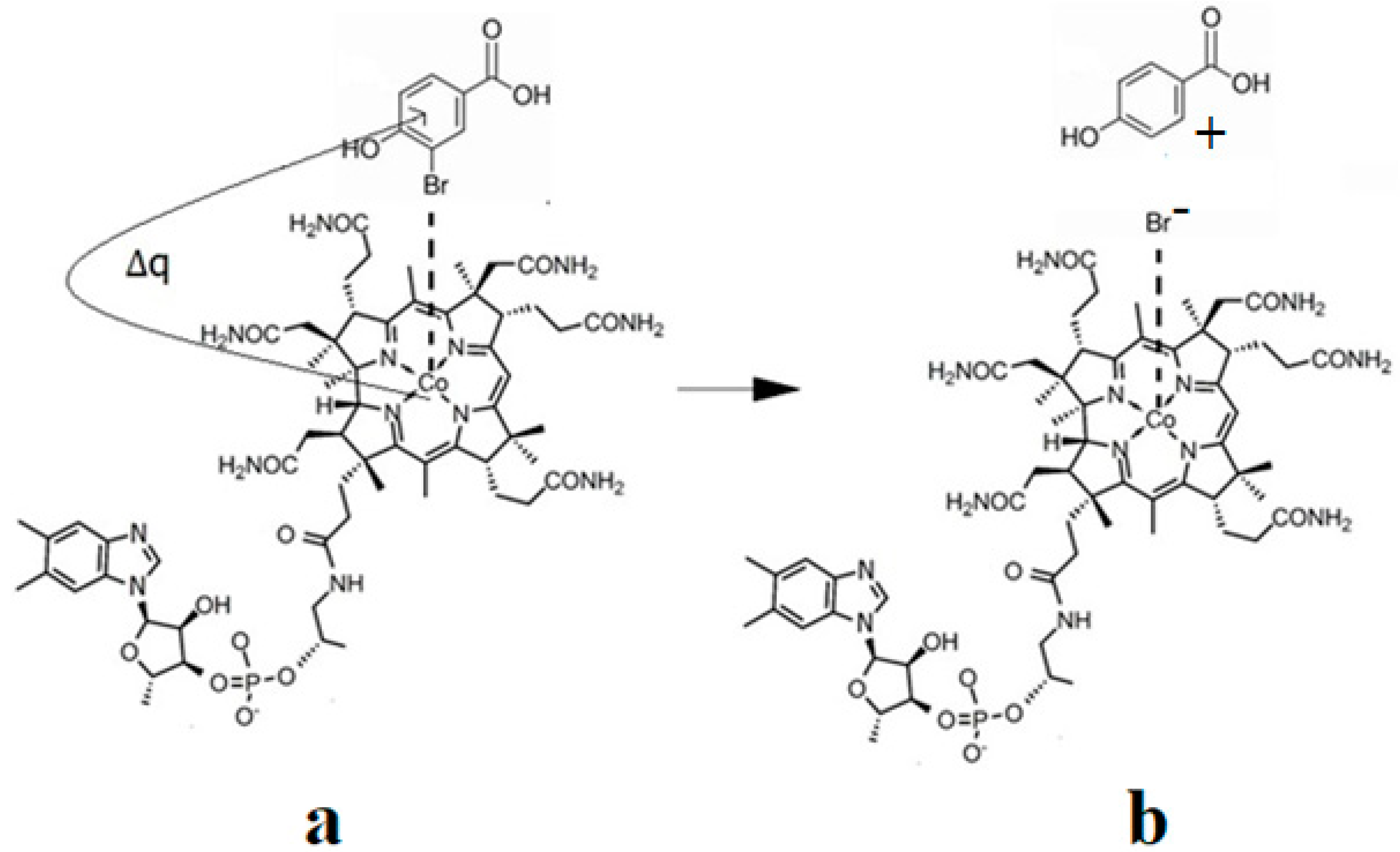
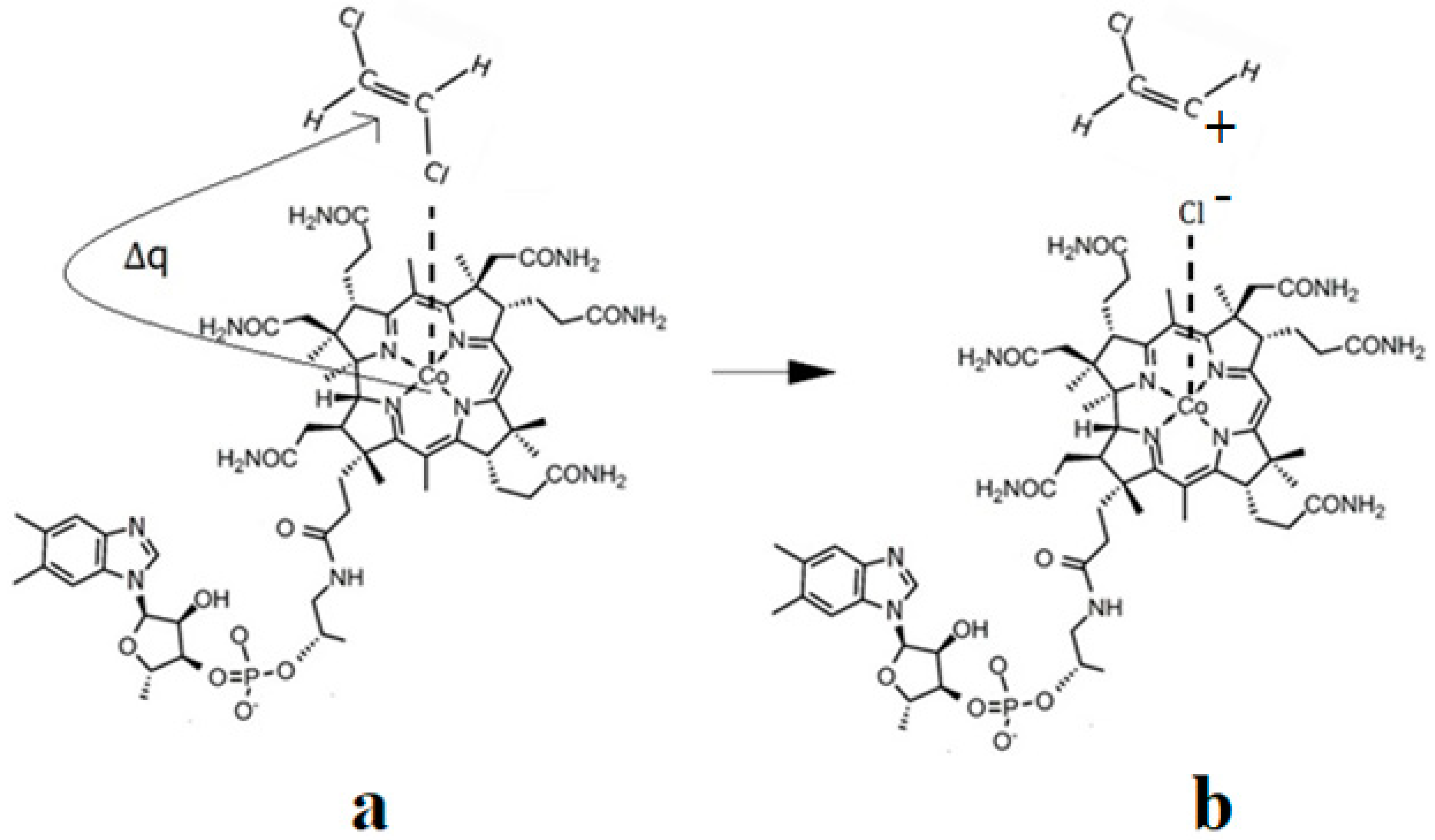
Martin and Finke [154] studied the electrochemical process of the methylcobalamin cofactor in more detail. They concluded that the rate of the Co-C cleavage reaction depends largely on the nature of the solvent in which the reaction takes place. Therefore, the authors proposed a mechanism of the Co-C bond cleavage in the methylcobalamin cofactor that introduced a so-called cage in which the reactant particles were coated with a layer of molecules of the solvent used in the experiment (Figure 7b). Finally, Birke and colleagues [89,90] used electrochemical modeling software and experimental electrochemical experiments to study the Co-C cleavage reaction in a number of compounds. These compounds included methylcobalamin cofactor derivatives as well as the methylcobalamin cofactor molecule. They proposed a more sophisticated electrochemical reaction mechanism of the methylcobalamin cofactor Co-C bond cleavage (Figure 7c), in which they took into account both the electrochemical research data mentioned above and the data obtained in their own findings.
Unfortunately, all of the mechanisms deduced from these electrochemical experiments cannot be used to study the more complex process of the methylcobalamin cofactor Co-C bond cleavage in the in vivo Methionine Synthase process due to at least two reasons: 1. Experimental electrochemical processes are severely impaired by the nature of the solvent in which they occur; 2. The mechanisms proposed by the authors of this research were intentionally constructed so that the Co-C bond breaks only in the base-off compound of methylcobalamin, in which the Co-N bond is already missing. This excludes the possibility that the Co-C bond cleavage may occur in the base-on structure of methylcobalamin.
The Co-C cleavage in methylcobalamin and adenosylcobalamin also was studied by using thermo- [155] and photo-chemistry [155,156,157,158,159]. Researchers looked for factors that would promote the breakdown of the Co-C bond in adenosylcobalamin cofactor-dependent processes. Many triggering factors and mechanistic details of Co-C bond cleavage were proposed as a result of these experimental studies. Such effects as axial ligand trans-influence, steric effects, structural strain, and protein influence [74,75,76,77,78,79,80,81,82,83,84,85,86] were proposed as decisive factors in the Co-C cleavage process during the vitamin B12 cofactor-dependent bioprocesses. The research community’s subsequent experimental and theoretical data mostly did not confirm the Co-C bond cleavage mechanisms and the triggering factors. The only confirmed proposed key factor determining the Co-C bond cleavage in adenosylcobalamin cofactor-dependent bioprocesses was the hydrogen transfer from the substrate to the 5′-deoxy-5′-adenosyl ligand [55,66,67,68,69,70,71,72,73]. Although this was deduced in subsequent experimental studies [14,15,16], its role in breaking the Co-C bond in adenosylcobalamin cofactor-dependent processes was not demonstrated experimentally.
Based on the above, the mechanism of vitamin B12-dependent processes can only be schematically determined from experimental data. The details of the methyl radical transfer from 5-methyltetrahydrofolate to the cob(I)alamin cofactor in the first phase of the methylcobalamin cofactor-dependent process were not revealed in experimental data. In its second phase, the transfer of the methyl radical from the methylcobalamin cofactor to homocysteine to obtain methionine cannot be studied in detail via the electrochemical method. Unfortunately, this method also cannot contribute to the in vivo mechanisms determination for several reasons: 1. There is no common opinion among the authors of these studies about the electrochemical Co-C bond cleavage reaction mechanism; 2. There is a dependence of the mechanism and of the constant rate of these electrochemical processes on the nature of the solvent; 3. The mechanistic details of these electrochemical processes were built on the idea that the Co-C chemical bond can be broken only in the case of methylcobalamin’s base-off species, neglecting the case of the base-on methylcobalamin species. Similarly, the details of the Co-C bond cleavage reaction mechanism in adenosylcobalamin-dependent processes cannot be determined exclusively from experimental data. Experimental results have determined that the Co-C bond cleavage takes place and that a hydrogen atom transfers from the substrate to the 5′-deoxy-5′-adenosyl ligand during the adenosylcobalamin processes; however, the relationship between these two was not determined. Is the actual mechanism concerted or stepwise? What is the nature of substrate participation along with the Co-C bond cleavage process? Does the hydrogen transfer from the substrate to 5′-deoxy’-5′-adenosyl-ligand influence the Co-C bond cleavage in the studied bioprocesses? All these mechanistic questions are not and (perhaps) cannot be answered solely by using experimental data. The experimental data must be complemented by theoretical results based on approximations (including credible and correct geometric structural models) appropriate to the nature of the studied vitamin B12-dependent processes.
3. DFT and QM/MM (Based on DFT) Calculations
3.1. General Considerations
A brief history of the DFT and QM/MM method based on density functional theory and the most summary data on the results of theoretical calculations of vitamin B12 cofactors by using these methods are presented in this section. A 1964 paper by Pierre Hohenberg and Walter Kohn [160] laid the groundwork for a revolutionary application of quantum mechanics to physics, chemistry, and biology. It described how multielectronic ground states could be determined by electron density within three geometric dimensions. The practical application of density functional theory (DFT) became possible only at the end of the 20th century following the theoretical development of functionals, basis sets, and the use of computers in DFT calculations. By using fewer computational capabilities, calculations based on DFT began to give much more accurate results than the Hartree–Fock (HF) method and its modifications. Due to this, researchers became enthusiastic about DFT applications in various fields. Here is a lengthy quote illustrating this optimism of the time:
“Density functional theory (DFT) finds increasing use in applications related to biological systems. Advancements in methodology and implementations have reached a point where predicted properties of reasonable to high quality can be obtained. Thus, DFT studies can complement experimental investigations, or even venture with some confidence into experimentally unexplored territory. Many properties can be calculated with DFT, such as geometries, energies, reaction mechanisms, and spectroscopic properties. A wide range of spectroscopic parameters is nowadays accessible with DFT, including quantities related to infrared and optical spectra, X-ray absorption, and Mössbauer, as well as all of the magnetic properties connected with electron paramagnetic resonance spectroscopy except relaxation times. Density functional theory is considered an extremely successful approach for the description of ground-state properties of metals, semiconductors, and insulators. The success of DFT not only encompasses standard bulk materials but also complex materials such as proteins and carbon nanotubes”[161]
The quantum mechanics/molecular mechanics (QM/MM) method [162] is a different computational method that can and often does incorporate DFT approximation. It was invented to calculate particularly large systems. The central part of the system that is particularly important for the proposed purpose is usually processed by the DFT method (QM for quantum mechanics). Meanwhile, the other less relevant part of the system (usually that of some substrates) is treated through a simpler molecular mechanics (MM) method. The molecular mechanics method is an approach of parametric fitting of the electronic structure, which requires fewer computational resources. In fact, QM/MM (with the QM part based on DFT) is the same methodology as DFT but can possibly use the influence of large substrates on the main computing core, which is usually treated with the same DFT method. Calculations using the QM/MM method basically have results that usually have improved parameters but are along the same theoretical line as the DFT method, hence the increasing enthusiasm for the use of these methods among the wider masses of researchers. Not surprisingly, due to this enthusiasm, most computational studies of electronic structure and vitamin B12-dependent mechanisms were conducted using DFT or QM/MM based on DFT. A large number of properties of cobalamins and of vitamin B12-dependent bioprocesses were studied in this way: the structural properties of cob(I)alamin cofactor [100], the stereoelectronic properties of cobalamins [101], the dissociation energy of the Co-C bond in cobalamins [102,103,104], the analysis of the electronic structure and breaking energy of the Co-C bond in cobalamins [104,125,129,133,134,135], the trans-influence effect in cobalamins [112], the force-field and vibrational analysis in methylcobalamin [106,107], the electronic spectra of cobalamins [114,115,116], the synthesis of methionine from homocysteine [113,114,128], the activation of the Co-C bond in methylcobalamin [120,121,122], the transfer of the methyl radical to cobalamin and the formation of the Co-C bond [125], and the study of the mechanism of the B12-dependent Methylmalonyl-CoA Mutase process and of the adenosylcobalamin cofactor-dependent biochemical processes [132,133,134,135,136,137,138,139,140].
As can be seen in Figure 2, the Methionine Synthase bioprocess consists of two stages. In the first stage, the cobalamin cofactor accepts the methyl radical from the 5-methyl tetrahydrofolate and forms the Co-C bond. In the second stage, the Co-C bond breaks to transfer the methyl radical from the methylcobalamin to the homocysteine molecule by synthesizing methionine. Interestingly, in the case of adenosylcobalamin cofactor-dependent processes, the underlying reaction is similar regarding the cleavage of the Co-C bond. In addition, it should be noted that the Co-C bond is quite strong, and the rate of the in vivo Co-C cleavage reaction is believed to be 12 orders higher than the breaking of the same bond in the in vitro reaction [56,59]. Therefore, the study of the process involving the breaking of the Co-C bond has aroused lively interest in the scientific community studying vitamin B12 cofactors. In principle, the Co-C chemical bond cleavage process can be rationalized in two ways: 1. An SN1 reaction that breaks the Co-C bond without any influence of other molecules or ions; or 2. An SN2 reaction that breaks the Co-C bond as a result of the methylcobalamin cofactor interacting with a substrate.
A remarkable number of authors have studied the Co-C bond cleavage in vitamin B12 cofactors using DFT by following either the SN1 or the SN2 reaction mechanism. Many other DFT calculations of the vitamin B12 cofactors were done but are less relevant to this mechanistic review. These calculations were performed to study properties not directly related to the mechanisms of the vitamin B12 cofactors catalyzed processes. Some such properties were found using: (a) a comparative analysis of the application of different basis sets and functionals with more complex methods of electronic structure calculations; (b) an electronic and vibrational spectra analysis; and (c) minor effects of the influence of the trans effect and side chains on the Co-C bond dissociation process [104,105,106]. Indeed, one can observe that these studies are not particularly relevant to the mechanisms of the vitamin B12-dependent processes and therefore are outside the scope of this paper. Instead, electronic structure calculations of the Co-C cleavage reaction in the methylcob(II)alamin cofactor, for instance, are relevant to studying the mechanisms of the real methylcobalamin and adenosylcobalamin cofactor-dependent processes.
3.2. Calculations Involving Methylcobalamin
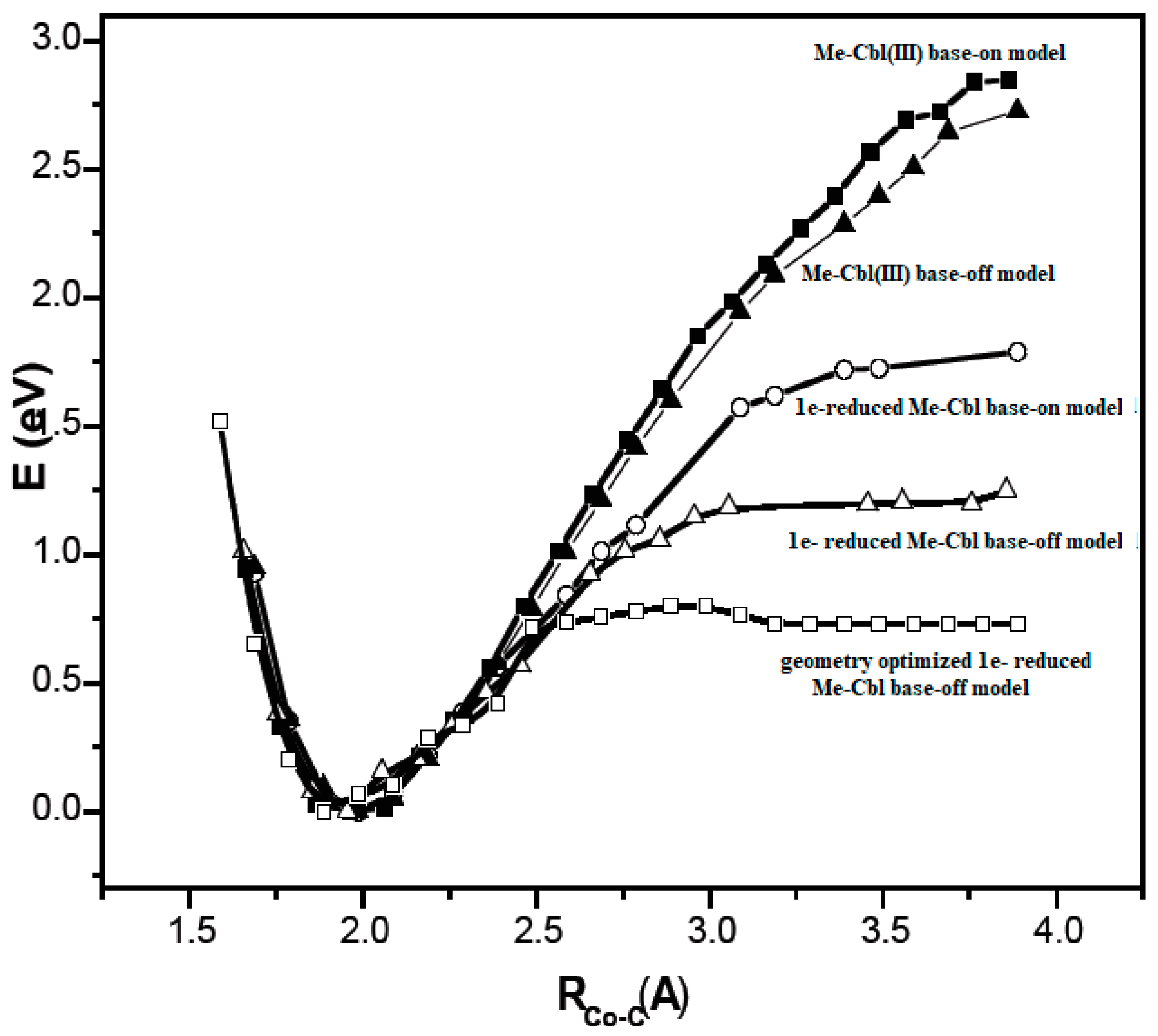
A critical analysis of the DFT and QM/MM calculations involving the methylcobalamin cofactor is presented here. According to the experimental data, the methylcobalamin cofactor can exist in two structural compositions: base-on (Figure 8b) and base-off (Figure 8c). It is also known that outside of biological processes, the methylcobalamin cofactor has a central coordinating ion with a +3 oxidation state. It has also been inferred from experimental data that the central cobalt ion in the methylcobalamin cofactor is first reduced by one electron in the Methionine Synthase biochemical process. Therefore, the total energy curves of the Co-C bond-breaking reaction path in the methylcobalamin cofactor following the SN1 reaction mechanism were determined by using DFT calculations for its base-off and base-on species and for the +3 and +2 oxidation states of the central cobalt coordinating ion [126]. In the electronic structure DFT calculations, the axial ligand histidine was replaced with the imidazole ligand, and hydrogens replaced the side chains. The B3LYP functional and LANDL basis sets were used throughout all calculations. As can be seen in Figure 9, the Co-C cleavage reaction for both the base-on and base-off species of methylcob(III)alamin have the highest total energy barriers. According to this indication as well as the experimental data, methylcob(III)alamin would be unlikely to take part in the mechanism involving the Methionine Synthase bioprocess if the Co-C cleavage reaction follows the SN1 mechanism. Of the two species of the methylcobalamin cofactor with the oxidation state of the central cobalt ion equal to +2, the Co-C bond-breaking reaction in the base-off structure has a lower total energy barrier compared to that of the base-on species (Figure 9). Therefore, according to DFT theory, the Co-C cleavage reaction in base-off methylcob(II)alamin would be the most likely when considering the SN1 scenario. The total energy barrier in this case is about 0.7 eV. The Boltzmann population at the level of 0.7 eV at room temperature is equal to only about 1.47 e−12. Therefore, only an insignificant amount of the reagents can be transformed into products during the Methionine Synthase process according to DFT calculations. This contradicts all the experimental data. Either the DFT approximation is inadequate for methylcobalamin cofactor calculations, the SN1 mechanism is inadequate for Co-C bond cleavage during the Methionine Synthase process, or both of these [126]. DFT calculations of the same reaction but with different functional and other basis sets lead to similar results [162], which proves that the functional or basis set is not the cause of the result failing to agree with the experimental data.
Unfortunately, some authors considered these DFT results adequate for in vivo processes, comparing them with in vitro electrochemical experimental data [163]. Indeed, Lexa and Savéant calculated the Co-C bond-breaking reaction barrier in the base-off species from cyclic voltammetry experiments in a 1:1 DMF/1-PrOH solution and found that the value of the energy barrier was equal to about 17 kcal/mol [88]. Since the DFT calculations gave a barrier equal to about 24 kcal/mol, the authors considered that they reached a reasonably good agreement between the DFT and experimental data. Therefore, they concluded that the base-off methylcobalamin cofactor is the active species in the biological processes. This agreement between theory and experiment is doubtful even in the case of in vitro experimental data since the rate constant and therefore the total energy barrier of the Co-C bond cleavage reaction depends strongly on the nature of the solvent [89,90]. For instance, the rate constant of this reaction is greater in DMS solvent than in water by about a thousand times. Therefore, any comparison of an energy barrier found using an unnatural solvent with the natural process is suspect. Furthermore, the choice of the base-off species of methylcob(II)alamin as the active particle in the Methionine Synthase process was based on DFT calculations that showed that the total energy barrier of the Co-C bond-breaking reaction was minimal among various methylcobalamin species. On the other hand, the experimental energy barrier of the Co-C bond cleavage reaction involving the methylcobalamin base-on species in similar conditions (in 1:1 DMF/MeOH solution) was much lower compared to the total energy barrier of the same reaction with the base-off species (having a value of only about 7.6 kcal/mol [90]). Moreover, as experimentally proved [89], the Co-C bond cleavage reaction energy barrier in similar conditions but in 1:1 DMF/1-PrOH solution must have even a lower value when taking into account the dependence of rate constants of this reaction on the polarity of the solvent [89]. DFT shows that the total energy barrier of the Co-C bond cleavage reaction in the base-on species of the methylcobalamin cofactor is higher by about 2 eV compared to the same barrier for the base-off species. In contrast, experimental data show that the total energy barrier of the Co-C bond cleavage reaction in the base-on species of methylcobalamin is lower by about 0.4 eV of the total energy by comparing the same barrier in the base-off species.
Unfortunately, some authors insist that the base-off variant is the only active particle in the Methionine Synthase process based on the DFT results. Numerous DFT calculations counter this assertion. DFT calculations [114,115] and QM/MM [125,128] (based on DFT) electronic structure calculations of the SN2 reaction transfer of methyl radical from methylcob(II)alamin to homocysteine and of methyl radical transfer from methyltetrahydrofolate to cob(I)alamin showed significant total energy barriers [113,114,125,128]. These energy barriers would lead to low and incomplete methyl radical transfers in the Methionine Synthase bioprocess. This is in contrast to the fact that the turnover of this process in the presence of the AdoMet substrate can, in principle, last without limits [141]. It should also be noted that DFT calculations and QM/MM calculations (based on DFT) of methyl radical transfer mechanisms of both the methylcob(II)alamin to homocysteine and that of methyltetrahydrofolate substrate to the cob(I)alamin cannot explain the complete methyl radical transfers in these reactions. According to the experimental pieces of evidence (Figure 6) and the international research community’s general understanding of the Methionine Synthase mechanism, these calculations cannot show the reverse electron transfers that take place during these biochemical processes.
3.3. Calculations Involving Adenosylcobalamin
A critical analysis of the DFT and QM/MM calculations involving the adenosylcobalamin cofactor is presented here. In addition to the numerous methylcobalamin-related calculations discussed above, several authors have performed calculations of the Co-C bond-breaking reaction in adenosylcobalamin-dependent bioprocesses by using the DFT and QM/MM methods [133,134,135,136,137,138,139,140]; however, due to difficulties, DFT and QM/MM (based on DFT) have been used to study the mechanism of the adenosylcobalamin cofactor-dependent processes to a lesser extent. The applications of these methods to the study of the Co-C and Co-N cleavages of adenosylcobalamin generally result in significant [133,134,135,136,137,138,139,140] energy barriers, in contradiction with experimental data that show a close-to-unity equilibrium constant, e.g., with the close-to-zero energy barrier for the Co-C bond cleavage reaction [55,132]. Assumptions that would lessen the energy barrier of these reactions were studied. One such assumption was to consider that the electron reduction of adenosylcobalamin occurred before the catalytic process took place [138]; however, the energy barrier of the Co-C bond cleavage obtained in these calculations was still significant. Moreover, all experimental data show that the reduced adenosylcobalamin species appears only due to its catalytic activity and not beforehand. Another assumption that was considered was that the energy barrier of the Co-C bond cleavage reaction is overcome by the gain in energy in the substrate’s various processes; however, the substrate energy processes were calculated using the MM [132], the energy of which might not be commensurate with the DFT energy due to different theoretical backgrounds. In addition, the QM/MM method inevitably ignores a series of interactions at the boundary between the QM and MM structures, so the total sum of energies is not convincing. On the other hand, the whole system, e.g., all mentioned processes, must work synchronously as a single interactive block to obtain the total energy directly, not separately. Also, factors like the deformation of the corrin ring are not convincing and were not reported or confirmed subsequently by any experimental or theoretical data. Furthermore, the relationship between the Co-C bond cleavage and the hydrogen transfer from the substrate to the 5′-deoxy-5′-adenosyl radical, which takes place in these processes, has not been completely resolved [133,134,135,136,137,138,139,140] by using the DFT or the QM/MM method based on DFT. These inconveniences are explained by the fact that the molecular orbital mixing, e.g., the Pseudo-Jahn–Teller Effect contribution, is crucial for these mechanisms’ reaction behavior. Therefore, the correct determination of adenosylcobalamin reaction mechanisms is beyond the limits of DFT-based methods.
3.4. Summary Considerations
All calculations of vitamin B12 cofactor-dependent processes performed using the DFT or the QM/MM method based on the DFT have failed to explain these bioprocesses or contained problematic issues. It has been shown on several occasions that the Pseudo-Jahn–Teller Effect is very prominent in the vitamin B12 cofactors [126,127,141,142,143,144]. In particular, the explanation for the failure of DFT in studying the vitamin B12-dependent bioprocesses can be drawn from work by Isaac Bersuker [164], who showed that the DFT method could not handle compounds in which Pseudo-Jahn–Teller is prominent. We must mention that the researchers’ enthusiasm toward using the DFT method is indeed based on the amazing qualities of the DFT method, which gave results very close to the experimental ones based on the use of modest computational resources. But any meritorious method has its limits of use. If the orbital mixing is significant and influences the properties of the studied compounds, the DFT method is no longer suitable for their study since it cannot take the orbital mixing into account. We do not know what the results would be if the nature of its scientific foundation were violated and the orbital mixing, which has no scientific basis, was artificially introduced. On the other hand, if these orbital mixings are missing, then all the MCSCF methods would turn into a simple Hartree–Fock method, which produces results less close to the experimental ones than the DFT.
4. Pseudo-Jahn–Teller Effect and MCSCF Calculations: Methionine Synthase Process
4.1. Pseudo-Jahn–Teller Effect and MCSCF Method: General Considerations
The basics of the Pseudo-Jahn–Teller Effect and its relation to the MCSCF method are presented in this section. The idea of the Pseudo-Jahn–Teller Effect, also known as the second-order Jahn–Teller Effect, originates from a 1957 paper by U. Öpik and M.H.L. Pryce [165]. The paper shows that a small splitting of degenerate states can still cause instability in molecular systems connected to the Jahn–Teller Effect if the vibronic coupling between states is strong enough. The theory of instability of molecular systems based on the Pseudo-Jahn–Teller Effect led to contributions by R.F.W. Bader [166], in which formulas related to second-order perturbation theory were developed. Robert L. Fulton and Martin Gouterman introduced the actual notion of the Pseudo-Jahn–Teller Effect [167] in a 1961 paper. However, a correlation between the Pseudo-Jahn–Teller Effect theory and a major observation of its effect on experimental properties was not obtained for a long time. The first person to solve the major physical problem as an observable Pseudo-Jahn–Teller Effect was Bersuker I.B., who revealed the origin of dipolar distortion leading to spontaneous polarizations and ferroelectricity in perovskite-type crystals [168]. Along with Stavrov S.S., he also solved a major biological problem, namely the mechanism of hemoglobin oxygenation, using the Pseudo-Jahn–Teller Effect for the first time [169]. In principle, the modern development and application of the Pseudo-Jahn–Teller Effect belong to Isaac B. Bersuker and coauthors, who explored these effects on molecular systems in several books [170,171] around the turn of the 21st century. The Pseudo-Jahn–Teller Effect was treated primarily as a two-state problem [165,166,167,168,169,170,171].
The development and motivation of the Pseudo-Jahn–Teller Effect is well known and described (for example, in the publications signed by Bersuker I.B.) and can be described shortly as follows [170,171]. Generally, the geometric equilibrium of a molecular system corresponds to the minimum point of the adiabatic potential adiabatic surface, with the first derivative equal to zero and the second derivative being positive if the molecular system does not have degenerate energy levels. At this minimum point, the curvature K of the total energy in the directions of the natural displacements G is equal to KG = (∂2E/∂QG2)o and is positive (K > 0). The total energy of a system is expressed by the general formula E = <ᴪo|H|ᴪo>, where H is the Hamiltonian while ᴪo is the ground state wave function. By introducing the total energy formula into the curvature formula K, we obtain K = <ᴪo|(∂2H/∂Q2)o|ᴪo> + 2<ᴪo|(∂H/∂Q)o|ᴪo’>. Here, the G natural directions are omitted for simplicity. The ᴪo’ is equal to (∂ᴪo/∂Q)o. For clarity, the formula of total energy curvature can be simplified to K = Ko + K’, where Ko = <ᴪo|(∂2H/∂Q2)o|ᴪo> while K’ = −2Σp|<ᴪo|(∂H/∂Q)o|ᴪp >|2/Ep − Eo if we present K’ in terms of the second-order perturbation theory. Here, ᴪp represents the exited states’ wave functions and Ep the exited states’ energy levels. It can be seen that the K value is negative and is all the greater; the smaller the energy Ep-Eo difference, the larger the |<ᴪo|(∂H/∂Q)o|ᴪp >|2 matrix element’s values. If the difference Ep-Eo between the energies of excited states and the ground state is small enough and/or the |<ᴪo|(∂H/∂Q)o|ᴪp >|2 matrix elements are large enough, then the K’ value, which is negative, can transform the general K curvature value of the total energy into a negative value, and the initial minimum energy point turns rather into a saddle point. In this case, the studied molecular system’s initial geometric equilibrium is no longer stable. The Pseudo-Jahn–Teller Effect theory is generally known elsewhere and has been used to explain many phenomena, such as the destabilization of some symmetrical geometric structures, the trans effect in coordination compounds, or multiple spectroscopic data. Unfortunately, this version of the Pseudo-Jahn–Teller Effect analysis suffers from two limitations. First of all, the perturbation of the geometric molecular system must be small enough to allow perturbation theory to obtain formulas that can be used in molecular or crystal structure analysis. Therefore, this development of the Pseudo-Jahn–Teller Effect can only be used to establish deviations in the studied molecular structures from the assumed geometric equilibrium state that are not too large and are usually based on their symmetry, not in the case where the Pseudo-Jahn–Teller Effect leads to breaking or forming new chemical bonds. Secondly, the use of this theoretical development of the Pseudo-Jahn–Teller Effect phenomenon is usually used in the case of systems with only two energy levels, which mix due to this effect. The phenomenon can be studied only at a general qualitative level if several electronic levels are mixed.
In 2002, M.J. Bearpark, L. Blancafort, and M.A. Robb showed that the CASSCF method, based on MCSCF theory, is nothing but the application of the Pseudo-Jahn–Teller Effect at the multistate level [172] due to the multistate orbital coupling during the CASSCF wave function-mixing procedure. The perturbation deviation is also not limited in this case. In principle, these allow the use of the Pseudo-Jahn–Teller Effect through CASSCF method geometry optimization to determine the geometric modifications of the molecular structures, including the structural changes when the breaking or forming of new chemical bonds is taking place.
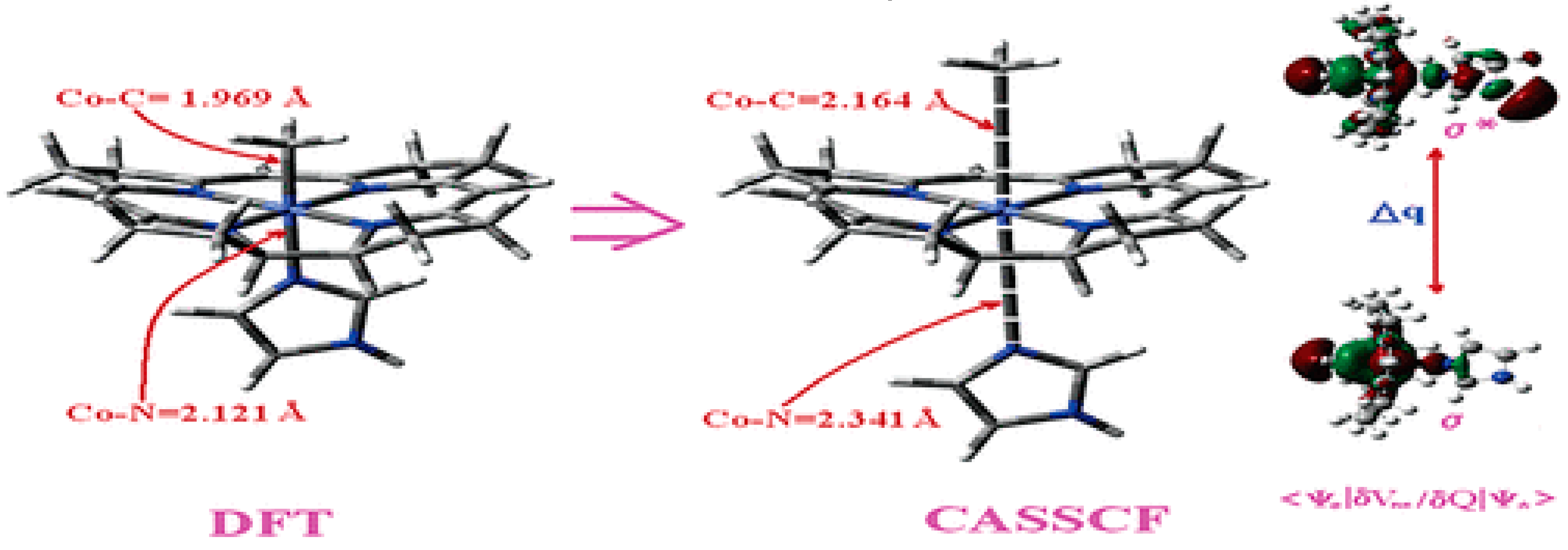
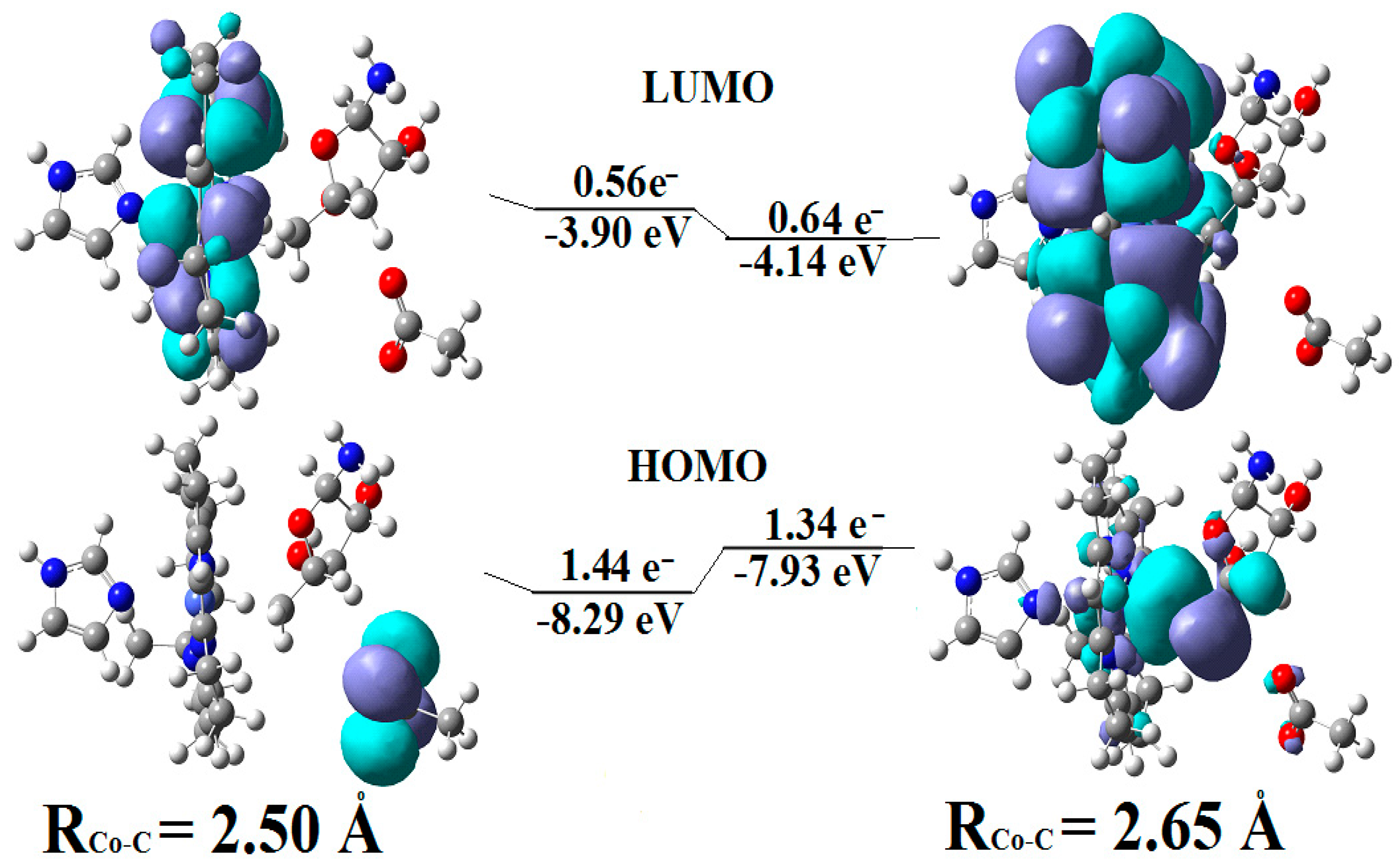
It has been shown that the Pseudo-Jahn–Teller Effect, for which DFT cannot account, is active in the methylcobalamin cofactor [126]. Indeed, the DFT geometry optimization of the methylcobalamin models does not show any noticeable difference between the geometries of the versions with the +3 and +2 oxidation states on the central cobalt atom. Despite this, the experimental X-ray data show a difference in the lengths of the Co-N axial bonds for the actual analogous vitamin B12 species [10,11,12,13,14,173] (about 2.12 Å for the Co+3 compound and between 2.35 Å and 2.50 Å for a Co+2 compound). On the other hand, the CASSCF geometry-optimization procedure of the same models showed that as the number of electrons and orbitals included in the active zones of the CASSCF method increases, so does the length of the Co-C and Co-N axial bonds. It can be inferred that the cause of such Co-C and Co-N bonds increasing is the strong mixing of the σ and σ* axial orbitals during the CASSCF procedure, i.e., the Pseudo-Jahn–Teller Effect. In this case, it is a two-state effect in which the value of the second-order mixing perturbation term (see Figure 10) is a cause of the geometry destabilization of the calculated model. So, a significant part of the electron density is transferred from the bonding σ HOMO to the antibonding σ* LUMO. Therefore, the lengths of the axial bonds (Co-C and Co-N) are “softened” and lengthened (Figure 10). After reaching the maximum active zone with 13 electrons and 13 orbitals, the Co-C and Co-N bond distances cease to grow [141] during the CASSCF geometry-optimization procedure. This is in full agreement with the experimental data. This demonstrates that the Pseudo-Jahn–Teller Effect largely governs the molecular structure of the one-electron-reduced methylcobalamin cofactor.
4.2. MCSCF: Methylcobalamin and Methionine Synthase Process
The most general information on the MCSCF method is provided here. The Multiconfigurational Self-Consistent Field (MCSCF) [174] procedure is not a new method for calculating chemical compounds. Still, it is one of the most general ones for calculating the electronic structure of the analyzed systems. MCSCF is usually based on the Hartree–Fock (HF) method. It builds a linear combination of configuration state functions in the form of the configuration determinants to express the electronic wave function of a molecule. It is one of the most sophisticated methods of calculating the electronic structure; depending on the approximation used, it uses a huge number of determinants (depending on the size of the active zone taken into account). Note that the DFT and HF methods only use the solution of a single determinant. As a result, the MCSCF method requires extremely large computational resources. Therefore, until recently, it was used mostly with systems of a few atoms. With the development of powerful computational resources, the MCSCF method began to be used with medium-sized compounds but very rarely in the case of larger compounds, such as, for example, vitamin B12 cofactors. The Complete Active Space Self-Consistent Field (CASSCF) method is the first approximation variation of the MCSCF method in which (in the multiconfigurational interaction process) one set of orbitals is used, which are usually responsible for the chemical changes in the calculated compounds. For this reason, CASSCF is likewise usually used with medium chemical compounds and less often in the case of larger compounds, such as Vitamin B12 cofactors.
4.3. The Methyl Radical Transfer from Methylcob(II)alamin to Homocysteine (SN1 Reaction)
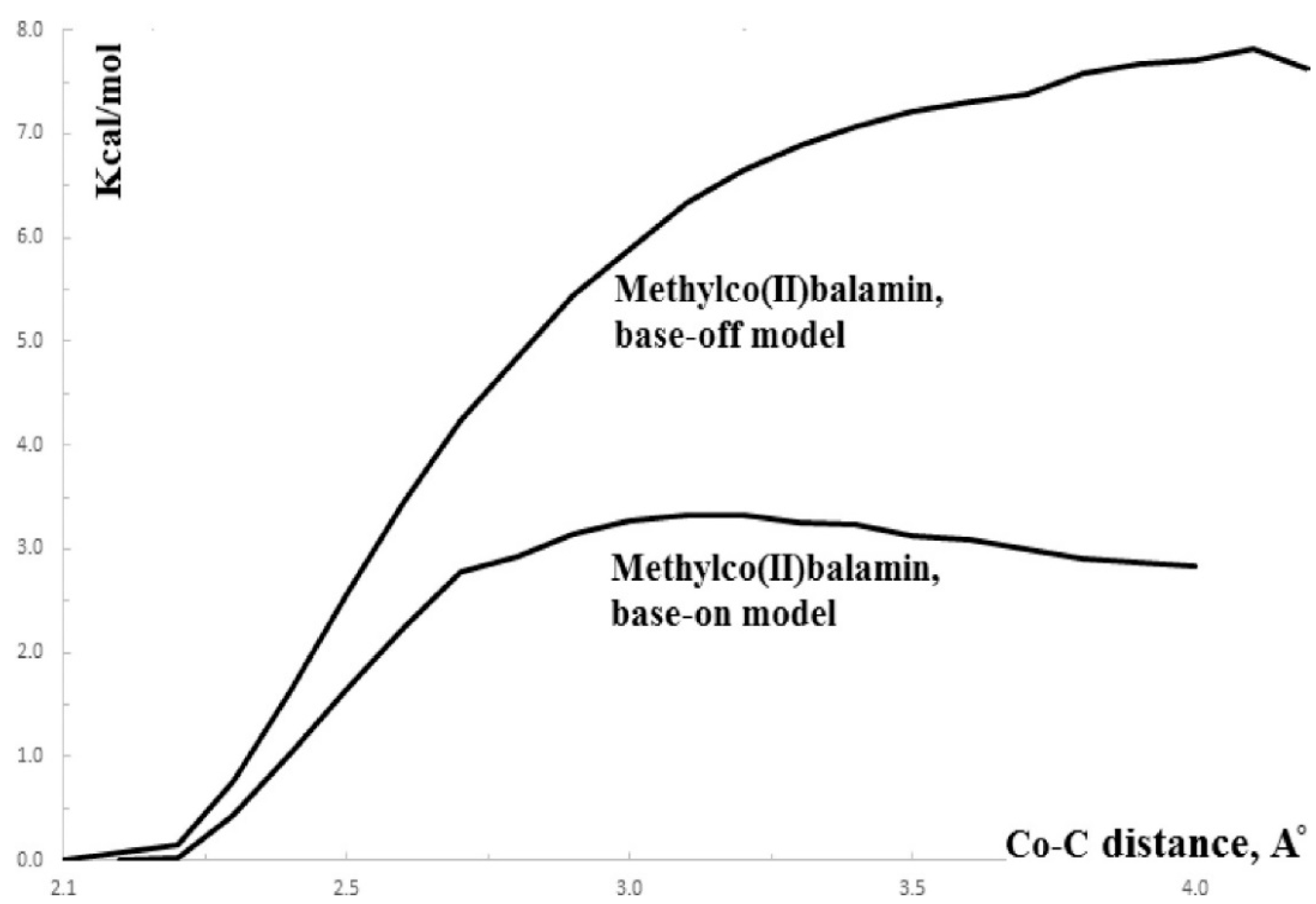
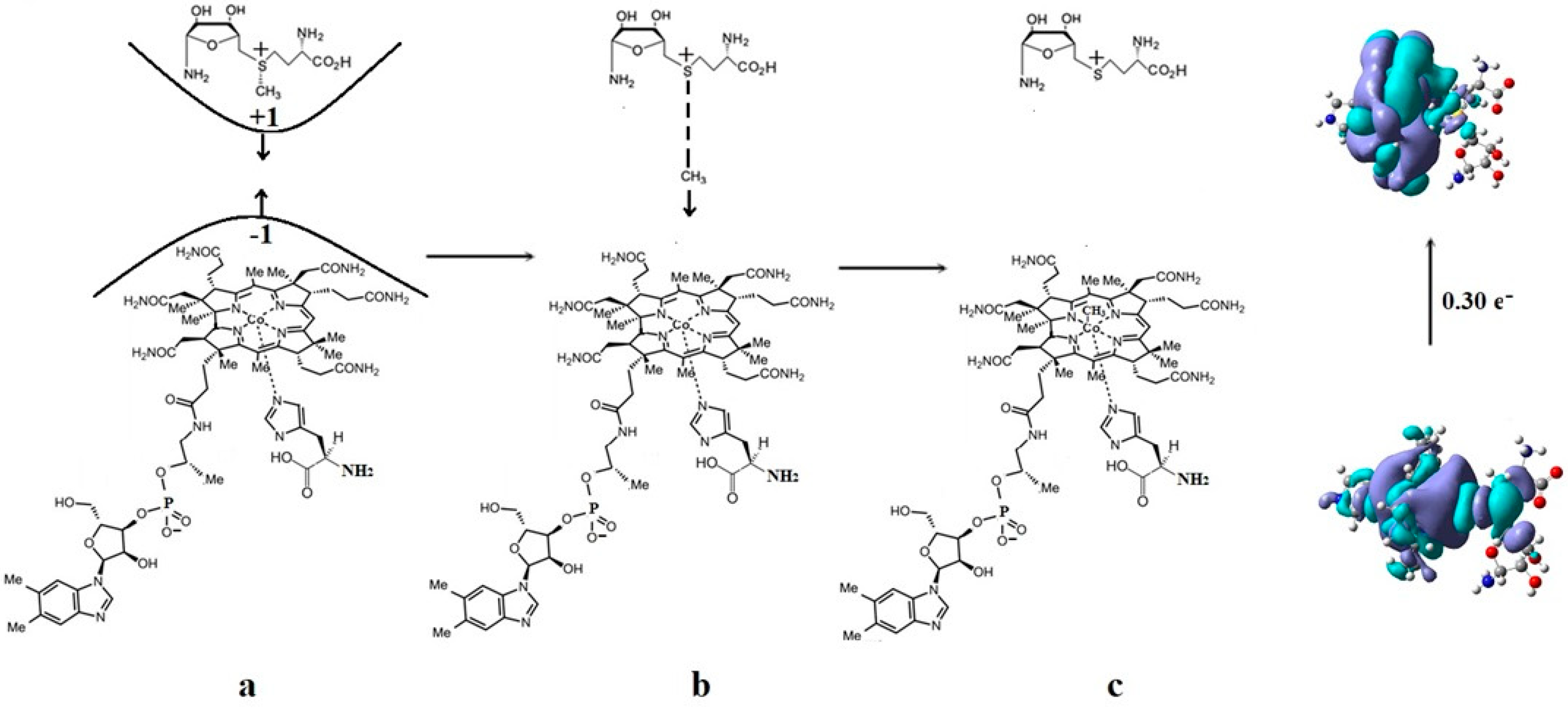
The SN1 mechanism of the methyl radical transfer from the methylcob(II)alamin cofactor to homocysteine in the frame of the MCSCF method is analyzed here. As discussed previously, in the first phase of the Methionine Synthase process, the methylcob(III)alamin is reduced by one electron. In the second phase of the process, the Co-C bond cleavage takes place with the transfer of the methyl radical from methylcob(II)alamin particle to homocysteine (Figure 2 and Figure 7). The transfer of the methyl radical from the methylcob(II)alamin to homocysteine can, in principle, take place via an SN1 or SN2 reaction mechanism. The SN1 reaction mechanism first involves breaking the Co-C bond in the methylcob(II)alamin and then transferring the methyl radical to homocysteine via two stepwise reactions, while the SN2 reaction mechanism involves the transfer of the methyl radical from the methylcob(II)alamin directly to the homocysteine during their simultaneous interaction. As a means to compare the two reaction scenarios, geometric optimization was performed at each point of the length of the Co-C bond by gradually shifting the methyl radical Co-C bond length from 2.00 Å to 4.00 Å at equal intervals of 0.1 Å. This was performed by using the CASSCF method, in which an active area of 13 orbitals and 13 electrons was considered [141]. The calculations were performed for both base-on and base-off species models (Figure 8b,c) of the methylcob(II)alamin cofactor. The same active area of 13 wave functions and 13 electrons was used to ensure the integrity of the calculations in terms of the CASSCF total energy barriers of the SN1 reaction [142]. As can be seen in Figure 11, the Co-C bond cleavage reaction has a total energy barrier of about 7.82 eV for the base-off species and about 3.32 eV for the base-on species. Therefore, the total energy barrier value is 4.50 eV lower for the base-on species of the methylcob(II)alamin cofactor compared with that of the base-off species. These results complement the data of in vitro experiments that found that the energy barrier of the Co-C bond cleavage reaction in the methylcob(II)alamin base-off species is much higher than in the base-on species [88,89,90]. There is an agreement on the correct orders of magnitude of the theoretical and experimental energy barriers between these two species even though the in vitro experiments took place in the presence and with the participation of the solvent. On the other hand, the DFT results demonstrate a reversal of the total energy barriers of the Co-C bond cleavage reaction for the base-off and base-on species of the methylcob(II)alamin cofactor. The causes of these erroneous data in the DFT electronic structure calculations of the Co-C bond cleavage reaction in the methylcobalamin have already been discussed above. This demonstrates the inability of the DFT method to resolve the vitamin B12-dependent reactions in which the Pseudo-Jahn–Teller Effect mixing of orbitals takes place [141,143]. The experimental data and MCSCF calculations of the Co-C bond cleavage reaction show that the active particle of the methylcob(II)alamin is not the base-off species but is the base-on species, given that the total energy barrier value of the Co-C bond cleavage reaction of the base-on species is much lower compared to the total energy barrier value of the same reaction in the case of the base-off species (Figure 11).
It should be noted that the Methionine Synthase bioprocess in the presence of the active substrate AdoMet can take, in principle, an unlimited number of turnovers according to experimental data [41]. The CASSCF electronic structure calculations showed that the base-on particle had the lowest total energy barrier of the Co-C bond breaking, approximately equal to 3.32 eV. The Boltzmann population at such an energy level is equal to about 0.367% of the total number of base-on species present in the biochemical medium of the Methionine Synthase process. This means that only 0.367% of the total amount of methylcobalamin base-on cofactor successfully participates in the Co-C bond cleavage reaction in the case of the SN1 reaction mechanism. This starkly contrasts with the experimental data [41], which shows that the Methionine Synthase bioprocess in the presence of the active substrate S-adenosyl-L-methionine (AdoMet) can take, in principle, an unlimited number of turnovers.
Conclusions. Independent methyl radical full lysis from methylcob(II)alamin is not likely to exist in vivo due to the significant energy barriers. Therefore, the SN1 mechanism of the transfer of the methyl radical from methylcobalamin to homocysteine is contraindicated by CASSCF calculations data and is unlikely [141].
4.4. The Methyl Radical Transfer from Methylcob(II)alamin to Homocysteine, SN2 Reaction
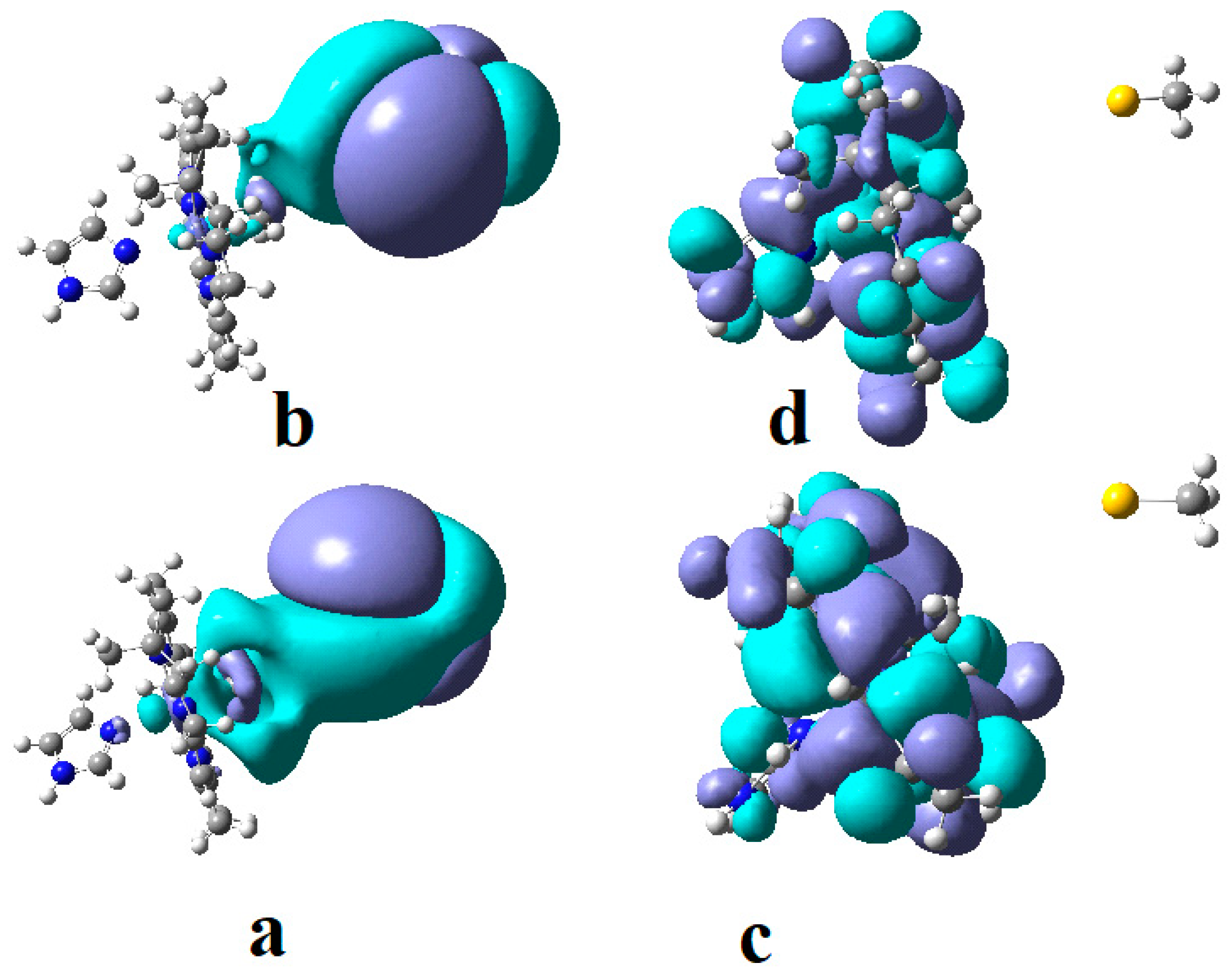
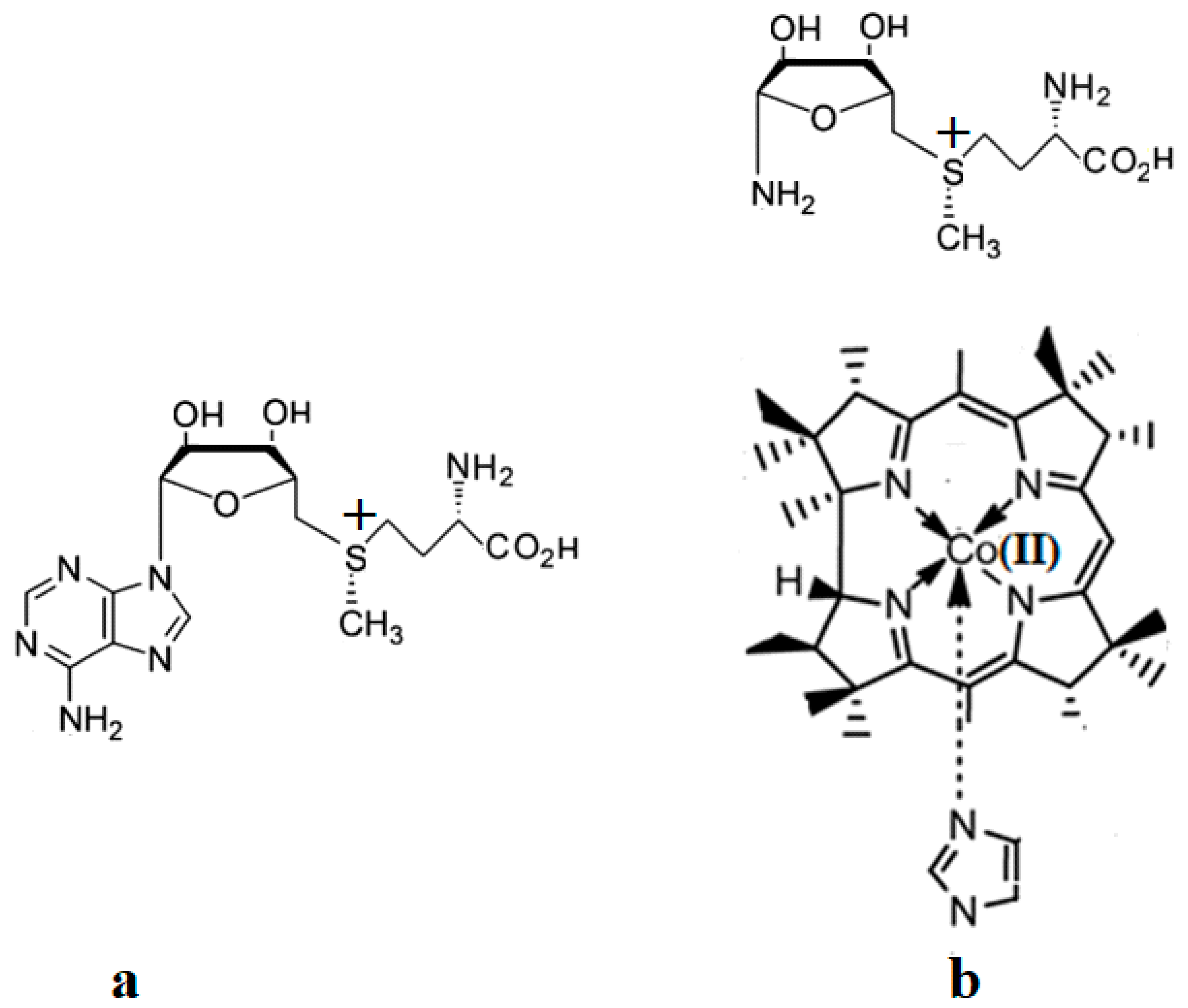
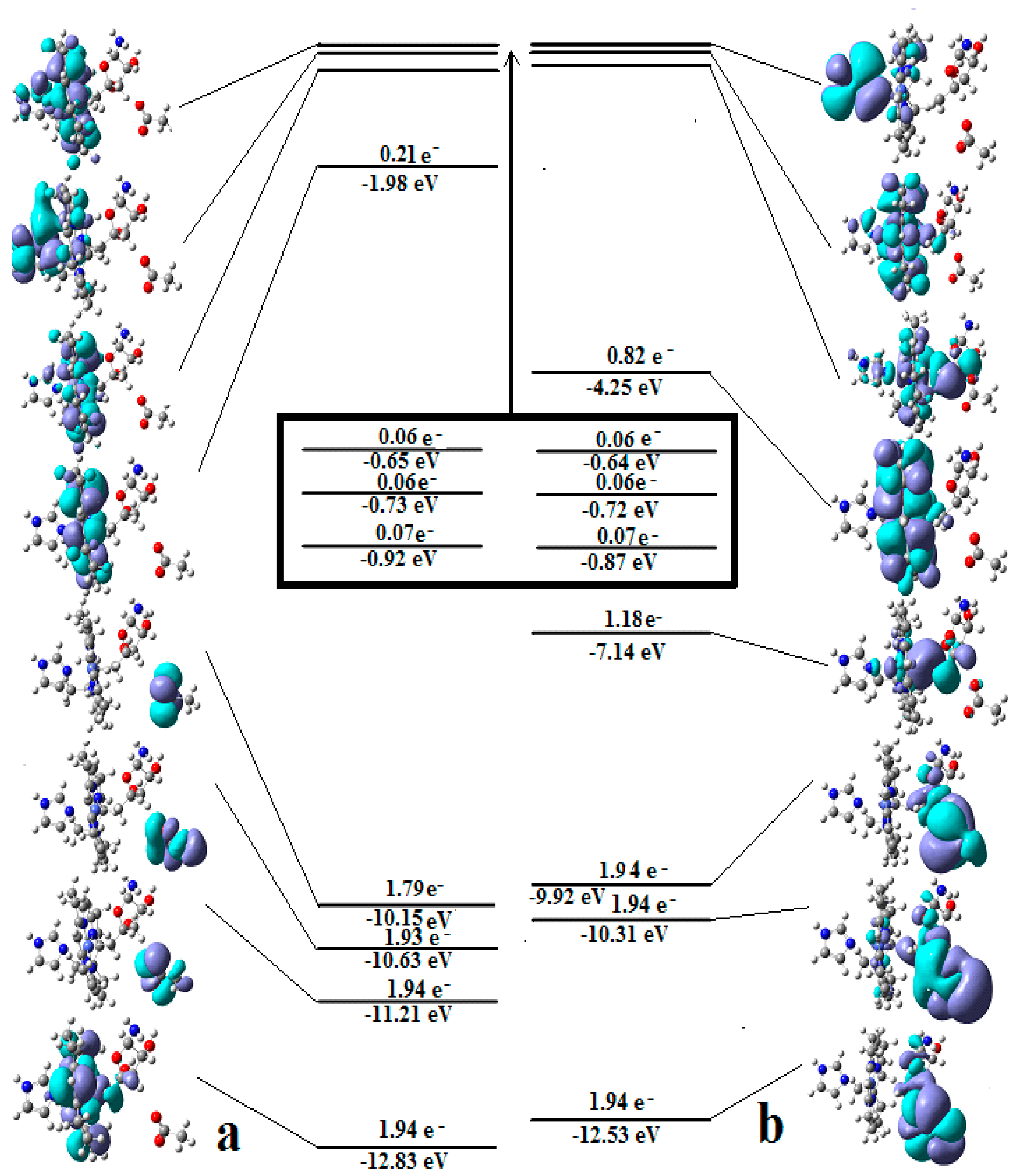
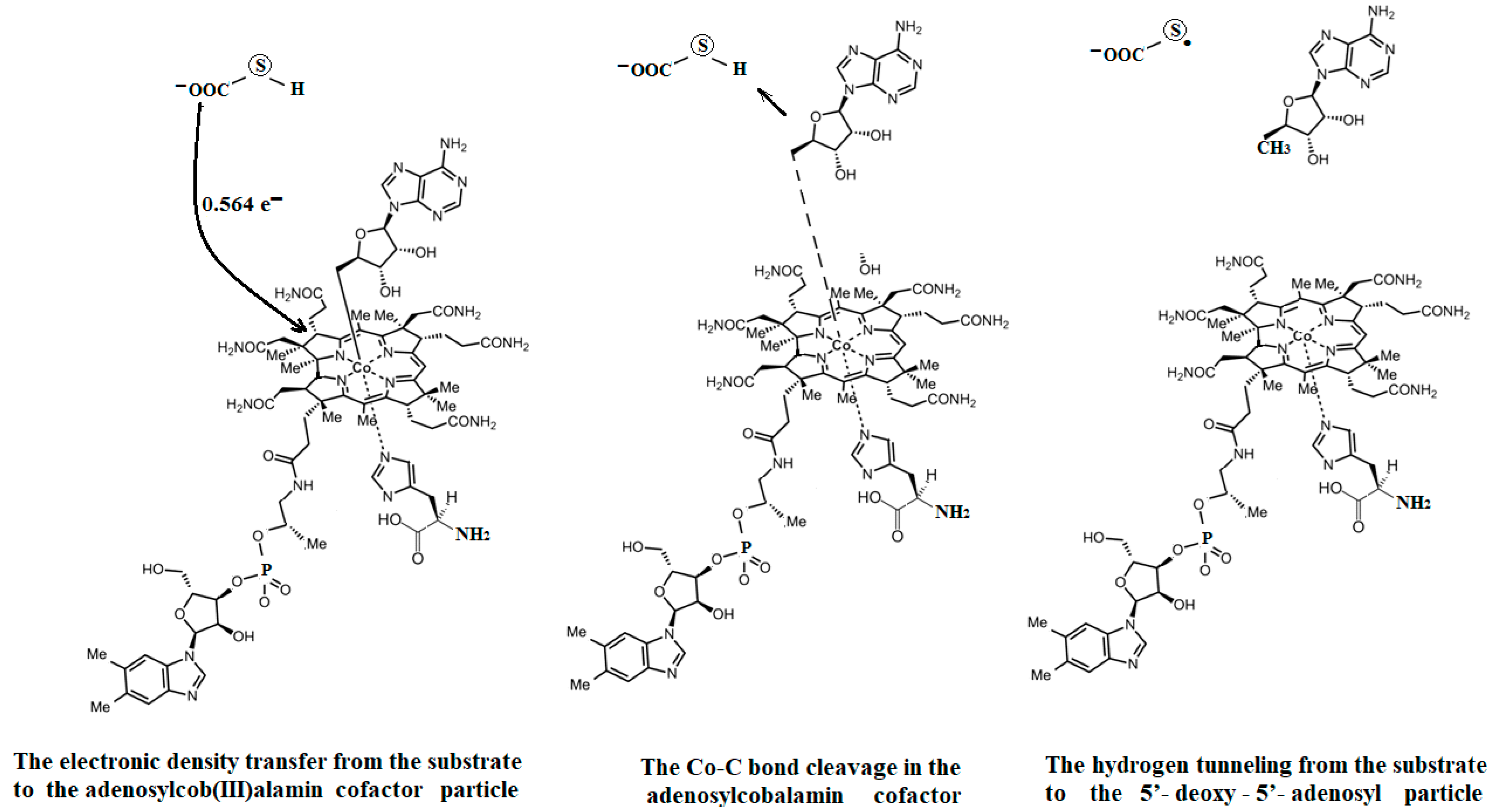
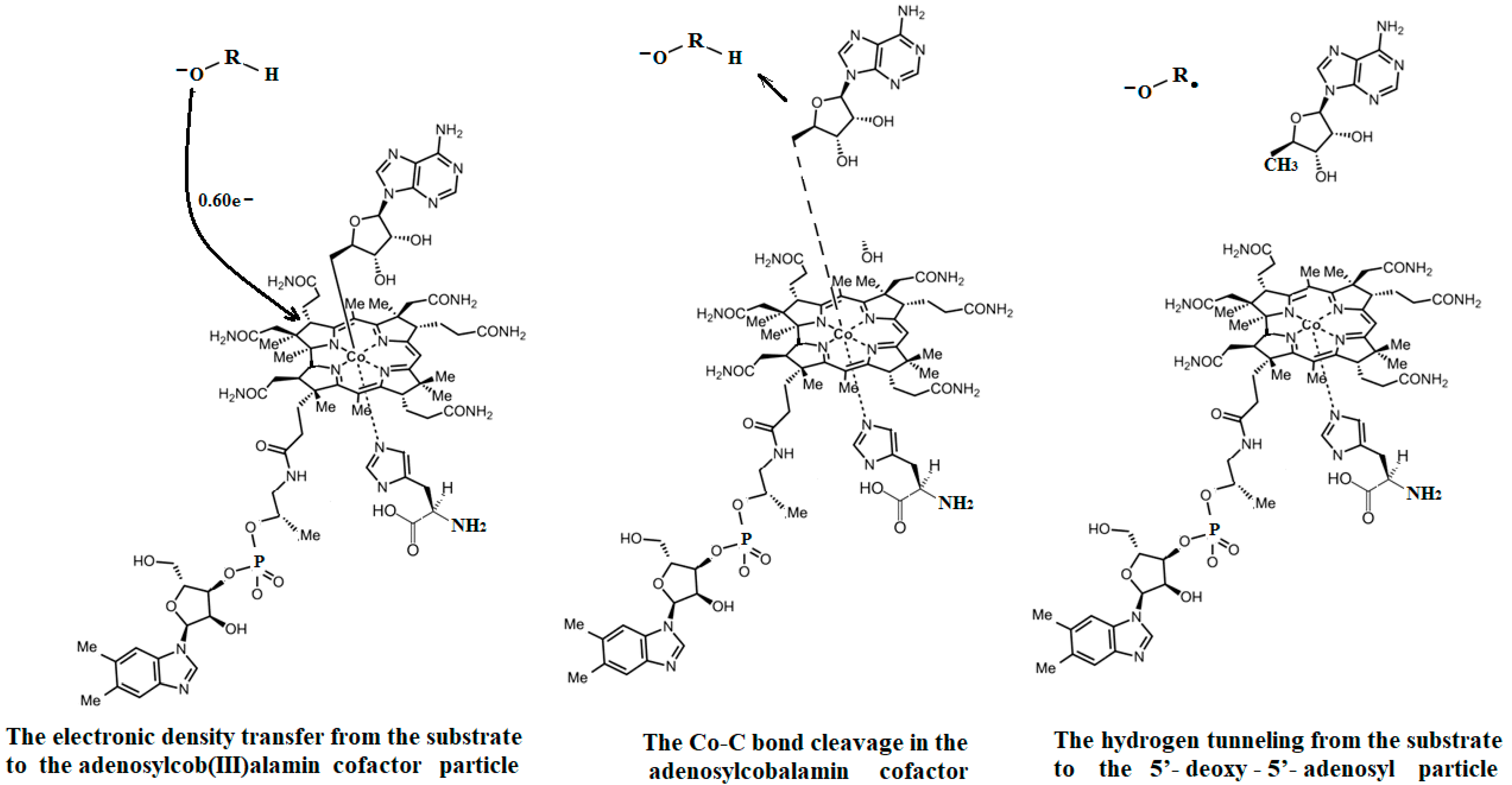
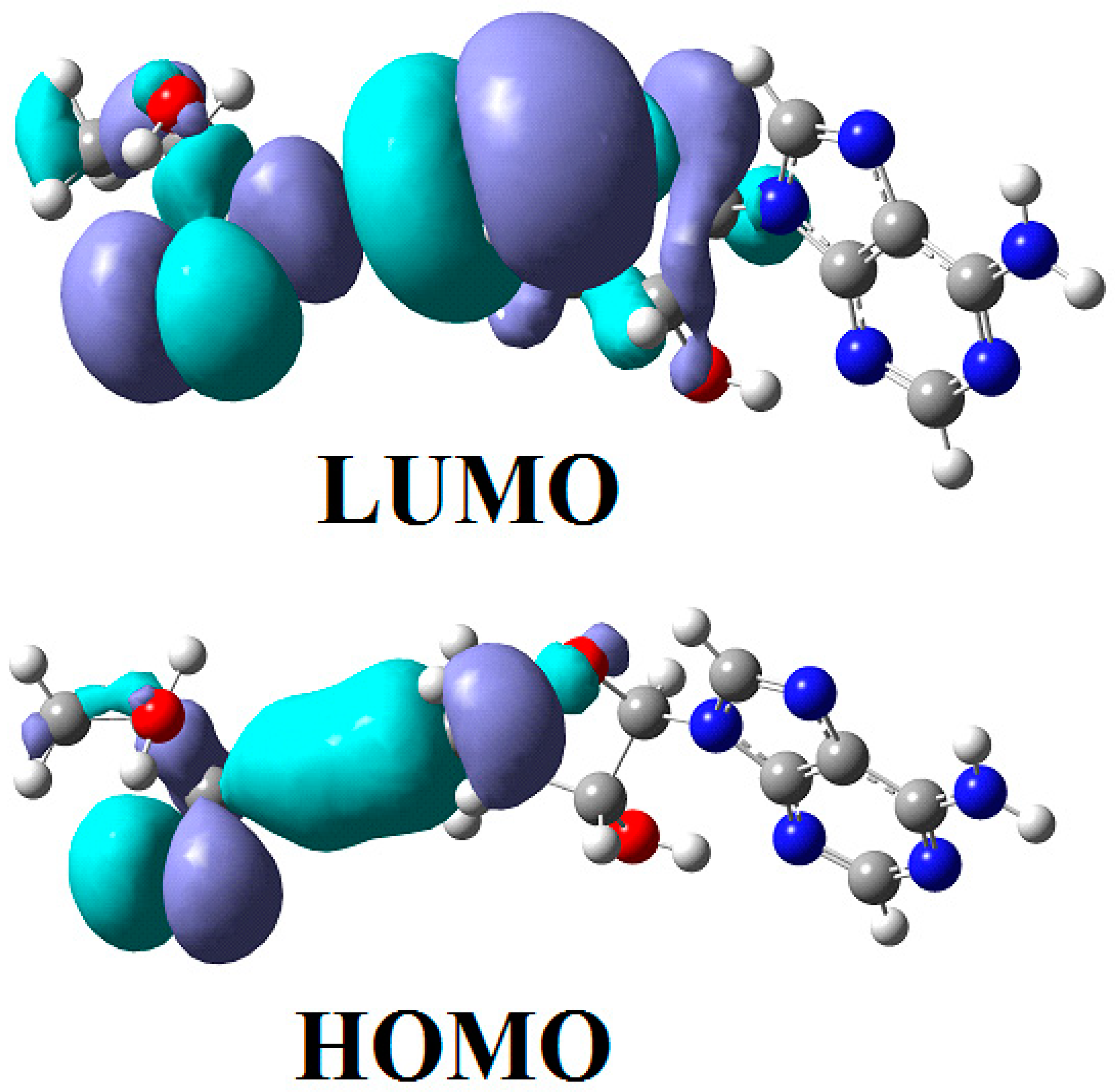
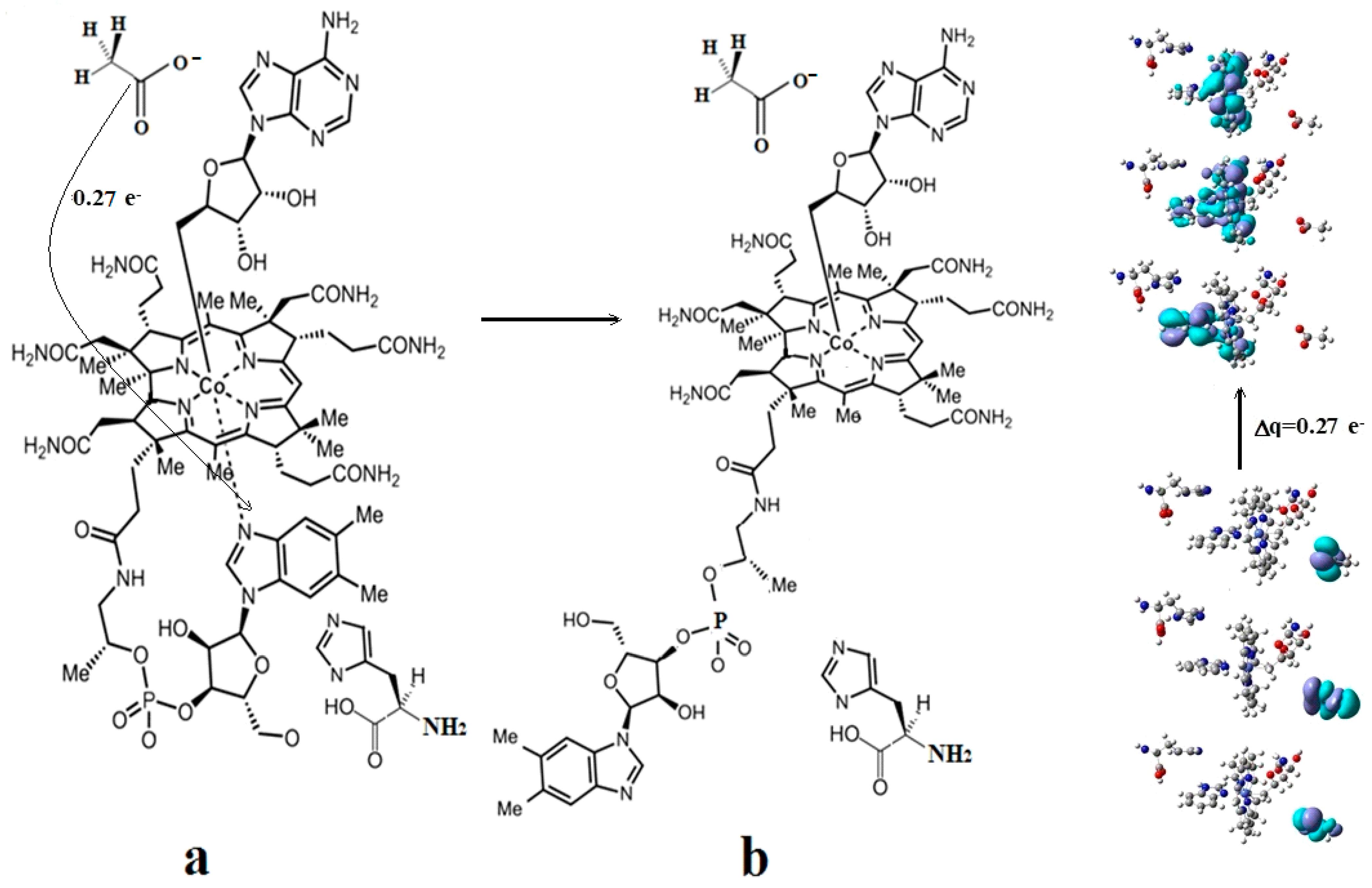
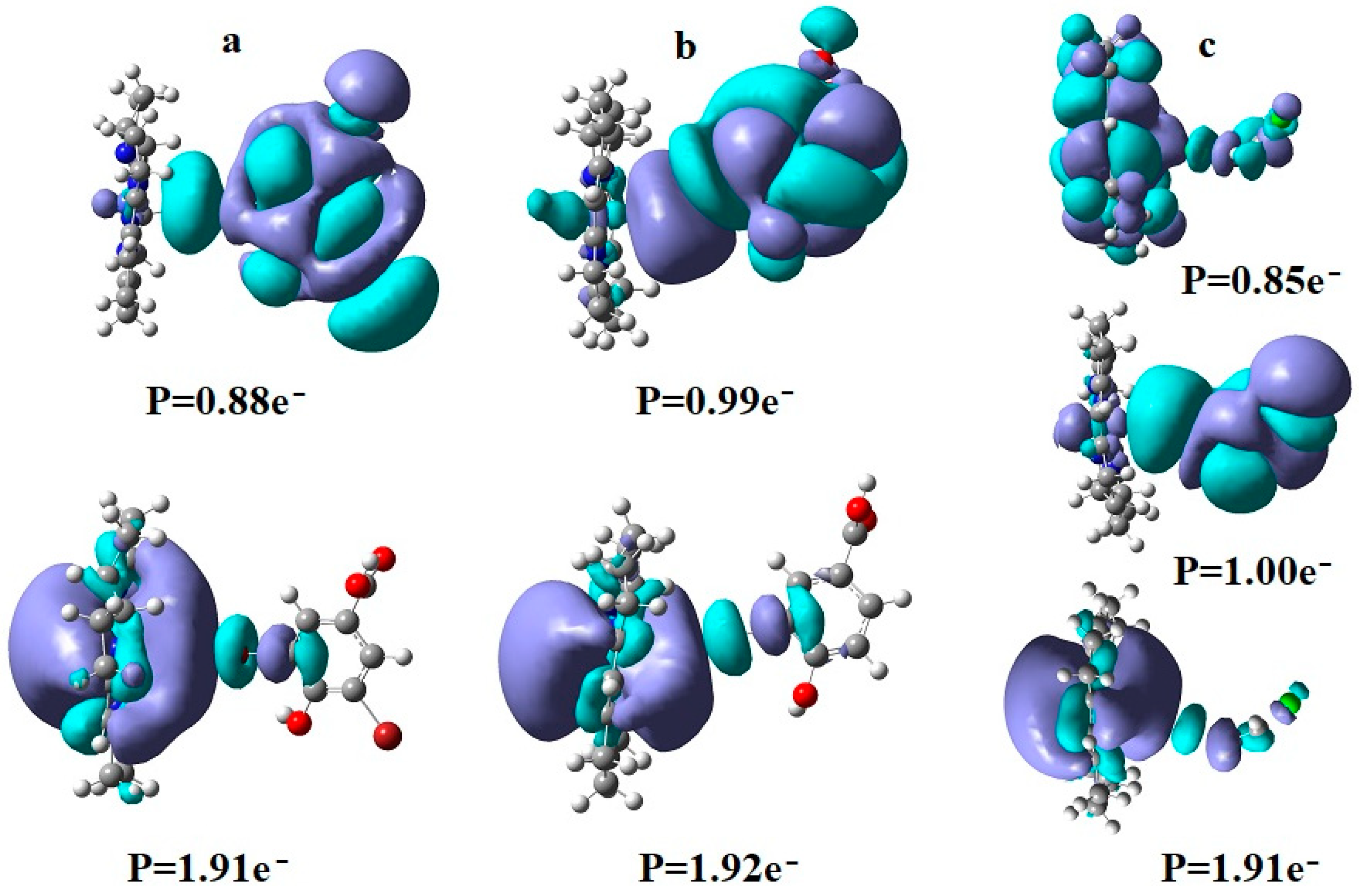
The SN2 mechanism of the methyl radical transfer from the methylcob(II)alamin cofactor to homocysteine in the frame of the MCSCF method is analyzed in this section. X-ray experimental methods have shown that a coordinating sphere of Zn+2 contains a series of negative homocysteine radicals in the immediate vicinity of the methylcobalamin cofactor in the Methionine Synthase process [40,44,47]. On the other hand, it has been theoretically demonstrated that these coordinating species are unstable. They modify their geometric structures, releasing the homocysteine-ion-ligand in the immediate vicinity of the upper part of the methylcobalamin [131] cofactor structure, where, according to the common experimental evidence, the Co-C bond is broken, and the transfer of the methyl radical to homocysteine takes place. Not surprisingly, CASSCF calculations show that only the negative homocysteine ion is active in transferring the methyl radical from methylcobalamin to homocysteine [141]. Models (Figure 12), including the methylcobalamin cofactor and the negative ion of the homocysteine (CH3S−), have been used in the CASSCF geometry-optimization procedure with an active area of 13 wave functions and 13 electrons, which guaranteed the integrity of the calculations in terms of the CASSCF total energy value of the used model [141].
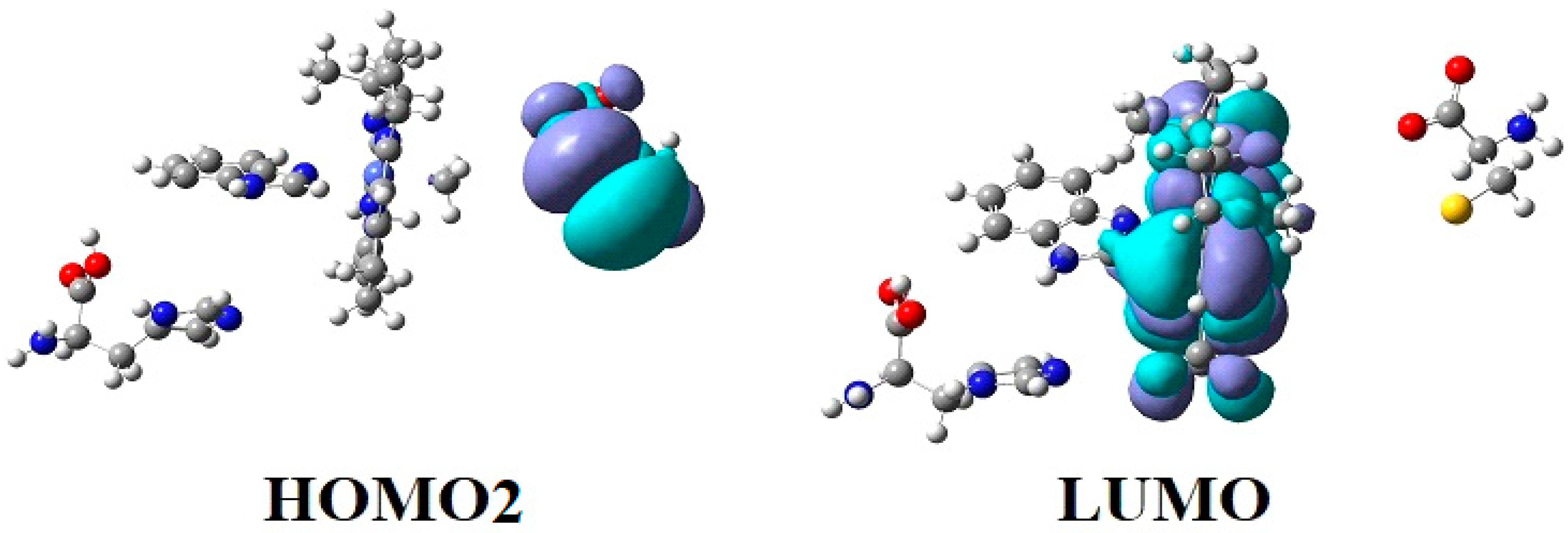
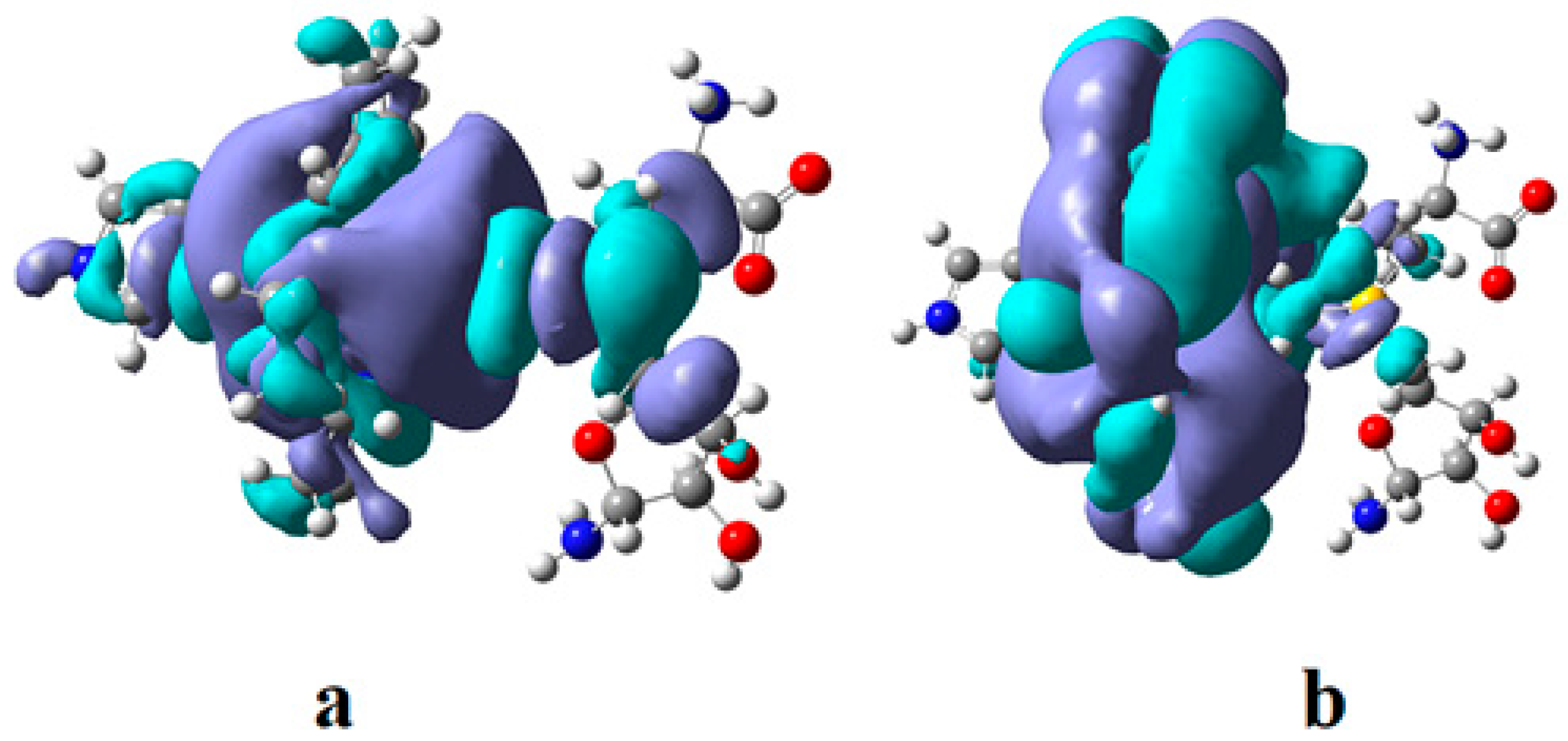
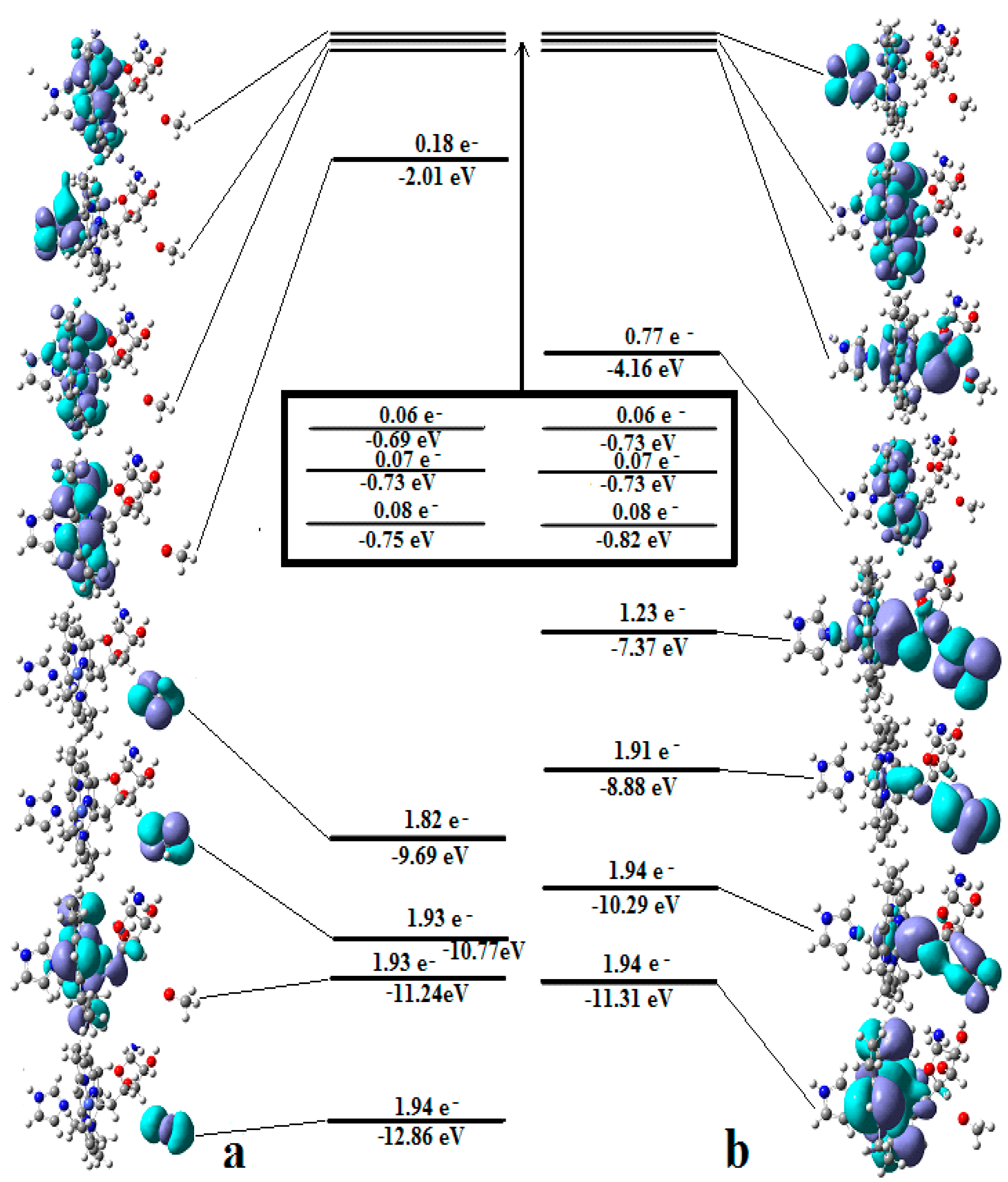
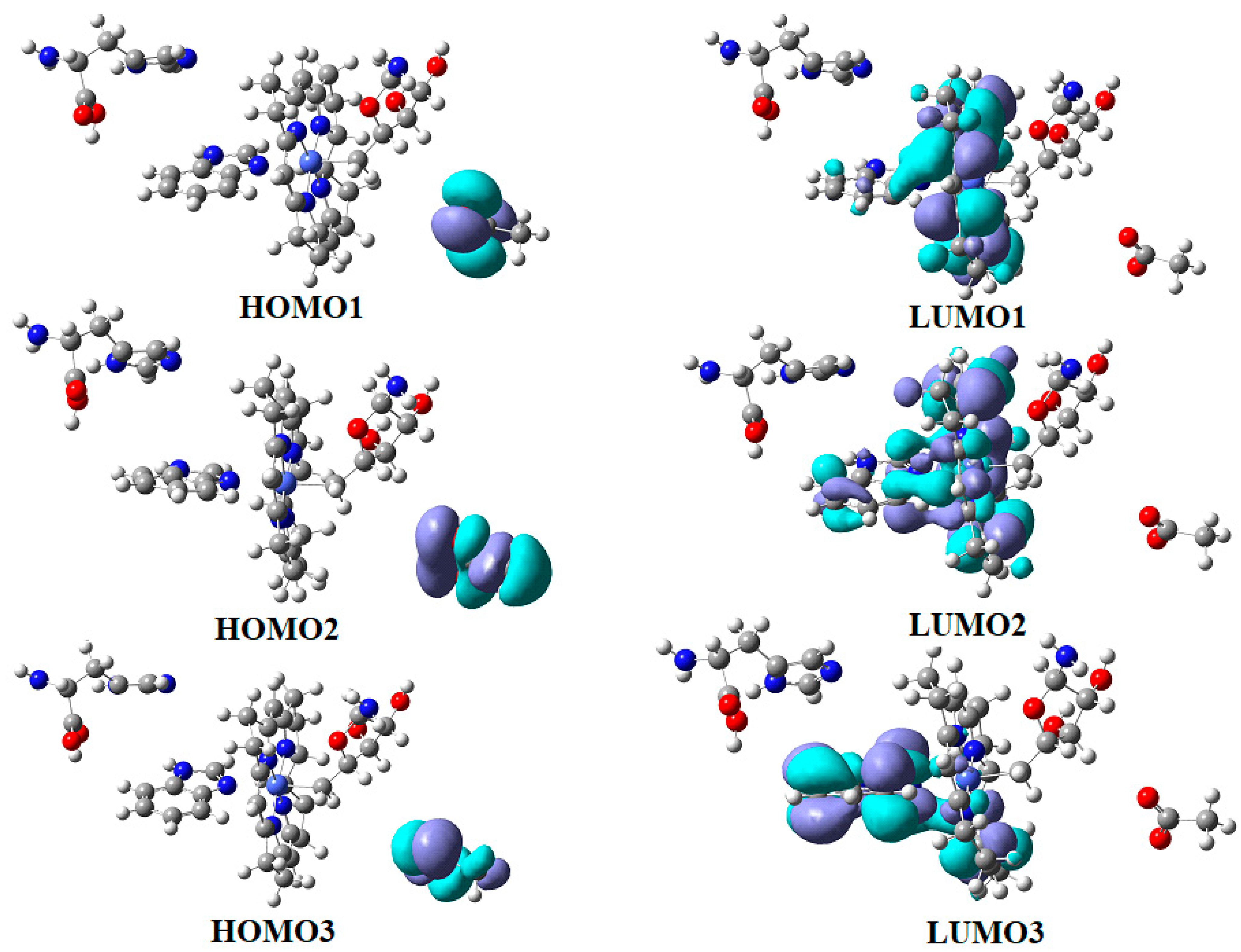
In the results of the MCSCF calculations, formally, there is no HOMO and LUMO since the orbital population is fractional. Also, the term “charge transfer” seems inappropriate for analyzing the populations of molecular orbitals. However, considering that the CASSCF method is nothing but the Pseudo-Jahn–Teller Effect [172] and that the Pseudo-Jahn–Teller Effect analysis consists of the use of both the HOMO and LUMO terms and the term “charge transfer”, which means the transfer of electron density from HOMO to LUMO, we will use all these terms. HOMO will mean the highest orbital populated with at least one electron or more, and LUMO will mean the lowest populated orbital with less than one electron density. We will name the other molecular orbitals accordingly, and the term “charge transfer” will have the same meaning as in the Pseudo-Jahn–Teller Effect analysis, i.e., the transfer of electron density from HOMOs to LUMOs here and below. The Co-C bond cleavage reaction is determined in this model by the formation of the two highest occupied molecular orbitals (HOMO2 and HOMO3) and the two lowest occupied molecular orbitals (LUMO1 and LUMO2) from the very beginning of the CASSCF geometry optimization (Figure 13) [141]. As can be seen in Figure 13, the two occupied molecular orbitals (HOMO2 and HOMO3) are formed exclusively from the atomic orbitals of the homocysteine ion model, and the two unoccupied molecular orbitals are formed exclusively from the atomic orbitals of atoms belonging to the methylcob(II)alamin cofactor model. At the beginning of the CASSCF geometry-optimization procedure, HOMO2 and HOMO3 are populated with 1.17 electrons and 1.53 electrons, respectively, and LUMO1 and LUMO2 are populated with 0.83 and 0.47 electrons, respectively. It is very clear that at the beginning of CASSCF geometry optimization, at least one electron is transferred from the homocysteine negative ion substrate to the methylcob(II)alamin cofactor. Thus, the CASSCF calculations [141] fully confirm the experimental data, according to which there is a reverse transfer of one electron from the substrate to the methylcob(II)alamin cofactor (Figure 6) concomitant with the transfer of the methyl radical from the last to the first [36,37]. These values of the electron density transfer from HOMO2 and HOMO3 to LUMO1 and LUMO2 are not only maintained during the CASSCF optimization procedure but are even slightly increased. This demonstrates that the reverse transfer of an electron from the active substrate to the methylcob(II)alamin cofactor indeed takes place. The transfer of an electron from the substrate to the methylcob(II)alamin cofactor changes the oxidation state of the central cobalt atom from +2 to +1 at the beginning of the CASSCF geometry-optimization procedure. As is commonly known, the cobalt ion in the +1 oxidation state coordinates only four ligands, and therefore the penta-coordinated and six-coordinated structures of the methylcobalamin cofactor species are not stable—consequently, the Co-C bond must be cleaved. Indeed, during CASSCF geometry optimization, the Co-C bond distance increases continuously until it becomes fully cleaved. The methyl radical is transferred and completely bound to homocysteine (Figure 14), which becomes methionine [141].
Conclusions. The transfer of an electron from the substrate to the methylcob(II)alamin cofactor triggers the Co-C bond cleavage and the methyl transfer from methylcobalamin to homocysteine. Therefore, the methyl radical transfer from methylcob(II)alamin cofactor to homocysteine is an SN2 non-barrier reaction with a minimum of total energy for its products.
4.5. The N-C Bond Cleavage and Methyl Radical Transfer from 5-methyltetrahydrofolate to Cob(I)alamin
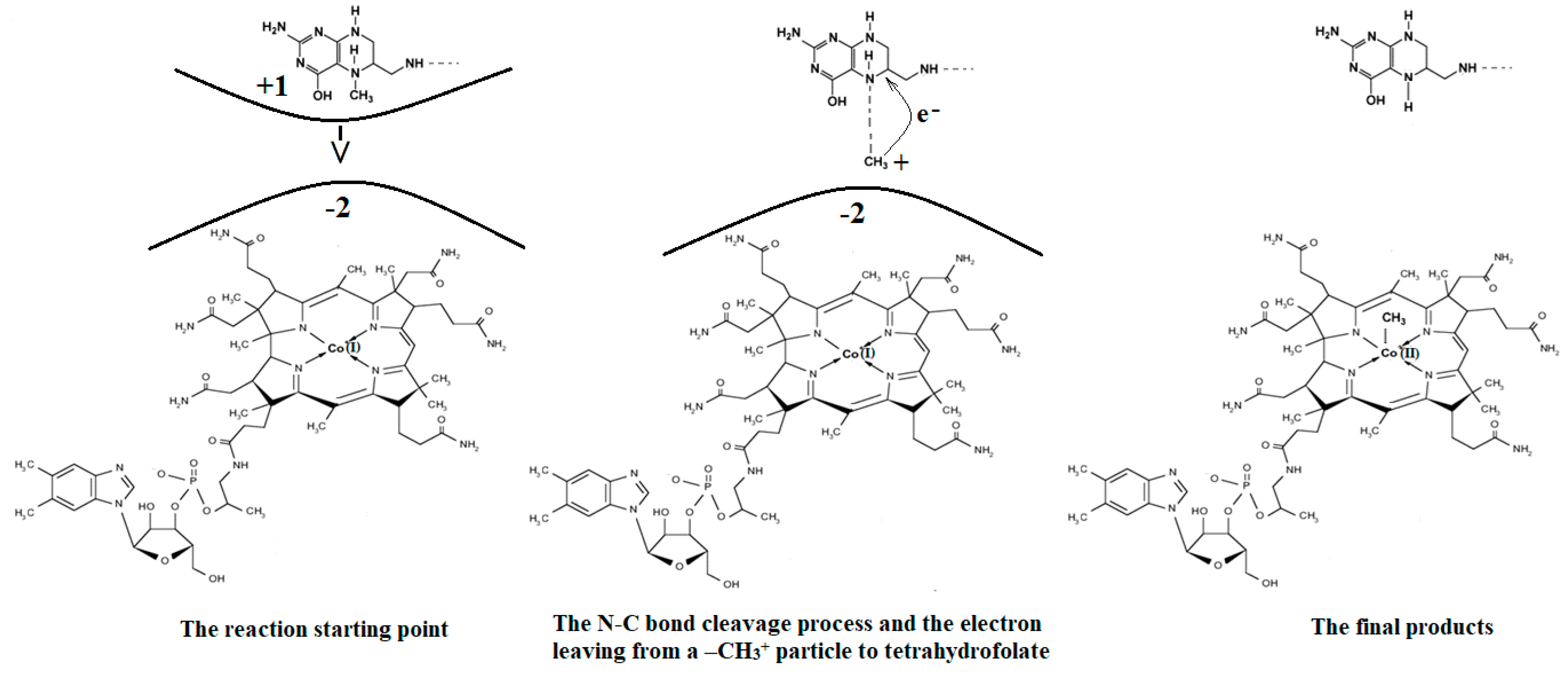
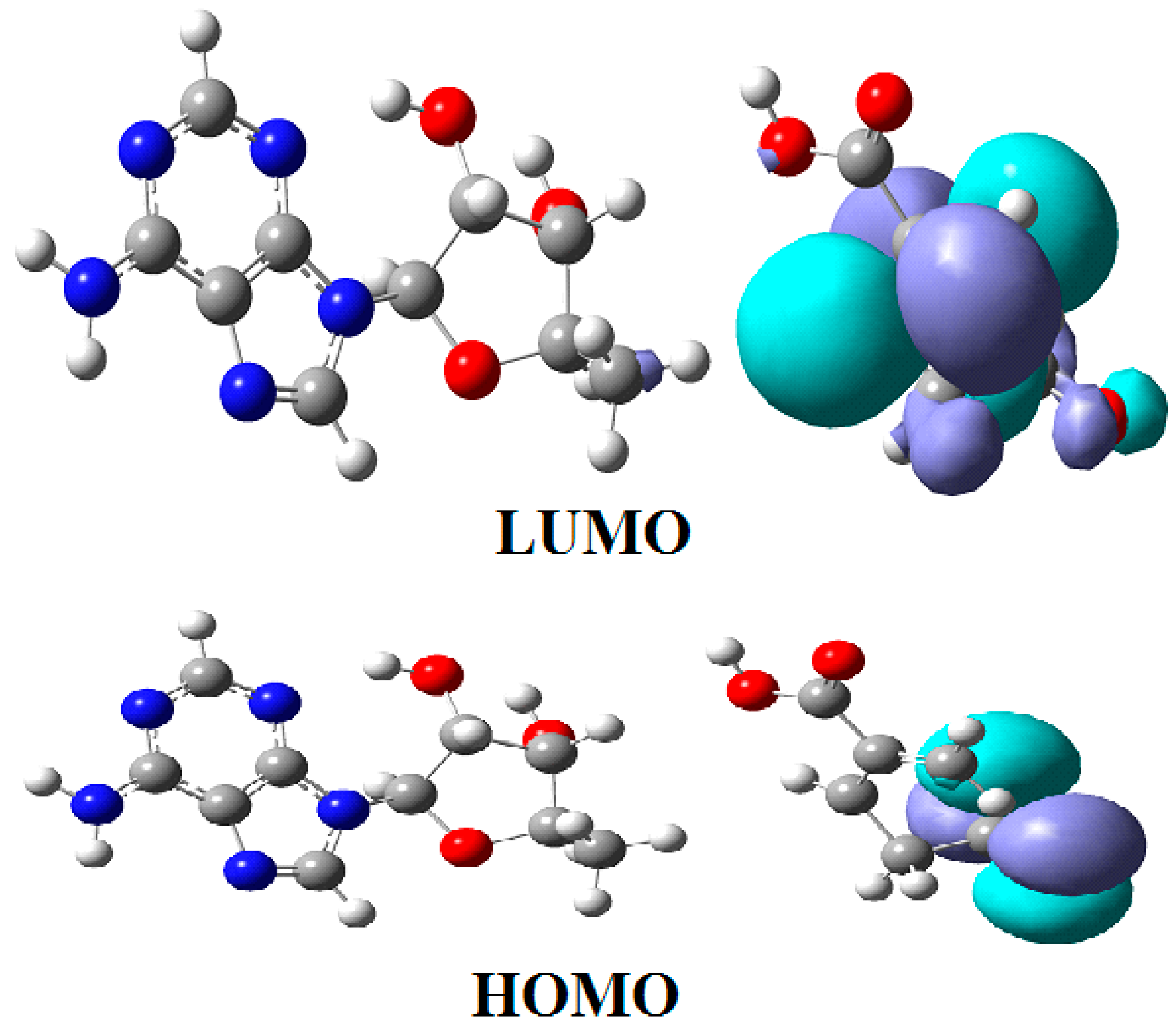
The mechanism of the methyl radical transfer from 5-methyltetrahydrofolate cob(I)alamin in the frame of the MCSCF method is analyzed here. As can be seen in Figure 3 and Figure 6, a second step in the Methionine Synthase process is the transfer of the methyl radical from 5-methyltetrahydrofolate to cob(I)alamin along with the transfer of an electron in the reverse direction. However, the CASSCF calculation of the electronic structure of the common model of 5-methyltetrahydrofolate and the cob(I)alamin cofactor shows a complete lack of orbital mixing or any interaction between reactants. Moreover, no common orbitals are formed between the reagents. It follows that no exchange, correlation, or configuration interactions occur, and only coulombic interactions are possible between them. Therefore, the coulombic interaction must trigger the N-C bond cleavage in the 5-methyltetrahydrofolate molecule and transfer the methyl radical to the cob(I)alamin cofactor. On the other hand, it has been shown that there is a protonation of the nitrogen atom, whose N-C bond must be cleaved in 5-methyltetrahydrofolate during the methyl radical transfer to the cob(I)alamin cofactor [44]. Indeed, the CASSCF calculations show that the nitrogen atom has a pyramidal geometric configuration in exactly the same geometric structure as the NH3 molecule, with a lone pair of electrons on the nitrogen atom. Its protonation is obvious according to generally accepted data. Thus, the protonation of the nitrogen atom is accepted both experimentally and theoretically in the Methionine Synthase process.
The protonated 5-methyltetrahydrofolate is a positive ion, and the value of its charge depends on the number of protonated nitrogen atoms, which are several in the molecule. On the other hand, the cob(I)alamin cofactor has a −2 charge. Therefore, the positively charged protonated 5-methyltetrahydrofolate and the negatively charged cob(I)alamin are attracted to each other as oppositely charged particles. Additionally, the CASSCF electronic structure calculations show that the central part of the cob(I)alamin cofactor carries a charge greater than −2. The 5-methyltetrahydrofolate protonate ion was used to study the influence of the charge of cob(I)alamin and its central −2 charged part on its behavior. The CASSCF calculations of the electronic structure of the 5-methyltetrahydrofolate protonate ion were performed at a distance of 5.00 Å from the −2 charged cob(I)alamin anion. At the beginning of the CASSCF geometry optimization, the 5-methyltetrahydrofolate protonate ion particle begins to move in the direction of the −2 charge. At the same time, the distance of the C-N chemical bond begins to increase abruptly, and the methyl radical becomes more and more isolated from the positive ion of the protonated 5-methyltetrahydrofolate structure. At a distance of about 3.50 Å from the cobalt atom, the N-C bond in the 5-methyltetrahydrofolate protonated ion is completely broken, and the methyl radical is completely dissociated from the 5-tetrahydrofolate particle, so it can be bonded to the cob(I)alamin cofactor molecule. At this distance, the total Mulliken summary charge of the methyl radical atoms becomes equal to +1, thus transferring an electron to the 5-tetrahydrofolate molecule. Interestingly, the more nitrogen atoms are protonated in the 5-methyltetrahydrofolate positive ions, the more abruptly the N-C bond breaks, releasing the methyl radical to the cobalt atom.
Conclusions. The charge of the cob(I)alamin cofactor negative ion perturbs [141] the electronic structure of the 5-methyltetrahydrofolate protonate ion in such a way that under its influence, the C-N bond breaks and the transfer of the positive CH3+ ion to the cob(I)alamin cofactor occurs. This perturbation in the electronic structure of 5-methyltetrahydrofolate protonate ion is under the influence of the −2 charge of the cob(I)alamin ion, and its central part is that of the triggering factor. This leads to the N-C bond cleavage and to the methyl radical ion transfer from the 5-methyltetrahydrofolate protonate ion to the cob(I)alamin negative ion. The C-N bond breaks heterolytically, and the reaction is of the SN2 type. The fact that this transfer takes place in the process of CASSCF geometry optimization demonstrates that the reaction takes place in the absence of a total energy barrier. All these results are in full agreement with the experimental data, according to which the transfer of the methyl radical from 5-methyltetrahydrofolate to the cob(I)alamin cofactor takes place simultaneously with the reverse transfer of an electron (Figure 15).
4.6. The Co-N Bond Cleavage and the Role of the Dimethylbenzimidazole Ligand in the Methionine Synthase Process
The Co-N bond cleavage in the methylcobalamin cofactor under the influence of the substrates in the frame of the MCSCF method is presented here. All experimental results, including X-ray data, show that the dimethylbenzimidazole ligand is only bound to the central cobalt atom in ex situ environments. Instead, within in situ biological environments, the Co-N bond of this ligand is cleaved, and in its place, another ligand (usually histidine) is attached to the central cobalt atom. This phenomenon clearly separates the in vitro from the in vivo experiments involving the methylcobalamin cofactor behavior, which seems very strange at first sight. Therefore, certain factors found only in the natural biological environment led to the breaking of the Co-N bond of the dimethylbenzimidazole ligand. To find these factors, a number of models were constructed that included the electron-reduced methylcob(II)alamin cofactor and two or three biological components presented within the cavity of the methylcobalamin cofactor. These models were used in CASSCF geometry determination to determine the substrates under whose influence would break the Co-N bond in the methylcobalamin cofactor.
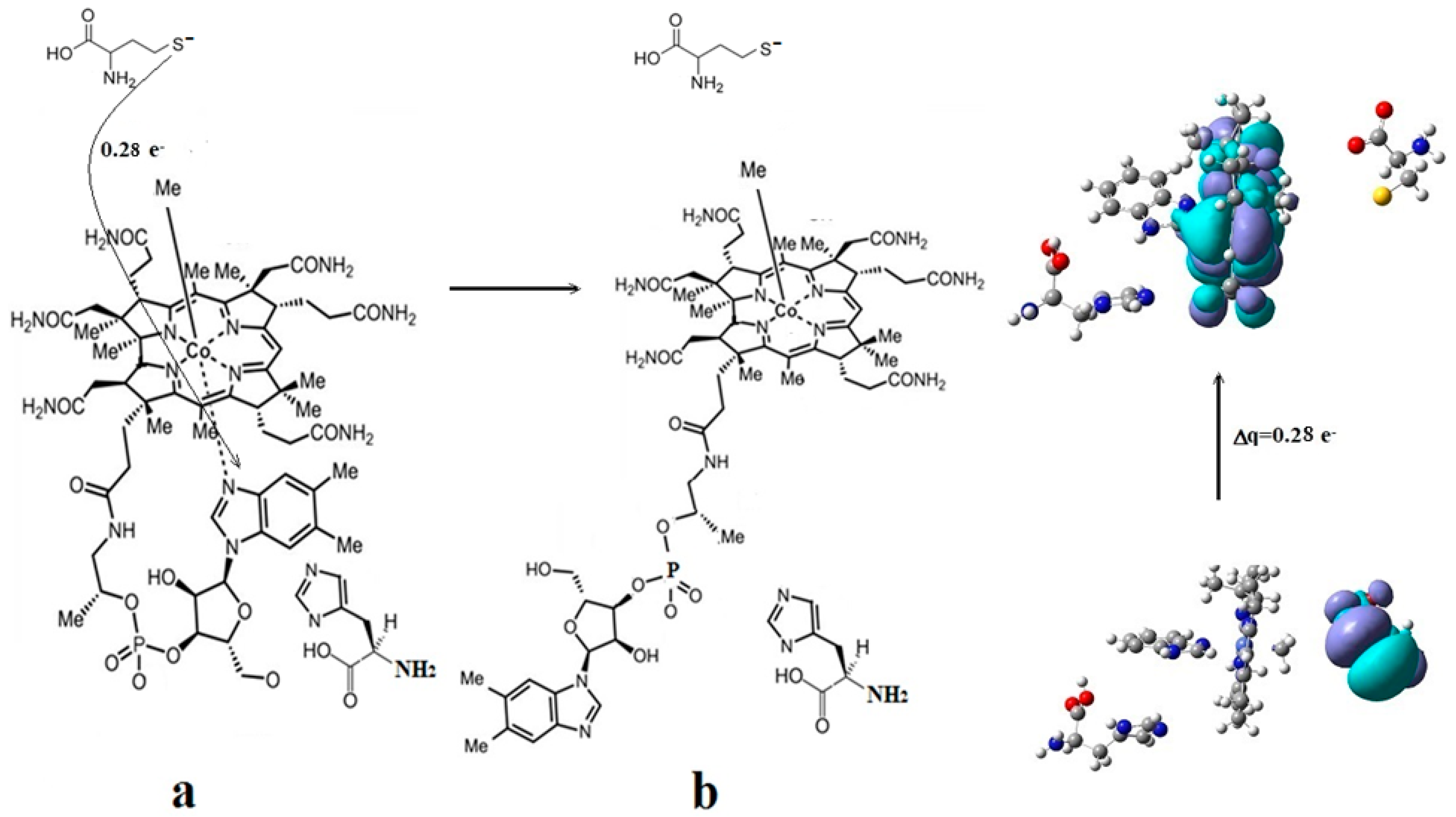
CASSCF geometry optimization of models built from the methylcob(II)alamin cofactor and substrates did not lead to a rupture in the Co-N bond except for one, which is presented in Figure 12b [143]. CASSCF calculations showed that most HOMOs have a population of electron density that does not differ dramatically from 1 or 2, while most LUMOs have a population of insignificant value. So, they could not significantly influence the chemical Co-N bond behavior. In contrast, HOMO2 and LUMO (Figure 16) have a population of electron density of approximately 1.72 e− and 0.28 e−, respectively, showing a significant mixture of these two orbitals and an electron density transfer from the first to the second one. It should be noted that the HOMO2 orbital consists only of the atomic orbitals of the substrate. The LUMO orbital is an antibonding molecular orbital formed by the atomic orbitals of the corrin ring and that of the Co-N chemical bond. It turns out that the transfer of electron density from HOMO2 to LUMO leads to a significant weakening of the Co-N bond due to the substrate-to-methylcobalamin electron density transfer. Furthermore, the Co-N chemical bond is the weakest chemical bond in both methylcobalamin and adenosylcobalamin cofactors, with a length of 2.12 Å in in vitro experiments [173,175] and between 2.35 Å and 2.50 Å in in vivo experiments [10,11,14].
These two factors—the transfer of electron density from HOMO2 of the substrate to the LUMO antibonding of the corrin ring and the weakness of the Co-N bond—lead to a rupture in the Co-N chemical bond and the removal of the dimethylbenzimidazole ligand from the central cobalt atom. CASSCF geometry optimization of the one-electron-reduced methylcobalamin cofactor, histidine, and homocysteine joint model (Figure 12b) started at the Co-C and Co-N distances [143] equal to 2.08 Å and 2.35 Å, respectively. From the start and then throughout the CASSCF geometry optimization, the Co-N bond increased continually until reaching a distance equal to 4.00 Å. At this distance, the Co-N bond is completely cleaved so that the further behavior of the dimethylbenzimidazole ligand no longer depends on its interaction with the central cobalt atom but on the interaction with various substrates, which are out of contact with the methylcobalamin cofactor. The mechanism of the Co-N bond cleavage and the removal of the dimethylbenzimidazole ligand from the corrin ring and from the cobalt atom is presented in Figure 17.
At this stage, the central atom of the methylcobalamin cofactor is ready for bonding with the histidine molecule, which plays a dual role: (a) participation in breaking the Co-N bond and removing the dimethylbenzimidazole ligand from the cobalt atom and from the corrin ring; and (b) binding to the cobalt atom so that together they can participate in the Methionine Synthase process. In this context, it seems that the role of the dimethylbenzimidazole ligand is to keep the methylcobalamin cofactor unaltered until the Methionine Synthase process starts and then to serve as an “anchor”, which, due to its interaction with nearby substrates, holds and supports the stability of the methylcobalamin cofactor.
Conclusions. The rupture in the Co-N axial bond and the removal of the axial ligand dimethylbenzimidazole takes place under the joint influence of the histidine molecule on the one hand and of the negative ion of the homocysteine on the other hand. The process of the Co-N axial bond breaking occurs under the influence of HOMO2–LUMO mixing, leading to the electron density transfer from the substrate negative ion atoms to the methylcobalamin cofactor in chemical interpretation. A major role is playing the fact that LUMO represents antibonding molecular π-orbitals composed of the atomic orbitals of the corrin ring and cobalt atom.
4.7. The Role of S-adenosyl-L-methionine (AdoMet) in the Methionine Synthase Process
The reactivation of the inactive cob(II)alamin particles by transferring the methyl radical from AdoMet in the frame of the MCSCF method is presented here. As discussed already, in the presence of the S-adenosyl-L-methionine (AdoMet) substrate, the Methionine Synthase process can, in principle, undergo an unlimited number of turnovers. Undoubtedly, if the AdoMet substrate was excluded from this process, its role in the Methionine Synthase process could be elucidated. Matthew and her group [41] ran the Methionine Synthase process in the absence of the AdoMet substrate to study how the biological environment influences the process. The turnovers of the Methionine Synthase process began at the usual rate but gradually slowed until reaching 2000 total turnovers and halted. The reactive cavity accumulated a significant number of inactive particles of the cofactor with the oxidation state of +2 on the central cobalt atom. One should mention that the experiment took place in a protective atmosphere. Moreover, the experiment occurred in the presence of dithiothreitol, a strong reducing agent that usually neutralizes oxidizing particles in the reactive medium. To explain this situation, the authors assumed that a portion of the cob(I)alamin cofactors are oxidized by flavodoxin back to Co+2 at each turnover. This was a logical hypothesis made in the absence of electronic structure data and was not supported or confirmed by any experimental or theoretical data. To solve this, we must recognize that even though the mechanism of this transfer is SN2, the SN1 mechanism plays a role. The total energy barrier of the Co-C bond cleavage reaction via the SN1 mechanism (Figure 11) is equal to 3.32 kcal/mol for the base-on cofactor compound compared to 7.82 kcal/mol for the base-off cofactor [143]. It has been shown that in biological systems, the relative Gibbs free energy can be approximated to the total energy of the biosystems [176]. The Co-C bond cleavage reaction rate and the methyl radical release are quite low, even in the base-on species of the methylcobalamin cofactor, which has a barrier of 3.32 kcal/mol of total energy. Only about 0.367% of this species has the Boltzmann Gibbs energy distribution at a room temperature of 298.15 K (temperature of the experiment) necessary to participate in this reaction. Thus, 0.367% of these cobalamin particles transform into bioinactive particles in this process during the first Methionine Synthase turnover. Accordingly, 99.633% of the methylcobalamin cofactor base-on particles do not participate in the SN1 Co-C cleavage reaction and are able to participate in the actual SN2 methyl radical transfer reaction from the methylcobalamin cofactor to the homocysteine. This 99.633% of the base-on particles of the methylcobalamin cofactor go through a series of biochemical transformations and collisions until they reach the same base-on structure with the cobalt atom in the same oxidation state and are ready to participate in the second turnover of the Methionine Synthase process. In this second Methionine Synthase turnover, another portion of 0.367% of base-on particles of the methylcob(II)alamin cofactor out of the remaining 99.633% of these particles participate in the SN1 reaction of the Co-C bond breaking and turn into inactive methylcob(II)alamin particles. Thus, in each Methionine Synthase turnover process, 0.367% of the active population of the base-on species of the methylcob(I)alamin cofactor is transformed into bioinactive particles, gradually decreasing the number of available active particles. To calculate the percentage of active particles of the base-on species of methylcob(I)alamin, the formula 1 − (1 − A/100)m = B/100 can be used, where A is the percentage rate per Methionine Synthase turnover, m is the number of turnovers, and B is the total percentage of the inactive methylcob(II)alamin particles. The equation shows that after a number of 2000 of the Methionine Synthase turnovers, a negligible amount of 0.00064% of methylcobalamin particles remain active, and 99.9994% become inactive particles if its total energy barrier is equal to that of the SN1 reaction of the Co-C bond cleavage of the base-on species (3.32 kcal/mol). Accordingly, the whole process becomes practically inactive after 2000 turnovers of the process. This shows that the CASSCF total energy barrier calculations of the Co-C bond-breaking reaction are in total agreement with the experimental data (in contrast with the DFT calculation data). As an illustrative point, if there is a small increase in the total energy barrier of the SN1 reaction to 4.00 kcal/mol., then after 2000 turnovers of the Methionine Synthase process, there would still be a number of active particles of methylcobalamin (equal to about 0.1% of the population) that would be observed in the experiment. All of this leads to the conclusion that the base-on species is the active particle of methylcobalamin in the Methionine Synthase process. Indeed, since the base-off species has an SN1 Co-C bond cleavage, the total energy barrier is equal to 7.82 kcal/mol, then after 2000 turnovers of the Methionine Synthase process, the number of active particles would be equal to 63% of the initial total of particles. Total turnovers would extend past 2000 in this scenario, in total contradiction with actual experimental data.
As shown above, in the absence of the AdoMet substrate, each turnover of the Methionine Synthase process accumulates 0.367% of the base-on particles of cob(II)alamin. This occurs due to the relatively low energy barrier of the SN1 Co-C bond cleavage and methyl radical loss. The accumulated inactive particles are the same base-on species but without the axial -CH3 ligand. To study the role of the AdoMet substrate in the Methionine Synthase process, CASSCF geometry optimization was performed on the AdoMet molecule, its dissociated ion, or its zwitterion along with a model of the cob(II)alamin base-on inactive particle. Both the AdoMet substrate and its zwitterion carry a positive charge and are sulfonic cations, while the AdoMet substrate, which lost a proton, is a neutral compound. Our calculations show that in the case of the interaction of AdoMet particles with those of inactive cob(II)alamin, the intermolecular HOMO and LUMO are formed already at the Co-C (-CH3) distance of 4.00 Å, including the atoms of both reactants with the populations of electron density equal to 1.88 e− and 0.12 e−, respectively, the first having the bonding nature and the second having an antibonding nature. So, the transfer in electron density from the HOMO to the LUMO leads to the weakening in the S-C (-CH3) bond in the AdoMet substrate and to the strengthening in the new Co-C (-CH3) bond in the case of the formation of the new methylcob(II)alamin compound. In the case of the cationic particles of the AdoMet substrate, the coulombic attraction between the cations of the AdoMet substrate and the anion inactive cob(II)alamin particle is added to this HOMO-LUMO interaction. Therefore, the interaction of the AdoMet cation substrate with the inactive particle cob(II)alamin was analyzed (Figure 18) (the interaction between the zwitterion and inactive cob(II)alamin is similar). Figure 18 shows the full structure of the AdoMet cation and a common model, including the AdoMet cation model and the cob(II)alamin inactive particle model. NH2 substituted the benzimidazole in the AdoMet structure in the CASSCF calculations, and the side chains of the cob(II)alamin cofactor inactive particle were replaced with hydrogens (Figure 18b).
As stated above, at the initial Co-C distance equal to about 4.00 Å, the CASSCF calculations show that several mixing common molecular orbitals are formed, two of which are HOMO and LUMO, their nature being intermolecular molecular orbitals with a maximum mixing degree and a significant electron density transfer from HOMO to LUMO (Figure 19). These orbitals include both the S-C bond atoms, which must break to transfer the methyl radical from the AdoMet ion to the inactive cob(II)alamin particle, and the Co-C bond atoms, which should form to revitalize the cob(II)alamin cofactor inactive particle.
The CASSCF geometry optimizations of the studied model at different Co-C distances show that the coulombic interaction determines the movement of the reactants toward each other in the calculated model until the Co-C distance becomes equal to 2.50 Å. Then, the HOMO-LUMO interaction is the factor determining the behavior of the calculated model. The CASSCF geometry optimization of the studied model at the Co-C distances below 2.50 Å shows that the S-C (-CH3) bond increases until it breaches and the new Co-C bond forms in newly formed methylcob(II)alamin active in the Methionine Synthase process. At the same time, the remaining part of the AdoMet substrate moves away from the -CH3 radical until it reaches the S-C distance greater than 4.00 A; that is, the transfer of the -CH3 radical from the AdoMet substrate to the inactive cob(II)alamin particle takes place, forming a new particle of methylcob(II)alamin, which is active in the Methionine Synthase bioprocess. This demonstrates that the reactions of the S-C bond (-CH3) breaking in the AdoMet substrate and the formation of the Co-C chemical bond in the new particle formed by methylcob(II)alamin are concerted reactions and that the process of transferring the -CH3 radical from the AdoMet substrate to the inactive particle of the cob(II)alamin is an SN2-type reaction. Thus, both the coulombic interaction between the negative ion of cob(II)alamin inactive in the Methionine Synthase process and the positive ion of the AdoMet substrate as well as the HOMO-LUMO interaction of their common model leads to the transfer of the -CH3 radical from the second reactant to the first. In the process of the transfer of the methyl radical from the AdoMet substrate cation to the inactive cob(II)alamin particle, the electron density transferred from the HOMO to the LUMO increases, reaching the level of 0.30 e−. The AdoMet positive-ion substrate transfers the methyl radical to the inactive cob(II)alamin particle so that the particle becomes active in the Methionine Synthase process. This is the role of the AdoMet substrate in the Methionine Synthase process. At all distances in the model’s CASSCF geometry-optimization procedures (including the inactive cob(II)alamin particle and the AdoMet-ion-substrate), the structures of the HOMO and LUMO orbitals are reminiscent of the HOMO and LUMO orbitals of the methylcob(II)alamin particle. At the end of the CASSCF geometry-optimization procedures, they become similar to those of the methylcob(II)alamin cofactor species. The formation of these frontier orbitals at the very starting distance of the CASSCF geometry-optimization procedures takes place even if the distance between the reagents is equal to about 4.00 A. This is similar to the behavior of the radical compounds [177,178]. Among other things, it confirms the generally known biochemical activity of the AdoMet substrate to be a methyl radical donor in biochemical processes. The mechanism of the methyl radical transfer from the AdoMet dissociate ion to the cob(II)alamin inactive particle is shown in Figure 20.
The MCSCF electronic structure of the model, which includes the inactive cob(II)alamin base-on particle and the AdoMet positive ion, is similar to the MCSCF electronic structure of the base-on species of the methylcob(II)alamin cofactor. This shows that the oxidation state of the central cobalt atom is +2 throughout the methyl radical transfer reaction from the AdoMet substrate to the inactive cob(II)alamin particle. This also proves that breaking the S-C (-CH3) bond in the AdoMet substrate is homolytic, which explains why the AdoMet substrate does not transfer the methyl radical to the cob(I)alamin cofactor particle, which might be present at this stage of the Methionine Synthase process turnover. Indeed, such an eventual homolytic methyl transfer from AdoMet to cob(I)alamin particle is impossible since the cob(I)alamin particle does not bond with the methyl radical without oxidation of the central Co atom. The coulombic interaction of two interacting structures along with the strong two-state Pseudo-Jahn–Teller Effect, e.g., the mixing of HOMO and LUMO intermolecular orbitals during the interaction of the AdoMet ion substrate with the inactive particle of the cob(II)alamin cofactor, leads to the S-C bond cleavage in the AdoMet ion and creates the Co-C bond in the methylcob(II)alamin cofactor. This transforms the inactive particles of the cob(II)alamin cofactor into active particles of the methylcob(II)alamin cofactor, which are able to participate in the reaction with homocysteine. Furthermore, taking into account that the methyl radical transfer reaction from the positive ion of AdoMet to the inactive cob(II)alamin particle is due to both the coulombic interaction between the reactants and the orbital mixing during the CASSCF geometry-optimization procedure causing it, one can conclude that it occurs in the absence of a total energy barrier.
Conclusions. The AdoMet substrate cation forms several common HOMO and LUMO intermolecular orbitals, with the inactive cob(II)alamine cofactor anion particles starting with distances slightly greater than 4.00 Å. The interaction and proximity of the two oppositely charged systems is caused by two factors: the coulombic interaction between them, which is prevalent at the distances larger than 2.50 Ᾰ, and the mixing of their HOMO-LUMO orbitals, which are prevalent at 2.50 Ᾰ distance between the interaction particles and lower. The structure of the HOMO and LUMO is such that the process of the CASSCF geometry optimization of the inactive particle cob(II)alamin–AdoMet substrate anion common model leads to the breaking of the S-C bond in the AdoMet anion substrate and to the formation of the Co-C chemical bond in the newly formed methylcob(II)alamin cofactor particle. During the mixing of the HOMO–LUMO orbitals, their overlapping led to an impressive electron density transfer of up to 0.30 e− at average distances in the transition model. At the same time that the -CH3 radical approaches the cobalt atom, the remaining part of the AdoMet anion moves away from the same -CH3 radical. When the Co-C bond is formed, the remaining part of the AdoMet ion is at a greater distance than 4.00 Å from the -CH3 radical. Thus, the formation of the Co-C bond in the methylcob(II)alamin cofactor particle and the breaking of the S-C bond in the AdoMet substrate anion are concerted reactions.
4.8. The Complete Mechanism of the Methionine Synthase Process
The complete mechanism of the Methionine Synthase is drawn here. The complete mechanism of the Methionine Synthase process was not fully demonstrated before without taking into account the effect of the AdoMet substrate [141]. Later, this mechanism was successfully updated (Figure 21) [143]. In the above, the mechanisms of all the phases of the Methionine Synthase process, both mechanistically and in terms of the electronic structure of the reagents, are exhaustively laid out. The active particle in the process is the base-on species of the methylcob(II)alamin cofactor. The process needs the AdoMet substrate to have a complete and continuous evolution. All reactions of the Methionine Synthase process are governed by the Pseudo-Jahn–Teller Effect; in other words, by the orbital mixing process of the reagents. All the reactions of the Methionine Synthase process take place in the absence of energy barriers, which explains the development of this process (in principle) of an unlimited number of turnovers. The truth is that the formation of the Co-N chemical bond occurs only if the oxidation state of the central atom is equal to +3, as our DFT calculations show.
5. MCSCF Calculations: Adenosylcobalamin Cofactor-Dependent Bioprocesses
5.1. General Considerations
The model building for adenosylcobalamin-dependent process calculations in the frame of the MCSCF method is analyzed here. The structure of the adenosylcobalamin cofactor is shown in Figure 1, and the different types of adenosylcobalamin cofactor-dependent bioprocesses (Figure 3, Figure 4 and Figure 5) are classified according to the type of reactions that take place in the biochemical environment: skeleton mutase, aminomutase, and eliminase processes. For their CASSCF method electron-structure calculations, the reaction types were classified according to the functional groups of the substrates participating in the bioprocesses: substrates containing participating –COOH groups and substrates containing participating –OH groups [142]. Unfortunately, due to the hasty publication of article 142, two small errors were made by the technical staff. Figure 5 and Figure 8 were mistakenly placed as one instead of the other. So, Figure 5 is actually Figure 8 and vice versa in 142 Instead of “CASSCF HOMO-LUMO surfaces of the 2-methylene-glutarate and the 5′deoxy5′-adenosyl radical common model at the beginning of the hydrogen transfer process.”, it should have said “CASSCF HOMO-LUMO surfaces of the 2-methylene-glutarate and the 5′deoxy5′-adenosyl radical common model at the beginning of the possible reverse hydrogen transfer process in 142. The adenosylcobalamin cofactor-dependent processes in which the substrate contains a –COOH group are Glutamate Mutase, 2-Methylene-glutarate Mutase, Methylmalonyl-CoA Mutase, Ethylmalonyl-CoA Mutase, two Lysine-5,6 aminomutases, and D-ornithine-4, 5 aminomutase bioprocesses. Another bioprocess that belongs to this group is the ribonucleoside triphosphate reductase process, in which the active part is the negative cysteine ion that contains the –COO− functional group similar to –COOH. The remaining bioprocess that belongs to this group is the Isobutyryl-CoA Mutase process, whose substrate contains neither the –OH nor the –COOH group. Instead, the radical of the amino acid valine [87], which contains the –COO− radical resulting from the –COOH group, plays the decisive enzymatic role (Figure 22). The second grouping of adenosylcobalamin cofactor-dependent processes with active substrates containing the –OH functional group includes Hydroxybutyryl-CoA Mutase, Ethanolamine Ammonia Mutase, Glycerol Dehydrate, and Propane-1,2-Diol Dehydrate bioprocesses (Figure 3 and Figure 5).
The –COOH functional group has a pKa value that is usually less than 10. This is approximately equal to the pKa values of the primary alcohols, with the highest pKa value of the alcohols equal to about 18. It is also known that R-NH3+ groups have pKa values equal to about 35. It is also known that a large number of R-NH3+ groups are present in the cavity of vitamin B12 cofactors. So, it can be inferred that the –COOH and –OH groups usually lose a proton to become –COO− and –CO− groups in the biochemical environment of vitamin B12. Therefore, the active substrates of adenosylcobalamin-dependent processes are species that contain either the –COO− group or the –CO− group [142]. Furthermore, during the adenosylcobalamin cofactor-dependent processes, two reactions take place. One reaction is breaking the Co-C bond that releases the cob(II)alamin species with the cobalt atom in the +2 oxidation state. The other reaction is the transfer of hydrogen from the active substrate to the formed 5′-deoxy-5′-adenosyl radical. As discussed previously, the relationship between the two reactions remained unknown for many years. Neither experimental data nor DFT or QM/MM (based on DFT) calculations have yet elucidated this relationship.
To study the behavior of the adenosylcobalamin cofactor in the presence of the active substrates of its dependent processes, CASSCF geometry optimizations were used on the models depicted in Figure 23b,c. In addition to the adenosylcobalamin model, the CH3-COO−− or the CH3-O−− model is also included, corresponding to the two active substrate group types discussed above.
Hydrogens have replaced the side chains in the adenosylcobalamin cofactor model, while the histidine has been replaced with imidazole. Since two adenosylcobalamin cofactor-dependent processes occur during its activity (breaking the Co-C bond and transferring hydrogen from the active substrate to the 5′-deoxy-5′-adenosyl radical), two CASSCF model calculations could be studied. One option was with the hydrogen transfer before breaking the Co-C bond, and another option was after its cleavage; however, only the first calculation option was chosen (Figure 23).
5.2. The Adenosylcobalamin Cofactor-Dependent Bioprocesses with Active Substrates Modeled with CH3COO−
The adenosylcobalamin-dependent processes with the substrates, which can be modeled with CH3COO−, are analyzed below in the frame of the MCSCF method. At the beginning of the CASSCF geometry-optimization procedure, the electronic structure of the adenosylcobalamin cofactor shows that the oxidation state of the central cobalt atom equals +3. The HOMO-LUMO distance is quite large at this oxidation state of the central atom. Thus, orbital mixing is insignificant, e.g., the prominence of the Pseudo-Jahn–Teller Effect is insignificant in such structures. The presence of the substrate transforms the calculated model so that the MCSCF calculation method is necessary. A significant number of the highest molecular orbitals (HOMO1, HOMO2, and HOMO4), which belong only to the atoms of substrates, enter the orbital mixing with the three lowest unoccupied orbitals, which belong exclusively to the adenosylcobalamin atoms. At the beginning of the CASSCF geometry optimization, a significant portion of the electron density (0.34 e-) was transferred from HOMOs to LUMOs (Figure 24). It is known that the Co-C chemical bond decreases its breaking energy as the axial ligand, bound to the central cobalt atom, increases its length [89,90]. Furthermore, the Co-C chemical bond in the adenosylcobalamin cofactor is one of the weakest in this series of compounds. It is obvious that this significant transfer in electron density from HOMO to LUMO orbitals (from the substrate to the adenosylcobalamin cofactor) will increase the Co-C bond distance starting from the beginning of the CASSCF geometry-optimization procedure of the calculated model. The Co-C distance increases continuously throughout the geometry-optimization procedure until about 0.56 e− is transferred from the substrate to the adenosylcobalamin cofactor. Cobalt-coordinating compounds with a +2 oxidation state are known to be more stable if they are penta-coordinated. This explains why during the CASSCF geometry optimization, the Co-C bond distance increases until its cleavage. Between the distances of 2.50 Å and 2.65 Å of the Co-C bond, the electronic structure of the adenosylcobalamin cofactor (Figure 24 and Figure 25) shows that its central cobalt atom converts from Co+3 to Co+2. At this stage, all the highest occupied molecular orbitals and some of the lowest unoccupied molecular orbitals are becoming σ and σ* axial orbitals. HOMO1, HOMO2, and HOMO3 transfer electron density to LUMO1, LUMO2, and LUMO3, i.e., from the substrate to the adenosylcobalamin cofactor. In addition, due to the orbital mixing, this additionally contributes to the Co-C bond breaking, given that all HOMOs and some of the LUMOs are σ-orbitals, causing the σ-σ* Pseudo-Jahn–Teller Effect. The mixing of orbitals also leads to a much lower HOMO-LUMO separation, from about 8.17 eV to about 2.89 eV, which increases the orbital mixing and enforces the Pseudo-Jahn–Teller Effect. This leads to a continuous Co-C bond distance increase during the entire CASSCF geometry-optimization procedure.
In fact, starting with the 2.50 Ǻ and larger distances between the substrate and adenosylcobalamin cofactor model, CASSCF geometry optimization the electronic density transfer increases up to 0.56 e− and more from the first to the second parts of the common adenosylcobalamin–substrate model (Figure 25), stimulating more the Co-C bond cleavage. The Co-C bond distance increases due to the larger charge transfer from the substrate to the adenosylcobalamin compound and the stronger Pseudo-Jahn–Teller Effect. Noticeably, the Co-C bond distance increase is accompanied by a concomitant decrease in the distance between the substrate and the release of the 5′-deoxy-5′-adenosyl radical. In effect, at 3.17 Å of the Co-C bond length, the distance between the substrate and the 5′-deoxy-5′-adenosyl radical becomes equal to only 2.11 Å even if, at the beginning of the CASSCF optimization process, the shortest distance between the closest atoms of the two systems was equal to 5.00 Å. At this distance, the interaction between the substrate and the releasing 5′-deoxy-5′-adenosyl radical becomes so strong that HOMO1, HOMO2, and HOMO3 become 5′-deoxy-5′-adenosyl radical and substrate common molecular orbitals (Figure 25).
Conclusions. The adenosylcobalamin cofactor-dependent bioprocess models CASSCF geometry optimization led to Co-C bond cleavage without an energy barrier. The triggering factor of the Co-C bond cleavage process is the intermolecular electron density transfer from the substrates of the adenosylcobalamin-dependent processes to the adenosylco(III)balamin cofactor itself (Figure 26). The Co-C bond cleavage in studied adenosylcobalamin cofactor-dependent bioprocesses and the hydrogen transfer from their substrates to the 5′-deoxy-5′-adenosyl radical are mutually dependent concerted reactions that are in full agreement with the experimental data.
5.3. The Adenosylcobalamin Cofactor-Dependent Bioprocesses with Active Substrates Modeled with CH3-O−
The adenosylcobalamin-dependent processes with the substrates, which can be modeled with CH3C-O−, are analyzed below in the frame of the MCSCF method. The mechanism of the adenosylcobalamin cofactor-dependent processes, whose active substrates are modeled with CH3-O−, is similar to the above-discussed processes, whose substrates can be modeled [142] with CH3COO−. From the beginning of the CASSCF geometry-optimization procedure, the electronic structure of the adenosylcobalamin cofactor shows that the oxidation state of the central cobalt atom is equal to +3. The Co-C bond is stable without interaction with the substrate. The HOMO-LUMO separation is too large for orbital mixing to be significant in the absence of the substrates. When substrates are introduced, HOMO1, HOMO2, and HOMO4, which belong to the atoms of the substrates, enter the orbital mixing with three LUMOs, which belong to the adenosylcobalamin cofactor in the adenosylcobalamin cofactor–substrate model. An amount of electron density (0.31 e−) transfers from HOMOs to LUMOs at the beginning of the CASSCF geometry optimization (Figure 27). It is known that the Co-C chemical bond decreases its bond-breaking energy as the length of the ligand, which is bound to the central cobalt atom in compounds similar to vitamin B12 cofactors, increases [89,90]. It is also known that the Co-C chemical bond in the adenosylcobalamin cofactor is one of the weakest in the group of compounds. It is obvious that this significant transfer of electron density from HOMOs to LUMOs (from the substrate to the adenosylcobalamin cofactor) would increase the Co-C distance from the beginning of the CASSCF geometry-optimization procedure of the calculated model. Then, the electronic structure of the adenosylcobalamin cofactor (Figure 27) shows that its central cobalt atom converts from Co+3 to Co+2. Cobalt-coordinating compounds with a +2-oxidation state are known to be more stable if they are penta-coordinated. This central atom oxidation state conversion leads to an even faster increase in the Co-C distance in the CASSCF geometry-optimization procedure. At this stage, all the highest occupied molecular orbitals and some of the lowest unoccupied molecular orbitals are becoming σ and σ* axial orbitals. HOMO1, HOMO2, and HOMO4 transfer electron density to LUMO1, LUMO2, and LUMO3, i.e., from the substrate to the adenosylcobalamin cofactor. Mixing orbitals contributes to breaking the Co-C bond; all HOMOs and some of the LUMOs are σ-orbitals, causing the σ-σ* Pseudo-Jahn–Teller Effect. The mixing of orbitals also leads to a much lower HOMO-LUMO separation, moving from about 7.68 eV to about 3.21 eV, which in turn increases the orbital mixing and the Pseudo-Jahn–Teller Effect’s influence on the Co-C bond cleavage process. All of this leads to a continuous Co-C bond distance increase during the entire CASSCF geometry-optimization procedure.
Additionally, with the increasing distance between substrate and adenosylcobalamin cofactor model, during the CASSCF geometry optimization- the transferred electronic density from the substrate to the adenosylcobalamin is increasing up to 0.60 e− and more stimulating the Co-C bond cleavage. Noticeably, the Co-C bond distance increase is accompanied by a concomitant decrease in the distance between the substrate and the release of the 5′-deoxy-5′-adenosyl radical. At the 3.05 Å of the Co-C bond length, the distance between the substrate and the released 5′-deoxy-5′-adenosyl radical becomes equal to 1.86 Å even if, at the beginning of the CASSCF optimization process, the shortest distance between the closest atoms of the two systems was equal to 5.00 Å. At this distance, the interaction between the substrate and the released 5′-deoxy-5′-adenosyl radical becomes so strong that HOMO1, HOMO2, and HOMO3 become 5′-deoxy-5′-adenosyl radical and substrate common molecular orbitals (Figure 27). The hydrogen transfer from the substrate to the 5′-deoxy-5′-adenosyl radical is facilitated. Thus, the process of transferring hydrogen from the substrate to the 5′-deoxy-5′-adenosyl radical is conditioned by the reaction of the Co-C bond cleavage and vice versa. The whole process is schematically demonstrated in Figure 28.
Conclusions. The adenosylcobalamin cofactor-dependent bioprocess models of CASSCF geometry optimization led to Co-C bond cleavage without the energy barrier. The triggering factor of the Co-C bond cleavage process is the intermolecular electron density transfer from the substrates of the adenosylcobalamin-dependent processes to the adenosylco(III)balamin cofactor itself. The Co-C bond cleavage in the studied adenosylcobalamin cofactor-dependent bioprocesses and the hydrogen transfer from their substrates to the 5′-deoxy-5′-adenosyl radical are mutually dependent concerted reactions.
5.4. The Nature of the Hydrogen Transfer from the Substrates to the 5′-Deoxy-5′-Adenosyl Radical
The transfer of a hydrogen from the substrate to the 5′-deoxy-5′-adenosyl radical is analyzed here through the prism of existing theories based on the MCSCF results. It has been postulated [73] that there is a tunneling nature in the hydrogen transfer from the active substrates in the adenosylcobalamin cofactor-dependent processes to the 5′-deoxy-5′-adenosyl radical. The research community recognizes hydrogen tunneling as a widespread but complex effect [179]. Several theories have been set forth to explain this phenomenon, ranging from the semi-classical theory of transition states to the theories of quantum mechanics [179,180]. Hydrogen tunneling has been evaluated with the help of the Pseudo-Jahn–Teller Effect [181]. In general, quantum tunneling is a phenomenon in which the energy barrier of its transfer reaction has a much higher value than the sum of the energy values of the tunneling reaction components. Quantum tunneling requires that the wave function of the system under consideration must propagate throughout an energy barrier. Therefore, the semi-classical theory of transition states does not fit the general features of this phenomenon [179,180].
The total energy barrier of the common model, including each substrate of all adenosylcobalamin cofactor-dependent bioprocesses and the 5′-deoxy-5′-adenosyl radical, was determined using the CASSCF method with 10 electrons and 10 orbitals in the active zone and the 6-31G** basis set for all atoms [142]. According to quantum mechanics, the crucial condition accompanying a tunneling transfer is the propagation of the wave functions of the compounds in the space between the systems participating in the tunneling transfer. Interestingly, hydrogen transfer in all adenosylcobalamin cofactor-dependent biochemical processes occurs under similar conditions. Also, the electronic structures of all substrates that participate in the process, in terms of hydrogen transfer from each of them to the 5′-deoxy-5′-adenosyl radical, are similar. So, only one common model will be discussed here: the 1,2-propanediol and the 5′deoxy-5′-adenosyl radical. The HOMO and LUMO surfaces at the beginning of the hydrogen transfer process are depicted in Figure 29. First, it is important to note that both HOMO and LUMO are composed of the orbitals of atoms, which are involved in the transfer of hydrogen from one system to another, despite the fact that the carbon atoms between which the transfer of hydrogen takes place are greater than 4.00 Å apart. A second, no less important aspect is that the wave functions are located in the space between those carbons where the hydrogen transfer occurs. Both these particularities of HOMO and LUMO orbitals of the calculated systems strongly point to the tunneling nature of the hydrogen transfer from one system to another.
It is interesting to explore whether hydrogen transfer from the substrate to the 5′-deoxy-5′-adenosyl radical is reversible (more specifically, to explore whether the reverse transfer from the hydrogenated 5′-deoxy-5′-adenosyl radical to the dehydrogenated substrate is possible through the tunneling effect). Figure 30 shows the surfaces of the HOMO and LUMO for the de-hydrogenated 2-methylene-glutarate substrate and the hydrogenated 5′deoxy-5′-adenosyl radical common model at the beginning of the possible reverse hydrogen transfer process. As can be seen from Figure 30, the HOMO and LUMO of these compounds only include the atomic orbitals of the atoms situated away from the hydrogen transferring zone and do not include the atomic orbitals of the atoms, which would be directly involved in the reversible transfer of hydrogen from the hydrogenated 5′deoxy-5′-adenosyl radical to the de-hydrogenated 2-methylene-glutarate substrate. Obviously, the HOMO and LUMO of these compounds are situated away from the space where the reverse hydrogen transfer would occur.
This evidence strongly refutes the possibility of a reverse hydrogen transfer through quantum mechanical tunneling effects. In conclusion, an irreversible hydrogen transfer from substrates of the adenosylcobalamin cofactor-dependent processes to the 5′-deoxy-5′-adenosyl radical takes place in full accordance with the experimental data.
It is relevant to discuss the possible presence of the Pseudo-Jahn–Teller Effect in the two systems just analyzed. We previously discussed the HOMO and LUMO surfaces of the 1,2-propanediol and the 5′-deoxy-5′-adenosyl radical common model at the beginning of the hydrogen transfer process between them (Figure 29) as well as the HOMO and LUMO surfaces of the de-hydrogenated 2-methylene-glutarate substrate and the hydrogenated 5′deoxy-5′-adenosyl radical common model (Figure 30) at the beginning of the hypothetical reverse hydrogen transfer process. In the case of direct transfer from the substrate to the 5′-deoxy-5′-adenosyl radical, the HOMO and LUMO orbitals are very similar. However, the HOMO is a bonding orbital and the LUMO is an antibonding orbital (Figure 29). Their orbital mixing during the CASSCF procedure is at the maximum level, and the electron density transfer is also maximum so that both orbitals are populated with almost an entire electron. Therefore, the two-state Pseudo-Jahn–Teller Effect is extremely prevalent. It strongly contributes to the hydrogen bond breaking and to the formation of another hydrogen bond. On the other hand, in the case of a hypothetical reverse transfer from the hydrogenated 5′-deoxy-5′-adenosyl radical to the de-hydrogenated substrate, the HOMO and LUMO orbitals are not similar. They are not similar bonding and antibonding orbitals that would include the atoms involved with hydrogen transfer. Their orbital mixing during the CASSCF procedure is at the minimum level, and the electron density transfer from one to the other is also minimal and insignificant. So, the HOMO is populated with about two electrons, and the LUMO electron density population is close to zero. Therefore, the Pseudo-Jahn–Teller Effect is not present in the common model of the possible reverse hydrogen transfer and does not contribute to the hydrogen bond cleavage process.
Conclusion. An irreversible tunneling transfer of hydrogen occurs from the general point of view of quantum mechanics and from the Pseudo-Jahn–Teller Effect’s perspective between the substrates of adenosylcobalamin cofactor-dependent processes and the 5′-deoxy-5′-adenosyl radical released during the Co-C bond cleavage process, in full accordance with the experimental data.
5.5. The Co-N Bond Cleavage in the Adenosylcobalamin Cofactor
The Co-N bond cleavage analysis of the adenosylylcobalamin cofactor under influence of the substrates in the frame of the MCSCF method is presented here. As shown above, experimental data, including X-rays of biological materials [10,11,14], show that the dimethylbenzimidazole ligand is replaced by the histidine ligand in the adenosylcobalamin cofactor before the real adenosylcobalamin Glutamate Mutase and Methylmalonyl-CoA Mutase processes start (Figure 31). The question not only regards how histidine binds to the central cobalt atom if it has a free coordinating site; it mostly regards how this free site appears and how the Co-N axial bond in the adenosylcobalamin cofactor is cleaved, removing the dimethylbenzimidazole ligand from the central cobalt atom. In all X-ray data of ex situ non-biological materials, this bond and the dimethylbenzimidazole ligand are always present. Therefore, only in in situ biological materials is the Co-N bond cleaved. To investigate this situation, the substrates present in the adenosylcobalamin cavity must be taken into CASSCF geometry optimizations similar to how the study of the breaking of the Co-N bond in the methylcobalamin [143] cofactor in advance to the Methionine Synthase process has been studied. As can be seen in Figure 32, the common feature of the Glutamate Mutase and Methylmalonyl-CoA Mutase processes’ substrates is the -COO− active group. Therefore, CASSCF calculations have used the models comprising the adenosylcobalamin cofactor model, the histidine molecule, and the CH3COO− structure (which model the active substrates in the processes on the adenosylcobalamin cofactor-dependent Glutamate Mutase and Methylmalonyl-CoA Mutase bioprocesses) (Figure 33).
CASSCF geometry optimizations of the structures, which model the initial stage of the adenosylcobalamin cofactor-dependent processes (Figure 33) with the substrates modeled by CH3COO− are similar to the CASSCF geometry optimization of the initial stage model in the Methionine Synthase process (see above) but with some particularities.
Their CASSCF electronic structures showed that three HOMOs and three LUMOs (Figure 34) are involved in the orbital mixing effect, in contrast to the initial step in the Methionine Synthase process model, in which only HOMO2 and LUMO showed a significant orbital mixture. It should be noted that the HOMO1, HOMO2, and HOMO3 orbitals consist only of the atomic orbitals of the substrate, and the LUMO1, LUMO2, and LUMO3 orbitals are antibonding molecular orbitals that are formed by the atomic orbitals of the corrin ring and of the Co-N chemical bond atoms (Figure 34). The population of the total electron density of the HOMO1, HOMO2, and HOMO3 orbitals is equal to 1.73 e- in the case of the model in Figure 33. At the same time, the population of the total electron density of the orbitals LUMO1, LUMO2, and LUMO3 is equal to 0.27 e-.
It turns out that the transfer of electron density from three HOMOs to three LUMOs leads to a significant weakening in the Co-N bond in the model studied of the Glutamate Mutase and Methylmalonyl-CoA Mutase adenosylcobalamin cofactor-dependent bioprocesses. On the other hand, the Co-N chemical bond is the weakest chemical bond in the adenosylcobalamin cofactor, with a length of 2.24 Å in in vitro experiments [175] and between 2.35 Å and 2.50 Å in in vivo experiments [10,11,14,182]. These two factors lead to the rupture in the Co-N chemical bond and the removal of the dimethylbenzimidazole ligand from the central cobalt atom. CASSCF geometry optimization of the adenosylcobalamin cofactor, histidine, and substrates joint model (Figure 31) starts at the Co-C and Co-N distances of 2.03 Å and 2.30 Å, respectively, in accordance with the X-ray data for in vivo compounds [10,11,14]. From the beginning and during CASSCF geometry optimization, the Co-N bond increased continually until reaching a distance equal to 4.00 Å. At this distance, the Co-N bond is completely cleaved, and the dimethylbenzimidazole ligand is completely removed from the central cobalt atom so that the further behavior of the dimethylbenzimidazole ligand no longer depends on its interaction with the central cobalt atom. Its behavior is determined by its interactions with other substrates, leading it to the location shown in the X-ray diffraction data [10,11,14]. In this new location, the dimethylbenzimidazole ligand supports the position of the whole molecule of the adenosylcobalamin cofactor. The mechanisms of the Co-N bond cleavage and the removal of the dimethylbenzimidazole ligand from the corrin ring and from the cobalt atom are presented in Figure 35.
The role of dimethylbenzimidazole in the adenosylcobalamin-dependent processes, whose substrate is modeled by the CH3COO− model, is to preserve the coordinate site ready for the histidine molecule until the onset of adenosylcobalamin cofactor-dependent processes and to support the stable position of this cofactor during these processes.
It must be said that the histidine molecule and the dimethylbenzimidazole ligand have a well-developed structure of π orbitals, which allows them to interact by forming π–π Wander Wals bonds, keeping them at approximately 4.00 A from each other throughout the geometry-optimization procedure until the C-N bond breaks completely. Interestingly, this interaction lasts only during the Co-N bond-breaking reaction. At distances of 4.00 A and greater in this bond, the histidine molecule and the dimethyl benzimidazole ligand do not interact, and their behavior becomes independent. It is obvious that as a result of this fact, the subsequent behavior of the dimethylbenzimidazole ligand depends only on its interaction with other substrates present in or near the adenosylcobalamin cofactor. As a result of these interactions, it can move away from this cofactor by positioning itself in the environment of other substrates, or it can bind again to the cobalt atom. Our DFT calculations show us that this choice is free because the formation of the Co-N bond is unhindered because the oxidation state of the cobalt ion is +3, and its movement among the substrates also does not encounter energy barriers. The histidine molecule has the same possibilities. Depending on the energy gain, one will unite with the cobalt atom, and another will settle among other substrates. Priority will be the position, leading to a lower total energy than the other.
A system that would include all the substrates near the adenosylcobalamin cofactor is enormously large and does not allow its calculation via the MCSCF method at this moment, especially since the experimental data on the state of this large system is still insufficiently studied experimentally for most of the 12 enzymatic processes dependent on the adenosylcobalamin cofactor. Fortunately, in the case of the Glutamate Mutase and Methylmalonyl-CoA Mutase processes, there are structural data [11,14] that show that in the case of these two bioprocesses, the most convenient state is the one in which the histidine molecule becomes the ligand of the cobalt atom. The dimethylbenzimidazole ligand interacts with other substrates in the area, moving away from the corrin ring and the cobalt atom and settling between those substrates. Interestingly, any substrate containing the COO− group can influence the adenosylcobalamin cofactor with the histidine molecule in the same way, according to the calculation model used by the author. In conclusion, the adenosylcobalamin cofactor in Glutamate Mutase and Methylmalonyl-CoA Mutase bioprocesses, which are active in the human body, lose their dimethylbenzimidazole axial ligand in the preliminary phase of the enzymatically active act.
Conclusions. The rupture in the Co-N axial bond and the removal of the axial ligand dimethylbenzimidazole take place under the joint influence of the histidine molecule and of negative ion of each bioactive substrate, which is participating in the enzymatic process. The process of the Co-N axial bond breaking occurs under the influence of partial electron density transfer from the highest occupied molecular orbitals of HOMO1, HOMO2, and HOMO3, which are composed only of the atomic orbitals of the negative ion atoms of the substrates to the lowest unoccupied molecular orbitals of LUMO1, LUMO2, and LUMO3, which represent antibonding molecular π-orbitals composed of the atomic orbitals of the corrin ring and dimethylbenzimidazole ligand.
6. The Disposal of the Toxic Organic Halides under the Catalytic Influence of Vitamin B12
The cleavage of the C-Hal bond in toxic organic halides under the influence of the cob(I)balamin cofactor in the frame of the MCSCF method is analyzed here. A large problem in modern society is organic halide compounds, which over the years have accumulated in all components of the environment, like soil and water basins, etc., in extraordinary quantities [183,184,185,186] in all countries, including the US [187]. Due to the toxicity of these compounds to animals, including humans, a decline in many fish and animal species has been observed [188]. The situation is much more complicated if we consider that these compounds are carcinogenic [189]. Reducing agents have been shown to be active in organic halides [185,189,190,191,192,193,194,195]. DFT calculations [196] showed that this is because the LUMOs of these compounds are antibonding orbitals containing the C-Hal bond orbitals, and their decomposition is sequential.
The direct action of vitamin B12 [195] or its action through microorganisms [197,198,199,200,201] leads to a degradation in organic compounds of different structures and compositions [202,203,204]. Interestingly, their mechanism of action is similar, assuming that it occurs when an electron passes from the cofactor cob(I)alamin of vitamin B12 to the molecules of organic halide compounds. In the case of organohalide-respiring microorganisms, this takes place in the process of using the energy of the decomposition of organic halide compounds for their growth and development [205,206,207,208,209]. When studying the action of Dehalococcoides bacteria, it was demonstrated that the active agent is the cofactor cob(I)alamin [205,206,207,208,209]. The decomposition process of organic halide compounds under the influence of Dehalococcoides mccartyi strain CBDB1 [199,209,210] was studied by using the DFT method. To explain the process of decomposition of organic halide compounds under the influence of the cofactor cob(I)alamin, the donor–acceptor method was used [199,200]. Unfortunately, the donor–acceptor process with the direct participation of HOMO and LUMO cannot be modeled by using the DFT method, so this process was studied in more depth using the MCSCF method [211]. Common models (Figure 36) of the cofactor cob(I)alamin and organic halide compounds, 3,5-dibromo-4-hydroxobenzoic [201] acid, 3-bromo-4-hydroxobenzoic acid [201], and 1,2-dichloroethene [198], for which there are experimental data on decomposition under the influence of vitamin B12, were used for the calculations. Hydrogens substituted the side chains, while OH−situated more than 10 Å far from the calculated models was used for substituting the =HPO4− group, balancing the total charge of the calculated models. The reactants were in an almost or completely perpendicular position to the organic compound halide on the corrin ring at approximately 4.00 Å from each other (Figure 36).
At the beginning of and during the geometry-optimization procedures in all models, intermolecular orbitals were created, the surfaces and the electron populations of which are shown in Figure 37. The electron density of the other molecular orbitals is insignificant in the active zone of the CASSCF procedures and cannot influence the behavior of the calculated models. These orbitals are formed by mixing the HOMO of the cob(I)alamine cofactor with the LUMO of the organic halide compounds, which is calculated in the approximation of single-determinant methods. Given the fact that their electronic population comes completely from the HOMOs of the vitamin B12 cofactor, it is obvious that their mixture with the LUMOs of halogenated organic compounds leads to the transfer in electron density from the former to the latter. Given the fact that these intermolecular orbitals are antibonding for C-Hal bonds from the organic halide compounds, they weaken and break during the geometry-optimization processes.
Indeed, the C-Hal connection increases during the geometry-optimization procedures until it breaks the heterolytic, releasing a negative ion of halogen, which binds the cobalt atom via the van der Waals force, while the other part of the organic halide molecule moves away from the halogen ion up to distances greater than 4.00 A. The van der Waals bond of the negative halogen ion turns into a chemical bond when the oxidation state of the cobalt ion changes from +1 to +2 or +3. These results are in agreement with the experimental data [199,202]. The catalytic processes of halogen ion transfer from organic halide compounds to the cofactor cob(I)balamin are shown in Figure 38, Figure 39 and Figure 40.
It must be said that breaking the C-Hal bond. occurs in organic halide molecules only if they interact with the central cobalt atom of the cofactor cob(I)alamin through one of its halogen atoms in a perpendicular or quasi-perpendicular manner to their position on the corrin ring plane. In all other cases, when they do not interact directly with the cobalt atom through its halogens or are parallel or quasi-parallel on the corrin ring plane, their interaction with the cofactor cob(I)alamin is much weaker.
Conclusions. The cleavage of the C-Hal bond in toxic organic halides and their disposal occurs under influence of the electron density transfer from HOMOs of the cob(I)alamin cofactor to the antibonding LUMOs of the toxic organic halides.
7. Concluding Remarks
The general mechanism of the vitamin B12 cofactor-dependent bioprocesses was established from the experimental data. The electronic structure calculations, based on the DFT and QM/MM method (based on DFT), found that the energy barrier of the Co-C bond cleavage is lower in the case of the base-off species of the methylcobalamin cofactor, which is in flagrant disagreement with the experimental data. This leads to the erroneous conclusion that the methylcobalamin base-off species is the active particle of the Methionine Synthase process. On the other hand, the high total energy barriers of the Co-C bond cleavage (one of the crucial reactions in all vitamin B12-dependent bioprocesses) obtained by using DFT and QM/MM (based on DFT) does not explain their unlimited turnover. In particular, it does not explain the experimentally proven possible unlimited number of Methionine Synthase turnovers. The limitations of DFT-based methods not taking into account the Pseudo-Jahn–Teller Effect and orbital mixing cause these disagreements between the experimental and DFT or QM/MM (based on DFT) data. Instead, the CASSCF method accounts for the Pseudo-Jahn–Teller Effect, which is central to the catalytic behavior of vitamin B12. The Co-C bond cleavage in methylcobalamin is triggered by a reverse electron transfer from the substrate to methylcobalamin during an SN2 reaction in the Methionine Synthase bioprocess. Meanwhile, the Co-C bond cleavage in adenosylcobalamin is triggered by the reverse electron density transfer from the substrates to the adenosylcobalamin cofactor in its dependent processes. The methyl radical positive ion is transferred from the protonated 5-methyltetrahydrofolate, whose electronic structure is perturbed by the -2 charge of the cob(I)alamin particle, to the cob(I)alamin base-off cofactor during the Methionine Synthase bioprocess. About 0.367% of the base-on methylcob(II)alamin particles lose the methyl radical due to the low total energy barrier of the SN1 Co-C bond cleavage reaction, converting into inactive ones during the Methionine Synthase process. The role of the AdoMet substrate is to transfer a methyl radical to the inactive cob(II)alamin particles to convert them back into active particles. The full mechanism of the Methionine Synthase process was proved and drawn for the first time recently. The active particle in the Methionine Synthase process is a base-on methylcobalamin cofactor with the dimethylbenzimidazole ligand substituted with histidine. The MCSCF electronic structure calculations show that the Co-C bond cleavage and hydrogen transfer from the active substrates in all adenosylcobalamin cofactor-dependent processes are mutually dependent concerted reactions. The HOMO of the vitamin B12 cob(I)alamin cofactor forms intermolecular orbitals mixing with the LUMO of toxic organic halides, populating them with one or two electrons. These intermolecular orbitals are of an antibonding nature to the C-Hal chemical bonds from organic halides, and their population with electrons leads to their weakening and breaking and to the annihilation of the organic halide compound’s toxicity. Therefore, vitamin B12 can serve as a solution for the disposal of organic halide toxic compounds and a remedy in case of intoxication with these chemicals.
All reactions of the vitamin B12-dependent reactions run in the absence of the total energy barrier, ensuring their long-living nature in the absence of outside influences. The Pseudo-Jahn–Teller Effect, e.g., the orbital mixing, governs all the reactions in the vitamin B12-dependent bioprocesses.
Funding
This research was supported in part by the National Science Foundation through TACC TeraGrid resources provided by the TeraGrid Science Gateways program under supercomputer grants CHE090082 and CHE140071.
Conflicts of Interest
The author declares no conflict of interest.
References
- Carme, R. Chapter 36: Megaloblastic anemias: Disorders of impaired DNA synthesis. In Wintrobe’s Clinical Hematology, 13th ed.; Greer, J.P., Ed.; Wolters Kluwer Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2014; pp. 468–470. [Google Scholar]
- Cohn, E.J.; Minot, G.R.; Alles, G.A.; Salter, W.T. The nature of the material in liver effective in pernicious anemia. J. Biol. Chem. 1938, 77, 325–358. [Google Scholar] [CrossRef]
- Elrod, J.M.; Karnad, A.B. William Bosworth Castle: Pioneer of haematological clinical investigation. Br. J. Haematol. 2003, 121, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Minot, G.R.; Murphy, W.P. Treatment of pernicious anemia by a special diet. J. Am. Med. Assoc. 1926, 87, 470–476. [Google Scholar] [CrossRef]
- Shorb, M.S. Annual Lecture for the Department of Animal & Avian Sciences; Archived from the original on December 12; University of Maryland: College Park, MD, USA, 2012. [Google Scholar]
- Hodgkin, D.C.; Kamper, J.; Mackay, M.; Pickworth, J.; Trueblood, K.N.; White, J.G. Structure of vitamin B12. Nature 1956, 178, 64–66. [Google Scholar] [CrossRef]
- Dodson, G. Dorothy Mary Crowfoot Hodgkin, 12 May 1910–29 July 1994. Biogr. Mem. Fellows R. Soc. 2002, 48, 181–219. [Google Scholar] [CrossRef]
- Khan, A.G.; Eswaran, S.V. Woodward’s synthesis of vitamin B12. Resonance 2003, 8, 8–16. [Google Scholar] [CrossRef]
- Eschenmoser, A.; Wintner, C.E. Natural product synthesis and vitamin B12. Science 1977, 196, 1410–1420. [Google Scholar]
- Drennanenan, C.L.; Huang, S.; Drummond, J.T.; Mathews, R.G.; Ludwig, M.L. How a protein binds B12: A 3.0 Å X-ray structure of B12-binding domains of methionine synthase. Science 1994, 266, 1669–1680. [Google Scholar] [CrossRef]
- Mancia, F.; Keep, N.M.; Nakagawa, A.; Leadlay, P.F.; McSweeney, S.; Rasmussen, B.; Bosecke, P.; Diat, O.; Evans, P.F. How coenzyme B12 radicals are generated: The crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 1996, 4, 339–350. [Google Scholar] [CrossRef]
- Koutmos, M.; Datta, S.; Pattridge, K.A.; Janet, L.S.; Matthews, R.G. Insights into the reactivation of cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2009, 106, 8527–18532. [Google Scholar] [CrossRef]
- Hagemeier, C.H.; Kruer, M.; Rudolf, K.; Thauer, R.K.; Eberhard, W.; Ermler, U. Insight into the mechanism of biological methanol activation based on the crystal structure of the methyl-cobalamin methyltransferase complex. Proc. Natl. Acad. Sci. USA 2006, 103, 18917–18922. [Google Scholar] [CrossRef]
- Reitzer, R.; Gruber, K.; Jogl, G.; Wagner, U.G.; Bothe, H.; Buckel, W.; Kratky, C. Glutamate mutase from clostridium cochlearium: The structure of a coenzyme B12-dependent enzyme provides new mechanistic insights. Structure 1999, 7, 891–902. [Google Scholar] [CrossRef]
- Gruber, K.; Reitzer, R.; Kratky, C. Radical shuttling in a protein: Ribose pseudorotation controls alkyl-radical transfer in the coenzyme B12 dependent enzyme glutamate mutase. Angew. Chem. Int. Ed. 2001, 40, 3377–3380. [Google Scholar] [CrossRef]
- Banerjee, R. Chapter 11: Spectroscopic and Molecular Genetic Characterization of the Two Mammalian Blz-dependent Enzymes. In Vitamin B12 and the B12 Proteins; Kräutler, B., Arigoni, D., Golding, B.T., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp. 189–197. [Google Scholar]
- Yamanishi, M.; Labunska, T.; Banerjee, R. Mirror base-off conformation of coenzyme B12 in human adenosyltransferase and its downstream target, methylmalonyl-CoA mutase. J. Am. Chem. Soc. 2004, 127, 526–527. [Google Scholar] [CrossRef]
- Schrauzer, G.N.; Lee, L.P. The molecular and electronic structure of vitamin B12r, cobaloximes(II), and related compounds. J. Am. Chem. Soc. 1968, 90, 6541–6543. [Google Scholar] [CrossRef]
- Schrauzer, G.N. Recent advances in the chemistry of vitamin B12 and vitamin B12 model compounds: Reductive cobalt-carbon bond cleavage reaction. Pure Appl. Chem. 1973, 33, 545–566. [Google Scholar] [CrossRef]
- Schrauzer, G.N. Cobaloximes give insight into reactions of B12. Chem. Eng. News 1968, 46, 42–44. [Google Scholar]
- Hogenkamp, H.P.C.; Bratt, G.T.; Sun, S. Methyl transfer from methylcobalamin to thiols: A reinvestigation. Biochemistry 1985, 24, 6428–6432. [Google Scholar] [CrossRef]
- Hogenkamp, H.P.C.; Bratt, G.T.; Kotchevar, A.T. Reaction of alkylcobalamins with thiols. Biochemistry 1987, 26, 4723–4727. [Google Scholar] [CrossRef]
- Pratt, J.M. Inorganic Chemistry of Vitamin B12; Academic Press: Cambridge, MA, USA, 1972. [Google Scholar]
- Banerjee, R.V.; Harder, S.R.; Ragsdale, S.W.; Matthews, R.G. Mechanism of reductive activation of cobalamin-dependent methionine synthase: An electron paramagnetic resonance spectroelectrochemical study. Biochemistry 1990, 29, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.V.; Frasca, V.; Ballou, D.P.; Matthews, R.G. Participation of cob(I)alamin in the reaction catalyzed by methionine synthase from escherichia coli: A steady-state and a rapid reaction kinetic analysis. Biochemistry 1990, 29, 11101–11109. [Google Scholar] [CrossRef]
- Gonzales, J.C.; Banerjee, R.V.; Huang, S.; Summer, J.S.; Matthews, R.G. Comparison of cobalamin-independent and cobalamin-dependent methionine synthase from escherichia coli: Two solutions to the same chemical problem. Biochemistry 1992, 31, 6045–6056. [Google Scholar] [CrossRef]
- Banerjee, R.V.; Matthews, R.G. Cobalamin–dependent methionine synthase. Fed. Am. Soc. Exp. Biol. J. 1990, 4, 1450–1459. [Google Scholar] [CrossRef]
- Retey, J. Enzymic reaction selectivity by negative catalysis or how do enzymes deal with highly reactive intermediates. Angew. Chem. Int. Ed. 1990, 29, 355–361. [Google Scholar] [CrossRef]
- Leutbecher, U.; Albracht SP, J.; Buckel, W. Identification of a paramagnetic species as an early intermediate in the coenzyme B12-dependent glutamate mutase reaction: A cob(II)amide. Fed. Eur. Biochem. Soc. Lett. 1992, 307, 144–146. [Google Scholar] [CrossRef]
- Michel, C.; Albracht, S.P.J.; Buckel, W. Adenosylcobalamin and cob(II)alamin as prosthetic groups of 2-methyleneglutarate mutase from clostridium barkeri. Eur. J. Biochem. 1992, 205, 767–773. [Google Scholar] [CrossRef]
- Stubbe, J. Radicals in biological catalysis. Biochemistry 1988, 27, 3893–3900. [Google Scholar] [CrossRef]
- Stubbe, J. Protein radical involvement in biological catalysis. Annu. Rev. Biochem. 1989, 58, 257–285. [Google Scholar] [CrossRef] [PubMed]
- Frey, P.A. Importance of organic radicals in enzymatic cleavage of unactivated C-H bonds. Chem. Rev. 1990, 90, 1343–1357. [Google Scholar] [CrossRef]
- Zhao, Y.; Such, P.; Retey, J. Radical intermediates in the coenzyme B12 dependent methyl-malonyl CoA mutase reaction shown by ESR spectroscopy. Angew. Chem. Int. Ed. 1992, 31, 215–216. [Google Scholar] [CrossRef]
- Stupperich, E. Recent advances in elucidation of biological corrinoid functions. Fed. Eur. Biochem. Soc. Microbiol. Rev. 1993, 12, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.G. Cobalamin-Dependent Methionine Synthase. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley: New York, NY, USA, 1999; pp. 681–706. [Google Scholar]
- Matthews, R.G. Cobalamin-dependent methyltransferases. Acc. Chem. Res. 2001, 34, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Koutmos, M.; Pejchal, R.; Bomer, T.M.; Matthews, R.G.; Smith, J.L.; Ludwig, M.L. Metal active site elasticity linked to activation of homocysteine in methionine synthases. Proc. Natl. Acad. Sci. USA 2005, 105, 3286–3291. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.G.; Koutmos, M.; Datta, S. Cobalamin-dependent and cobamide-dependent methyltransferases. Curr. Opin. Struct. Biol. 2008, 18, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Olteanu, H.; Banerjee, R. Human methionine synthase reductase, a soluble P-450 reductase-like dual-flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J. Biol. Chem. 2001, 276, 35558–35563. [Google Scholar] [CrossRef] [PubMed]
- James, T.; Drummond, J.T.; Sha, H.; Blumenthal, R.M.; Matthews, R.G. Assignment of enzymatic function to specific protein regions of cobalamin-dependent methionine synthase from escherichia coli. Biochemistry 1993, 32, 9290–9295. [Google Scholar]
- Hondorp, E.R.; Matthews, R.G. Oxidation of cysteine 645 of cobalamin-independent methionine synthase causes a methionine limitation in Escherichia coli. J. Bacteriol. 2009, 191, 3407–3410. [Google Scholar] [CrossRef]
- Smith, A.E.; Matthews, R.G. The protonation state of methyltetrahydrofolate in a binary complex with cobalamin-dependent methionine synthase. Biochemistry 2000, 39, 13880–13890. [Google Scholar] [CrossRef]
- Goulding, C.W.; Matthews, R.G. Cobalamin-dependent methionine synthase from escherichia coli: involvement of zinc in homocysteine activation. Biochemistry 1997, 36, 15749–15757. [Google Scholar] [CrossRef]
- Goulding, C.W.; Postigo, D.; Matthews, R.G. Cobalamin-dependent methionine synthase is a modular protein with distinct regions for binding homocysteine, methyltetrahydrofolate, cobalamin, and adenosylmethionine. Biochemistry 1997, 36, 8082–8091. [Google Scholar] [CrossRef]
- Bandarian, V.; Matthews, R.G. Quantitation of rate enhancements attained by the binding of cobalamin to methionine synthase. Biochemistry 2001, 40, 5056–5064. [Google Scholar] [CrossRef] [PubMed]
- Peariso, K.; Goulding, C.W.; Matthews, R.G.; Penner-Hahn, J. The role of zinc in the binding of homocysteine to cobalamin-dependent methionine synthase. J. Am. Chem. Soc. 1998, 120, 8410–8416. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Choi, C.Y.; Matthews, R.G. Changes in protonation associated with substrate binding and cob(I)alamin formation in cobalamin-dependent methionine synthase. Biochemistry 1997, 36, 15739–15748. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.G. Activation of methyltetrahydrofolate by cobalamin-independent methionine synthase. Biochemistry 2006, 45, 5092–5102. [Google Scholar]
- Pratt, J.M. The Roles of Co, Corrin, and Protein. I. Co-Ligand Bonding and the Trans Effect. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 73–112. [Google Scholar]
- Marsh, E.N.G. Review article coenzyme-B12-dependent glutamate mutase. Bioorganic Chem. 2000, 28, 176–189. [Google Scholar] [CrossRef]
- Verroust, P.J.; Christensent, E.I.; Moestrup, S.K.; Hammond, T.G.; Seetharam, B. Chapter 32: The Intrinsic Factor-Cobalamin Receptor Expressed by Yolk Sac and Proximal Tubule Epithelial Cells is the Target of Teratogenic Antibodies. In Vitamin B12 and the B12 Proteins; Kräutler, B., Arigoni, D., Golding, B.T., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp. 491–504. [Google Scholar]
- Babior, B.M.; Carty, T.J.; Abeles, R.H. The mechanism of action of ethanolamine ammonia-lyase, a B12-dependent enzyme: The reversible formation of 5′-deoxyadenosine from adenosylcobalamin during the catalytic process. J. Biol. Chem. 1974, 249, 1689–1695. [Google Scholar] [CrossRef]
- Toraya, T. Radical catalysis of B12 enzymes: Structure, mechanism, inactivation, and reactivation of diol and glycerol dehydratases. Cell. Mol. Life Sci. 2000, 57, 106–127. [Google Scholar] [CrossRef]
- Marsh, E.N.G.; Ballou, D.P. Coupling of cobalt−carbon bond homolysis and hydrogen atom abstraction in adenosylcobalamin-dependent glutamate mutase. Biochemistry 1998, 37, 11864–11872. [Google Scholar] [CrossRef]
- Chowdhury, S.; Banerjee, R. Thermodynamic and kinetic characterization of Co−C bond homolysis catalyzed by coenzyme B12-dependent methyl malonyl-CoA mutase. Biochemistry 2000, 39, 7998–8006. [Google Scholar] [CrossRef]
- Buckel, W.; Golding, B.T. Wiley Encyclopedia of Chemical Biology; Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 1–9. [Google Scholar]
- Banerjee, R.; Ragsdale, S.W. The many faces of vitamin B12: Catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 2003, 72, 209–247. [Google Scholar] [CrossRef]
- Brooks, A.J.; Vlasie, M.; Banerjee, R.; Brunold, T.C. Co−C bond activation in methylmalonyl-CoA mutase by stabilization of the post-homolysis product Co2+cobalamin. J. Am. Chem. Soc. 2005, 127, 16522–16528. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W.; Kratky, C.; Golding, B.T. Stabilization of methylene radicals by cob(II)alamin in coenzyme B12 dependent mutases. Chem. A Eur. J. 2006, 12, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Madhavapeddi, P.; Ballou, D.P.; Marsh, E.N.G. Pre-steady-state kinetic studies on the Glu171Gln active site mutant of adenosylcobalamin-dependent glutamate mutase. Biochemistry 2002, 41, 15803–15809. [Google Scholar] [CrossRef] [PubMed]
- Pallares, I.G.; Moore, T.C.; Escalante-Semerena, J.C.; Brunold, T.C. Spectroscopic studies of the salmonella enterica adenosyltransferase enzyme SeCobA: Molecular-level insight into the mechanism of substrate cob(II)alamin activation. Biochemistry 2014, 53, 7969–7982. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Li, J. Activation parameters for the carbon−cobalt bond homolysis of coenzyme B12 induced by the B12-dependent ribonucleotide reductase from lactobacillus leishmanial. J. Am. Chem. Soc. 1998, 120, 9466–9474. [Google Scholar] [CrossRef]
- Moore, T.C.; Newmister, S.A.; Rayment, I.; Escalante-Semerena, J.C. Structural insights into the mechanism of four-coordinate cob(II)alamin formation in the active site of the salmonella enterica ATP: Co(I)crinoid adenosyltransferase enzyme: Critical role of residues Phe91 and Trp93. Biochemistry 2012, 51, 9647–9657. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.N.G.; Meléndez, G.D.R. Adenosylcobalamin enzymes: Theory and experiment begin to converge. Biochim. Biophys. Acta 2012, 1824, 1154–1164. [Google Scholar] [CrossRef]
- Padmakumar, R.; Banerjee, R. Evidence that cobalt−carbon bond homolysis is coupled to hydrogen atom abstraction from substrate in methylmalonyl-CoA mutase. Biochemistry 1997, 36, 3713–3718. [Google Scholar] [CrossRef]
- Licht, S.S.; Booker, S.; Stubbe, J. Studies on the catalysis of carbon−cobalt bond homolysis by ribonucleoside triphosphate reductase: Evidence for concerted carbon−cobalt bond homolysis and thiyl radical formation. Biochemistry 1999, 38, 1221–1233. [Google Scholar] [CrossRef]
- Meier, T.W.; Thomä, N.H.; Evans, P.R.; Leadlay, P.F. Tritium isotope effects in adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry 1996, 35, 11791–11796. [Google Scholar] [CrossRef]
- Chih, H.W.; Marsh, E.N.G. Pre-steady-state kinetic investigation of intermediates in the reaction catalyzed by adenosylcobalamin-dependent glutamate mutase. Biochemistry 1999, 38, 13684–13691. [Google Scholar] [CrossRef] [PubMed]
- Bandarian, V.; Reed, G.H. Isotope effects in the transient phases of the reaction catalyzed by ethanolamine ammonia-lyase: Determination of the number of exchangeable hydrogens in the enzyme−cofactor complex. Biochemistry 2000, 39, 12069–12075. [Google Scholar] [CrossRef] [PubMed]
- Fink, R.G. Chapter 25: Coenzyme B12-Based Chemical Precedent for Co-C Bond Homolysis and Other Key Elementary Steps. In Vitamin B12 and the B12 Proteins; Kräutler, B., Arigoni, D., Golding, B.T., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp. 383–402. [Google Scholar]
- Banerjee, R. The Yin-yang of cobalamin biochemistry. Chem. Biol. 1997, 4, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.; Song, H.; Håkansson, K.; Neil, E.; Marsh, G. Hydrogen tunneling in adenosylcobalamin-dependent glutamate mutase: Evidence from intrinsic kinetic isotope effects measured by intra-molecular competition. Biochemistry 2010, 49, 3168–3173. [Google Scholar] [CrossRef] [PubMed]
- Hayward, G.C.; Hill, H.A.O.; Pratt, J.M.; Vanston, N.J.; Williams, R.J.P. The chemistry of vitamin B12: Part IV. the thermodynamic trans-effect. J. Chem. Soc. 1965, 6485–6493. [Google Scholar] [CrossRef]
- Grate, J.H.; Schrauzer, G.N. Chemistry of cobalamins and related compounds. 48. sterically induced, spontaneous dealkylation of secondary alkylcobalamins due to axial base coordination and conformational changes of the corrin ligand. J. Am. Chem. Soc. 1979, 101, 4601–4611. [Google Scholar] [CrossRef]
- Halpern, J. Mechanisms of coenzyme B12-dependent rearrangements. Science 1985, 227, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.N.G.; Drennan, C.L. Adenosylcobalamin-dependent isomerases: New insights into structure and mechanism. Curr. Opin. Chem. Biol. 2001, 5, 499–505. [Google Scholar] [CrossRef]
- Brown, K.L.; Zou, X.J. Thermolysis of coenzymes B12 at physiological temperatures: Activation parameters for cobalt-carbon bond homolysis and quantitative analysis of the perturbation of the homolysis equilibrium by the ribonucleoside triphosphate reductase from lactobacillus leishmania. Inorg. Biochem. 1999, 77, 185–195. [Google Scholar]
- Dong, S.L.; Padmakumar, R.; Banerjee, R.; Spiro, T.G. Co−C bond activation in B12-dependent enzymes: Cryogenic resonance raman studies of methylmalonyl-coenzyme A mutase. J. Am. Chem. Soc. 1999, 121, 7063–7070. [Google Scholar] [CrossRef]
- Bresciana-Pahor, N.; Forcolin, M.; Marzilli, L.G.; Randaccio, L.; Summers, M.F.; Toscano, P.J. Organocobalt B12 models: Axial ligand effects on the structural and coordination chemistry of cobaloximes. Coord. Chem. Rev. 1985, 63, 1–125. [Google Scholar] [CrossRef]
- Mancia, F.; Evans, P.R. Conformational changes on substrate binding to methyl malonyl CoA mutase and new insights into the free radical mechanism. Structure 1998, 6, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Vlasie, M.D.; Banerjee, R. Tyrosine 89 accelerates Co−C bond homolysis in methylmalonyl-CoA mutase. J. Am. Chem. Soc. 2003, 125, 5431–5435. [Google Scholar] [CrossRef] [PubMed]
- Thomä, N.H.; Meier, T.W.; Evans, P.R.; Leadlay, P.F. Stabilization of radical intermediates by an active-site tyrosine residue in methylmalonyl-CoA mutase. Biochemistry 1998, 37, 14386–14393. [Google Scholar] [CrossRef] [PubMed]
- Toraya, T.; Ishida, A. Acceleration of cleavage of the carbon-cobalt bond of sterically hindered alkylcobalamins by binding to apoprotein of diol dehydrase. Biochemistry 1988, 27, 7677–7682. [Google Scholar] [CrossRef]
- Zhu, L.; Kostic, N.M. Molecular orbital study of coenzyme B12: Activation of the cobalt-carbon bond by angular distortions. Inorg. Chem. 1987, 26, 4194–4197. [Google Scholar] [CrossRef]
- Stuart, S.L.; Lawrence, C.C.; Stubbe, J. Thermodynamic and kinetic studies on carbon−cobalt bond homolysis by ribonucleoside triphosphate reductase: the importance of entropy in catalysis. Biochemistry 1999, 38, 1234–1242. [Google Scholar]
- Vrijbloed, J.W.; Zerbe-Burkhardt, K.; Ratnatilleke, A.; Grubelnik-Leiser, A.; Robinson, J.A. Insertional inactivation of methylmalonyl coenzyme A (CoA) mutase and isobutyryl-CoA mutase genes in streptomyces cinnamonensis: Influence on polyketide antibiotic biosynthesis. J. Bacteriol. 1999, 181, 5600–5605. [Google Scholar] [CrossRef]
- Lexa, D.; Savéant, J.M. Electrochemistry of vitamin B12: Part 3: One-electron intermediates in the reduction of methylcobalamin and methylcobinamide. J. Am. Chem. Soc. 1978, 100, 3220–3222. [Google Scholar] [CrossRef]
- Spataru, T.; Birke, R.L. The effect of solvent on the electrode process of methylcobalamin as studied by cyclic voltammetry. J. Electroanal. Chem. 2006, 593, 74–86. [Google Scholar] [CrossRef]
- Birke, R.L.; Huang, Q.; Spataru, T.; Gosser Jr, D.K. Electroreduction of an alkylcobalamin: Mechanism of stepwise reductive cleavage of the Co-C bond. J. Am. Chem. Soc. 2006, 128, 1922–1936. [Google Scholar] [CrossRef] [PubMed]
- Scheffold, R.; Abrecht, S.; Ruf, H.R.; Stamouli, P.; Tinembart, O.; Walder, L.; Weymuth, C. Vitamin B12-mediated electrochemical reactions in the synthesis of natural products. Pure Appl. Chem. 1987, 59, 363–372. [Google Scholar] [CrossRef]
- Ogoshi, H.; Kikuchi, Y.; Yamaguchi, T.; Toi, H.; Aoyama, Y. Asymmetric induction in the nucleophilic cyclopropane ring cleavage reaction with vitamin B12s. Organometallics 1987, 6, 2175–2178. [Google Scholar] [CrossRef]
- Pattenden, G. Simonsen lecture. cobalt-mediated radical reactions in organic synthesis. Chem. Soc. Rev. 1988, 17, 361–382. [Google Scholar] [CrossRef]
- Baldwin, D.A.; Betterton, E.A.; Chemaly, S.M.; Pratt, J.M.J. The chemistry of vitamin B12.: Part 25: Mechanism of the β-elimination of olefins from alkylcorrinoids; evidence for initial homolytic fission of the Co–C bond. J. Chem. Soc. Dalton Trans. 1985, 8, 1613–1618. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Adlington, R.M.; Kang, T.W. Direct ring expansion of penicillins to 3-exomethylene cephalosporins. Tetrahedron Lett. 1991, 48, 7093–7096. [Google Scholar] [CrossRef]
- Paquette, L. (Ed.) Encyclopedia of Reagents for Organic Synthesis; Wiley: New York, NY, USA, 1995; pp. 5511–5514. [Google Scholar]
- Lee, E.R.; Lakomy, I.; Bigler, P.; Scheffold, R. Reductive radical cyclisations of bromo acetals and (bromomethyl) silyl ethers of terpenoid alcohols. Helv. Chim. Acta 1991, 74, 146–162. [Google Scholar] [CrossRef]
- Shey, J.; McGinley, C.M.; McCauley, K.M.; Dearth, A.S.; Young, B.T.; van der Donk, W.A. Mechanistic investigation of a novel vitamin B12-catalyzed carbon−carbon bond forming reaction, the reductive dimerization of arylalkenes. J. Org. Chem. 2002, 67, 837–846. [Google Scholar] [CrossRef]
- McGinley, C.M.; Relyea, H.A.; van der Donk, W.A. Vitamin B12 catalyzed radical cyclizations of arylalkenes. Synlett 2006, 2, 211–214. [Google Scholar] [CrossRef]
- Kumar, M.; Kozlowski, P.M. Electronic and structural properties of cob(I)alamin: Ramifications for B12-dependent processes. Coord. Chem. Rev. 2017, 333, 71–81. [Google Scholar] [CrossRef]
- Andruniow, T.; Zagierski, M.Z.; Kozlowski, P.M. Density functional theory analysis of stereoelectronic properties of cobalamins. J. Phys. Chem. B 2000, 104, 10921–10927. [Google Scholar] [CrossRef]
- Andruniow, T.; Zagierski, M.Z.; Kozlowski, P.M. Theoretical determination of the Co-C bond energy dissociation in cobalamins. J. Am. Chem. Soc. 2001, 123, 2679–2680. [Google Scholar] [CrossRef]
- Kumar, M.; Kozlowski, P.M. The cobalt-methyl bond dissociation: New benchmark analysis based on density functional theory and completely renormalized coupled-cluster calculations. J. Chem. Theory Comput. 2012, 8, 1870–1894. [Google Scholar]
- Dolker, N.; Maseras, F.; Liedos, A. A density functional study on the effect of the trans axial ligand of cobalamin on the homolytic cleavage of the Co−C bond. J. Phys. Chem. B 2001, 105, 7564–7571. [Google Scholar] [CrossRef]
- Jensen, P.; Ryde, U. The axial N-base has minor influence on Co-C bond cleavage in cobalamins. J. Mol. Struct. 2002, 585, 239–255. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Zagierski, M.Z. Electronic and steric influence of trans axial base on the stereoelectronic properties of cobalamins. J. Phys. Chem. B 2004, 108, 14163–14170. [Google Scholar] [CrossRef]
- Andruniow, T.; Zagierski, M.Z.; Kozlowski, P.M. DFT–SQM force field for cobalt corrinoids. Chem. Phys. Lett. 2000, 331, 502–518. [Google Scholar] [CrossRef]
- Andruniow, T.; Zagierski, M.Z.; Kozlowski, P.M. Vibrational analysis of methylcobalamin. J. Phys. Chem. A 2002, 106, 1365–1373. [Google Scholar] [CrossRef]
- Andruniow, T.; Kozlowski, P.M.; Zagierski, M.Z. Theoretical analysis of electronic absorption spectra of vitamin B12 models. J. Chem. Phys. 2001, 115, 7522–7533. [Google Scholar] [CrossRef]
- Kozlowski, P.M. Quantum chemical modeling of Co-C bond activation in B12-dependent enzymes. Curr. Opin. Chem. Biol. 2001, 5, 736–743. [Google Scholar] [CrossRef]
- Mealli, C.; Sabat, M.; Marzilli, L. Coenzyme B12 cobalt-carbon bond homolysis: Insights from qualitative molecular orbital theory. J. Am. Chem. Soc. 1987, 109, 1593–1594. [Google Scholar] [CrossRef]
- Andruniow, T.; Kuta, J.; Zgierski, M.Z.; Kozlowski, P.M. Molecular orbital analysis of anomalous trans effect in cobalamins. Chem. Phys. Lett. 2005, 410, 410–416. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Kamachi, T. Reductive elimination pathway for homocysteine to methionine conversion in cobalamin-dependent methionine synthase. J. Biol. Inorg. Chem. 2012, 17, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-L.; Blomberg MR, A.; Siegbahn, P.E.M. How Is a Co-methyl intermediate formed in the reaction of cobalamin-dependent methionine synthase? theoretical evidence for a two-step methyl cation transfer mechanism. J. Phys. Chem. B 2011, 115, 4066–4077. [Google Scholar] [CrossRef]
- Reig, A.J.; Conrad, K.S.; Brunold, T.S. Combined spectroscopic/computational studies of vitamin B12 precursors: Geometric and electronic structures of cobinamides. Inorg. Chem. 2012, 51, 2867–2879. [Google Scholar] [CrossRef] [PubMed]
- Liptak, M.D.; Fleischhacker, A.S.; Matthews, R.G.; Teiser, J.; Brunold, T.C. Spectroscopic and computational characterization of the base-off forms of cob(II)alamin. J. Phys. Chem. B 2009, 113, 5245–5254. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Brunold, T. Combined spectroscopic and computational analysis of the vibrational properties of vitamin B12 in its Co3+, Co2+, and Co1+. J. Phys. Chem. B 2013, 117, 5397–5410. [Google Scholar] [CrossRef]
- Govender, P.P.; Navizet, I.; Perry, C.B.; Marcus, H.M. DFT studies of trans and cis influences in the homolysis of the Co-C bond in models of the alkylcobalamins. J. Phys. Chem. A 2013, 117, 3057–3068. [Google Scholar] [CrossRef]
- Kornobis HS, K.; Ruud, K.; Kozlowski, P.M. Electronically excited states of vitamin B12 and methylcobalamin: Theoretical analysis of absorption, CD, and MCD data. J. Phys. Chem. B 2011, 115, 737–748. [Google Scholar]
- Ghosh, A.P.; Al Mamun, A.; Kozlowski, P.M. Light-induced activation of organo-metallic Co-C bond in MeCbl-dependent methionine synthase- QM/MM study. Biophys. J. 2019, 116, 67–68. [Google Scholar] [CrossRef]
- Ghosha, A.P.; AlMamuna, A.; Lodowski, P.; Jaworska, M.; Kozlowski, P.M. Mechanism of the photo-induced activation of Co-C bond in methylcobalamin-dependent methionine synthase. J. Photochem. Photobiol. B Biol. 2018, 189, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.P.; Al Mamun, A.; Kozlowski, P.M. How does the mutation in the cap domain of methylcobalamin-dependent methionine synthase influence the photoactivation of the Co–C bond? Phys. Chem. Chem. Phys. 2019, 21, 20628–20640. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Jaworska, M.; Lodowski, P.; Kumar, M.; Kozlowski, P. Electronic structure of cofactor-substrate reactant complex involved in the methyl transfer reaction catalyzed by cobalamin-dependent methionine synthase. J. Phys. Chem. B 2011, 115, 6722–6731. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Alfonso-Prieto, M.; Rovira, C.; Lodowski, P.; Jaworska, M.; Kozlowski, P.M. Role of the axial base in the modulation of the cob(I)alamin electronic properties: Insight from QM/MM, DFT, and CASSCF calculations. J. Chem. Theory Comput. 2011, 7, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kozlowski, P.M. Mechanistic insights for formation of an organometallic Co–C bond in the methyl transfer reaction catalyzed by methionine synthase. J. Phys. Chem. B 2013, 117, 16044–16057. [Google Scholar] [CrossRef] [PubMed]
- Spataru, T.; Birke, R.L. Carbon-cobalt bond distance and bond cleavage in one-electron reduced methylcobalamin: A failure of the conventional DFT method. J. Phys. Chem. A 2006, 110, 8599–8604. [Google Scholar] [CrossRef] [PubMed]
- Spataru, T.; Fernandez, F. The nature of the Co-C bond cleavage processes in the methylcob(II)alamin and adenosylcob(III)alamin. Chem. J. Mold. 2016, 11, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.P.; Ryde, U. Conversion of homocysteine to methionine by methionine synthase: A density functional study. J. Am. Chem. Soc. 2003, 125, 13970–13971. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Biarnés, X.; Kumar, M.; Rovira, C.; Kozlowski, P.M. Reductive cleavage mechanism of Co−C bond in cobalamin-dependent methionine synthase. J. Phys. Chem. B 2010, 114, 12965–12971. [Google Scholar] [CrossRef]
- Brooks, A.J.; Vlasie, M.; Banerjee, R.; Brunold, T.C. Spectroscopic and computational studies on the adenosylcobalamin-dependent methylmalonyl-CoA mutase: Evaluation of enzymatic contributions to Co-C bond activation in the Co3+ ground state. J. Am. Chem. Soc. 2004, 126, 8167–8180. [Google Scholar] [CrossRef]
- Abdel-Azeim, S.; Li, X.; Chung, L.W.; Morokuma, K. Zinc–Homocysteine binding in cobalamin-dependent methionine synthase and its role in the substrate activation: DFT, ONIOM, and QM/MM molecular dynamics studies. Comput. Chem. 2011, 32, 3154–3167. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.P.; Ryde, U. How the Co-C bond is cleaved in coenzyme B12 enzymes: A theoretical study. J. Am. Chem. Soc. 2005, 127, 9117–9128. [Google Scholar] [CrossRef] [PubMed]
- Kwiecien, R.A.; Khavrutskii, I.V.; Musaev, D.G.; Morokuma, K.; Banerjee, R.; Paneth, P. Computational insights into the mechanism of radical generation in B12-dependent methylmalonyl-CoA mutase. J. Am. Chem. Soc. 2006, 128, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chung, L.W.; Paneth, P.; Morokuma, K. DFT and ONIOM (DFT:MM) studies on Co−C bond cleavage and hydrogen transfer in B12-dependent methylmalonyl-CoA mutase. stepwise or concerted mechanism? J. Am. Chem. Soc. 2009, 131, 5115–5125. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Bucher, D.; Kozlowski, P.M. Mechanistic implications of reductive Co−C bond cleavage in B12-dependent methylmalonyl CoA mutase. J. Phys. Chem. B 2019, 123, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Liu, S.; Kozlowski, P.M. Charge separation propensity of the coenzyme B12–tyrosine complex in adenosylcobalamin-dependent methylmalonyl–CoA mutase enzyme. J. Phys. Chem. Lett. 2012, 3, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kozlowski, P.M. Role of tyrosine residue in the activation of Co−C bond in coenzyme B12-dependent enzymes: Another case of proton-coupled electron transfer? J. Phys. Chem. B 2009, 113, 9050–9054. [Google Scholar] [CrossRef]
- Pawel, M.K.; Kamachi, T.; Kumar, M.; Yoshizawa, K. Initial step of B12-dependent enzymatic catalysis: Energetic implications regarding involvement of the one-electron-reduced form of adenosylcobalamin. J. Biol. Inorg. Chem. 2012, 17, 293–300. [Google Scholar]
- Conrad, K.S.; Jordan, C.D.; Brown, K.L.; Brunold, T.C. Spectroscopic and computational studies of cobalamin species with variable lower axial ligation: Implications for the mechanism of Co–C bond activation by class I cobalamin-dependent isomerases. Inorg. Chem. 2015, 54, 3736–3747. [Google Scholar] [CrossRef]
- Brooks, A.J.; Fox, C.C.; Marsh, E.N.G.; Vlasie, M.; Banerjee, R.; Brunold, T.C. Electronic structure studies of the adenosylcobalamin cofactor in glutamate mutase. Biochemistry 2005, 44, 15167–15181. [Google Scholar] [CrossRef]
- Spataru, T. The complete electronic structure and mechanism of the methionine synthase process as determined by the MCSCF method. J. Organomet. Chem. 2021, 942, 181211–181221. [Google Scholar] [CrossRef]
- Spataru, T. The electronic structure and mechanism of the adenosylcobalamin-dependent bio-processes as determined by the MCSCF method. J. Med. Chem. 2021, 11, 595–606. [Google Scholar]
- Spataru, T. The First Step and the Cob(II)alamin Cofactor Inactive Particles Reactivation in the Updated Mechanism of the Methionine Synthase Process. Reactions 2023, 4, 274–285. [Google Scholar] [CrossRef]
- Spataru, T. The Co-N bond cleavage in the adenosylcobalamin cofactor in advance to Glutamete Mutase and Methylmalonyl-Co-A Mutase processes. Chem. J. Mold. 2023, 18, 96–104. [Google Scholar] [CrossRef]
- Li, Y.N.; Gulati, S.; Baker, P.J.; Brody, L.C.; Banerjee, R.; Kruger, W.D. Cloning, mapping and RNA analysis of the human methionine synthase gene. Hum. Mol. Genet. 1996, 5, 1851–1858. [Google Scholar] [CrossRef]
- Marmion, C.J. Interrelations Between Essential Metal Ions and Human Diseases. In Metal Ions in Life Sciences; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; Springer: Dordrecht, The Netherland, 2013; Volume 13, pp. 295–320. [Google Scholar]
- Wald, I.; Członkowska, A.; Dowżenko, A. Clinical Neurology; National Institute of Medical Publications: Warsaw, Poland, 1987; p. 451. (in Polish) [Google Scholar]
- Morris, M.C.; Evans, D.A.; Schneider, J.A.; Tangney, C.C.; Bienias, J.L.; Aggarwal, N.T. Dietary folate and vitamins B12 and B6 not associated with incident alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 435–443. [Google Scholar] [CrossRef]
- Siuda, J.; Gorzkowska, A.; Patalong-Ogiewa, M.; Krzystanek, E.; Czech, E.; Wiechuła, B.; Garczorz, W.; Danch, A.; Jasińska-Myga, B.; Opala, G. From mild cognitive impairment to alzheimer’s disease—Influence of homocysteine, vitamin B12 and folate on cognition over time: Results from one-year follow-up. Pol. J. Neurol. Neurosurg. 2009, 43, 321–329. (In Polish) [Google Scholar]
- Kivipelto, M.; Annerbo, S.; Hultdin, J.; Bäckman, L.; Viitanen, M.; Fratiglioni, L.; Lökk, J. Homocysteine and holo-transcobalamin and the risk of dementia and alzheimer’s disease: A prospective study. Eur. J. Neurol. 2009, 16, 808–813. [Google Scholar] [CrossRef]
- Kageyama, M.; Hiraoka, M.; Kagawa, Y. Relationship between genetic polymorphism, serum folate and homocysteine in Alzheimer’s disease. Asia-Pac. J. Public Health 2008, 20, 111–117. [Google Scholar]
- Prodan, C.I.; Cowan, L.D.; Stoner, J.A.; Ross, E.D. Cumulative incidence of vitamin B12 deficiency in patients with Alzheimer’s disease. J. Neurol. Sci. 2009, 284, 144–148. [Google Scholar] [CrossRef]
- Neil, E.; Marsh, G.; Patterson, D.P.; Lei, L. Adenosyl radical: Reagent and catalyst in enzyme reactions. ChemBioChem 2010, 11, 604–621. [Google Scholar]
- Martin, B.D.; Finke, R.G. Methylcobalamin’s full- vs. half-strength cobalt-carbon sigma bonds and bond dissociation enthalpies: A>1015 Co-CH3 homolysis rate enhancement following one-antibonding-electron reduction of methylcobalamin. J. Am. Chem. Soc. 1992, 114, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, H.; Vogler, A. Photolysis of methylcobalamin. nature of the reactive excited state. J. Organomet. Chem. 1993, 453, 269–272. [Google Scholar] [CrossRef]
- Walker LA, I.I.; Jarrett, J.T.; Anderson, N.A.; Pullen, S.H.; Matthews, R.G.; Sension, R.J. Time-resolved spectroscopic studies of B12 coenzymes: The identification of a metastable cob(III)alamin photoproduct in the photolysis of methylcobalamin. J. Am. Chem. Soc. 1998, 120, 3597–3603. [Google Scholar] [CrossRef]
- Luo, L.B.; Chen, H.L.; Fu, S.W.; Zhang, S.Y. Laser-induced photoacoustic calorimetric determination of enthalpy and volume changes in the photolysis of 5′-deoxyadenosylcobalamin and methylcobalamin. J. Chem. Soc. Dalton Trans. 1998, 12, 2103–2107. [Google Scholar] [CrossRef]
- Shiang, J.J.; Walker, L.A.; Anderson, N.A.; Cole, A.G.; Sension, R.J. The time-resolved spectroscopic studies of B12 coenzymes: The photolysis of methylcobalamin is wavelength dependent. J. Phys. Chem. B 1999, 103, 10532–10539. [Google Scholar] [CrossRef]
- Cole, A.G.; Yoder, L.M.; Shiang, J.J.; Andeson, N.A.; Walker, L.A.; Banaszak Holl, M.M.; Sension, R.J. Time-resolved spectroscopic studies of B12 coenzymes: A comparison of the primary photolysis mechanism in methyl-, ethyl-, n-propyl-, and 5′-deoxyadenosylcobalamin. J. Am. Chem. Soc. 2002, 124, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Orio, M.; Pantazis, A.D.; Neese, F. Density functional theory. Photosynth. Res. 2009, 102, 443–453. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymatic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Kozlowski, P.M.; Kuta, J.; Galezowski, W. Reductive cleavage mechanism of methylcobalamin: Elementary steps of Co-C bond breaking. J. Phys. Chem. B 2007, 111, 7638–7645. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, I.B. Limitations of density functional theory in application to degenerate states. J. Comput. Chem. 1997, 2, 260–267. [Google Scholar] [CrossRef]
- Öpik, U.; Pryce, M.H.L. Studies of the jahn-teller effect. I. a survey of the static problem. Proc. R. Soc. London. Ser. A Math. Phys. Sci. R. Soc. 1957, 238, 425–447. [Google Scholar]
- Bader, R.F.W. An interpretation of potential interaction constants in terms of low-lying excited states. Mol. Phys. 1960, 3, 137–151. [Google Scholar] [CrossRef]
- Fulton, R.L.; Gouterman, M. Vibronic coupling. I. mathematical treatment for two electronic states. J. Chem. Phys. 1961, 35, 1059–1071. [Google Scholar] [CrossRef]
- Bersuker, I.B. On the origin of ferroelectricity in perovskite-type crystals. Phys. Lett. 1966, 20, 589–590. [Google Scholar] [CrossRef]
- Bersuker, I.B.; Stavrov, S.S. Structure and properties of metalloporphyrins and hemoproteins: The vibronic approach. Coord. Chem. Rev. 1988, 88, 1–68. [Google Scholar] [CrossRef]
- Bersuker, I.B.; Polinger, V.Z. Vibronic Interactions in Molecules and Crystals; Springer Series in Chemical Physics; Springer: Berlin/Heidelberg, Germany, 1989; p. 49. [Google Scholar]
- Bersuker Issak, B. The Jahn–Teller Effect; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Bearpark, M.J.; Blancafort, L.; Robb, M.A. The pseudo-jahn—Teller effect: A CASSCF diagnostic. Mol. Phys. 2002, 100, 1735–1739. [Google Scholar] [CrossRef]
- Ouyang, L.; Rulis, P.; Ching, W.Y.; Nardin, G.; Randaccio, L. Accurate redetermination of the X-ray structure and electronic bonding in adenosylcobalamin. Inorg. Chem. 2004, 43, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Gerzberg, G.; Monfils, A. The dissociation energies of the H2, HD, and D2 molecules. J. Mol. Spectrosc. 1961, 5, 482–498. [Google Scholar] [CrossRef]
- Randaccio, L.; Furlan, M.; Geremia, S.; Šlouf, M.; Srnova, I.; Toffoli, D. Similarities and differences between cobalamins and cobaloximes. accurate structural determination of methylcobalamin and LiCl- and KCl-containing cyanocobalamins by synchrotron radiation. Inorg. Chem. 2000, 39, 3403–3413. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 enzymes: Their structure, reactivity, and selectivity modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.R.; Spataru, T.; Camaioni, D.M.; Lee, S.-J.; Li, G.; Choi, J.; Franz, J.A. Kinetics and mechanism of the hydrogenation of CpCr(CO)3•/[CpCr(CO)3]2 equilibrium to CpCr(CO)3H. Organometallics 2014, 33, 2496–2502. [Google Scholar] [CrossRef]
- Han, A.; Spataru, T.; Hartung, J.; Li, G.; Norton, J.R. Effect of double-bond substituents on the rate of cyclization of α-carbomethoxyhex-5-enyl, radicals. J. Org. Chem. 2014, 79, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A.; Pklinman, J. Hydrogen tunneling in biology. Chem. Biol. 1999, 6, R191–R198. [Google Scholar] [CrossRef] [PubMed]
- Lerner, T. Encyclopedia of Physics, 2nd ed.; VCH: New York, NY, USA, 1991; pp. 1308–1408. [Google Scholar]
- Gorinchoy, N.; Balan, I.; Polinger, V.; Bersuker, I. Pseudo jahn-teller origin of the proton-transfer energy barrier in the hydrogen-bonded [FHF]-system. Chem. J. Moldova. Gen. Ind. Ecol. Chem. 2021, 16, 115–120. [Google Scholar] [CrossRef]
- Shibata, N.; Tamagaki, H.; Hieda, N.; Akita, K.; Komori, H.; Shomura, Y.; Terawaki, S.; Mori, K.; Noritake Yasuoka, N.; Higuchi, Y.; et al. Crystal structures of ethanolamine ammonia-lyase complexed with coenzyme B12 analogs and substrates. J. Biol. Chem. 2010, 285, 26484–26493. [Google Scholar] [CrossRef]
- Dobrzyńska, E.; Pośniak, M.; Szewczyńska, M.; Buszewski, B. Chlorinated Volatile Organic Compounds Old, However, Actual Analytical and Toxicological Problem. Crit. Rev. Anal. Chem. 2010, 40, 41–57. [Google Scholar] [CrossRef]
- Lee, M.; Wells, E.; Wong, Y.K.; Koenig, J.; Adrian, L.; Richnow, H.H.; Manefield, M. Relative Contributions of Dehalobacter and Zerovalent Iron in the Degradation of Chlorinated Methanes. Environ. Sci. Technol. 2015, 49, 4481–4489. [Google Scholar] [CrossRef]
- Torrentó, C.; Palau, J.; Rodríguez-Fernández, D.; Heckel, B.; Meyer, A.; Domènech, C.; Rosell, M.; Soler, A.; Elsner, M.; Hunkeler, D. Carbon and Chlorine Isotope Fractionation Patterns Associated with Different Engineered Chloroform Transformation Reactions. Environ. Sci. Technol. 2017, 51, 6174–6184. [Google Scholar] [CrossRef]
- Palau, J.; Shouakar-Stash, O.; Hatijah Mortan, S.; Yu, R.; Rosell, M.; Marco-Urrea, E.; Freedman, D.L.; Aravena, R.; Soler, A.; Hunkeler, D. Hydrogen Isotope Fractionation during the Biodegradation of 1,2-Dichloroethane: Potential for Pathway Identification Using a Multielement (C, Cl, and H) Isotope Approach. Environ. Sci. Technol. 2017, 51, 10526–10535. [Google Scholar] [CrossRef]
- Carter, J.M.; Lapham, W.W.; Zogorski, J.S. Occurrence of Volatile Organic Compounds in Aquifers of the United States1. J. Am. Water Resour. Assoc. 2008, 44, 399–416. [Google Scholar] [CrossRef]
- Thakur, M.; Pathania, D. Chapter 12—Environmental fate of organic pollutants and effect on human health. In Abatement of Environmental Pollutants Trends and Strategies; Elsveier: Amsterdam, The Netherlands, 2020; pp. 245–262. [Google Scholar]
- Lacrampe-Couloume, G.; Edwards, E.A.; Sherwood Lollar, B. Large Carbon Isotope Fractionation during Biodegradation of Chloroform by Dehalobacter Cultures. Environ. Sci. Technol. 2012, 46, 10154–10160. [Google Scholar]
- Palau, J.; Shouakar-Stash, O.; Hunkeler, D. Carbon and Chlorine Isotope Analysis to Identify Abiotic Degradation Pathways of 1,1,1-Trichloroethane. Environ. Sci. Technol. 2014, 48, 14400–14408. [Google Scholar] [CrossRef]
- Hartenbach, A.; Hofstetter, T.B.; Berg, M.; Bolotin, J.; Schwarzenbach, R.P. Using Nitrogen Isotope Fractionation To Assess Abiotic Reduction of Nitroaromatic Compounds. Environ. Sci. Technol. 2006, 40, 7710–7716. [Google Scholar] [CrossRef]
- Nijenhuis, I.; Richnow, H.H. Stable isotope fractionation concepts for characterizing biotransformation of organohalides. Curr.Opin. Biotechnol. 2016, 41, 108–113. [Google Scholar] [CrossRef]
- Chen, G.; Shouakar-Stash, O.; Phillips, E.; Justicia-Leon, S.D.; Gilevska, T.; Sherwood Lollar, B.; Mack, E.E.; Seger, E.S.; Löffler, F.E. Dual Carbon-Chlorine Isotope Analysis Indicates Distinct Anaerobic Dichloromethane Degradation Pathways in Two Members of Peptococcaceae. Environ. Sci. Technol. 2018, 52, 8607–8616. [Google Scholar] [CrossRef]
- Zwank, L.; Elsner, M.; Aeberhard, A.; Schwarzenbach, R.P.; Haderlein, S.B. Carbon isotope fractionation in the reductive dehalogenation of carbon tetrachloride at iron (hydro)oxide and iron sulfide minerals. Environ. Sci. Technol. 2005, 39, 5634–5641. [Google Scholar] [CrossRef]
- Rodríguez-Fernández, D.; Torrentó, C.; Guivernau, M.; Viñas, M.; Hunkeler, D.; Soler, A.; Domènech, C.; Rosell, M. Vitamin B12 effects on chlorinated methanes-degrading microcosms: Dual isotope and metabolically active microbial populations assessment. Sci. Total Environ. 2018, 621, 1615–1625. [Google Scholar] [CrossRef]
- Spataru, T.; Fernandez, F.; Sista, J.; Spataru, P.; Povar, I. Disposal of Poisonous Organic Halides by Using the Electrochemical Method: DFT Simulation. Chem. J. Mold. 2016, 11, 93–98. [Google Scholar] [CrossRef]
- Lapeyrouse, N.; Liu, M.; Zou, S.; Booth, G.; Yestrebsky, C.L. Remediation of Chlorinated Alkanes by Vitamin B12 and Zero-Valent Iron. J. Chem. 2019, 2019, 7565464. [Google Scholar] [CrossRef]
- Heckel, B.; Elsner, M. Exploring Mechanisms of Biotic Chlorinated Alkane Reduction: Evidence of Nucleophilic Substitution (SN2) with Vitamin B12. Environ. Sci. Technol. 2022, 56, 6325–6336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lorenz, A.; Schüürmann, G. Interaction Mode and Regioselectivity in Vitamin B12-Dependent Dehalogenation of Aryl Halides by Dehalococcoides mccartyi Strain CBDB1. Environ. Sci. Technol. 2018, 52, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Heckel, B.; McNeill, K.; Elsner, M. Chlorinated Ethene Reactivity with Vitamin B12 Is Governed by Cobalamin Chloroethylcarbanions as Crossroads of Competing Pathways. ACS Catal. 2018, 8, 3054–3066. [Google Scholar] [CrossRef]
- Payne, K.P.A.P.; Quezada, C.P.; Fisher, K.; Dunstan, M.S.; Collins, F.A.; Sjuts, H. Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature 2015, 517, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Löffler, F.E.; Ritalahti, K.M.; Zinder, S.H. Dehalococcoides and reductive dechlorination of chlorinated solvents. In Bioaugmentation for Groundwater Remediation; Stroo, H.F., Leeson, A., Ward, C.H., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 39–88. [Google Scholar]
- Hendrickson, E.R.; Payne, J.A.; Young, R.M.; Starr, M.G.; Perry, M.P.; Fahnestock, S.; Ellis, D.E.; Ebersole, R.C. Molecular analysis of Dehalococcoides 16S ribosomal DNA from chloroethene-contaminated sites throughout North America and Europe. Appl. Environ. Microbiol. 2002, 68, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.A.; Rosner, B.M.; Von Abendroth, G.; Meshulam-Simon, G.; McCarty, P.L.; Spormann, A.M. Molecular identification of the catabolic vinyl chloride reductase from Dehalococcoides sp. strain VSand its environmental distribution. Appl. Environ. Microbiol. 2004, 70, 4880–4888. [Google Scholar] [CrossRef] [PubMed]
- Villemur, R. The pentachlorophenol-dehalogenating Desulf itobacterium haf niense strain PCP-1. Philos. Trans. R. Soc. B 2013, 368, 20120319. [Google Scholar] [CrossRef]
- Leys, D.; Adrian, L.; Smidt, H. Organohalide respiration: Microbes breathing chlorinated molecules. Philos. Trans. R. Soc. B 2013, 368, 20120316. [Google Scholar] [CrossRef]
- Hug, L.A.; Maphosa, F.; Leys, D.; Löffler, F.E.; Smidt, H.; Edwards, E.A.; Adrian, L. Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos. Trans. R. Soc. B 2013, 368, 20120322. [Google Scholar] [CrossRef]
- Schipp, C.J.; Marco-Urrea, E.; Kublik, A.; Seifert, J.; Adrian, L. Organic cofactors in the metabolism of Dehalococcoides mccartyi strains. Philos. Trans. R. Soc. B 2013, 368, 20120321. [Google Scholar] [CrossRef]
- Cooper, M.; Wagner, A.; Wondrousch, D.; Sonntag, F.; Sonnabend, A.; Brehm, M.; Schüürmann, G.; Adrian, L. Anaerobic microbial transformation of halogenated aromatics and fate prediction using electron density modeling. Environ. Sci. Technol. 2015, 49, 6018–6028. [Google Scholar] [CrossRef]
- Zhang, S.; Wondrousch, D.; Cooper, M.; Zinder, S.H.; Schüürmann, G.; Adrian, L. Anaerobic Dehalogenation of Chloroanilines by Dehalococcoides mccartyi Strain CBDB1 and Dehalobacter Strain 14DCB1 via Different Pathways as Related to Molecular Electronic Structure. Environ. Sci. Technol. 2017, 51, 3714–3724. [Google Scholar] [CrossRef]
- Spataru, T.; Dumitrescu, T.; Moraru, M.; Fernandez, F.; Spataru, P. The Mechanism of the Toxic Organic Halides Disposal under the Catalytic Influence of the Vitamin B12. J. Toxicol. Risk Assess. 2023, 9, 1–9. [Google Scholar]
Figure 1.
Biologically active vitamin B12 structures [23]: (a) Base-on structure; (b) base-on structure with dimethylbenzimidazole ligand substituted with a histidine molecule; (c) base-off structure.
Figure 1.
Biologically active vitamin B12 structures [23]: (a) Base-on structure; (b) base-on structure with dimethylbenzimidazole ligand substituted with a histidine molecule; (c) base-off structure.

Figure 2.
The methyl radical (-CH3) transfers in the Methionine Synthase process [141]. Cbl: cob(I)alamin; MeCbl(II): methylcob(II)alamin; MeTHF: 5-methyltetrahydrofolate, THF: tetrahydrofolate molecule through methylcobalamin itself.
Figure 2.
The methyl radical (-CH3) transfers in the Methionine Synthase process [141]. Cbl: cob(I)alamin; MeCbl(II): methylcob(II)alamin; MeTHF: 5-methyltetrahydrofolate, THF: tetrahydrofolate molecule through methylcobalamin itself.

Figure 3.
Biological reactions catalyzed by the skeleton mutase process [142].
Figure 3.
Biological reactions catalyzed by the skeleton mutase process [142].

Figure 4.
Biological reactions catalyzed by aminomutase processes [142].
Figure 4.
Biological reactions catalyzed by aminomutase processes [142].

Figure 5.
Biological reactions catalyzed by eliminase processes [142].
Figure 5.
Biological reactions catalyzed by eliminase processes [142].

Figure 6.
Methylcobalamin and adenosylcobalamin turnover sequences [127].
Figure 6.
Methylcobalamin and adenosylcobalamin turnover sequences [127].

Figure 7.
The three known methylcobalamin reductive electrochemical mechanism schemes: (a) Lexa and Savéant [88] (R = CH3); (b) Martin and Finke [154]; (c) Spataru and Birke [89,90].

Figure 8.
Methylcobalamin models used in DFT calculations [126]: (a) base-on methylcobalamin with histidine axial ligand; (b) base-on methylcobalamin without the side chains and side loop but with the imidazole axial ligand; (c) base-off methylcobalamin without the side chains, side loop, and base axial ligand.
Figure 8.
Methylcobalamin models used in DFT calculations [126]: (a) base-on methylcobalamin with histidine axial ligand; (b) base-on methylcobalamin without the side chains and side loop but with the imidazole axial ligand; (c) base-off methylcobalamin without the side chains, side loop, and base axial ligand.

Figure 9.
The Co-C bond cleavage total energy values (eV) for different methylcobalamin model species calculated by using the DFT method as a function of Co-C distance (Å) [126]. Each curve was accordingly aligned at the same total energy minimum.
Figure 9.
The Co-C bond cleavage total energy values (eV) for different methylcobalamin model species calculated by using the DFT method as a function of Co-C distance (Å) [126]. Each curve was accordingly aligned at the same total energy minimum.

Figure 10.
The differences in the geometries of a methylcobalamin model calculated by using DFT and CASSCF methods [126]. These are illustrative of the influence of the Pseudo-Jahn–Teller Effect.
Figure 10.
The differences in the geometries of a methylcobalamin model calculated by using DFT and CASSCF methods [126]. These are illustrative of the influence of the Pseudo-Jahn–Teller Effect.

Figure 11.
CASSCF total energy as a function of the Co-C bond distance for base-on and base-off methylcob(II)alamin models [141]: (base-on) imidazole methylcob(II)alamin; (base-off) methylcob(II)alamin.
Figure 11.
CASSCF total energy as a function of the Co-C bond distance for base-on and base-off methylcob(II)alamin models [141]: (base-on) imidazole methylcob(II)alamin; (base-off) methylcob(II)alamin.

Figure 12.
Models of base-on methylcob(II)alamin used in CASSCF geometry optimization [141,143]: (a) imidazole-methyl cob(II)alamin cofactor with a homocysteine ion dissociate ion common model; (b) dimethylbenzimidazole-methylcob(II)alamin with a homocysteine ion and histidine molecule substrate common model.
Figure 12.
Models of base-on methylcob(II)alamin used in CASSCF geometry optimization [141,143]: (a) imidazole-methyl cob(II)alamin cofactor with a homocysteine ion dissociate ion common model; (b) dimethylbenzimidazole-methylcob(II)alamin with a homocysteine ion and histidine molecule substrate common model.

Figure 13.
The MCSCF frontier molecular orbital surfaces of the imidazole-methylcob(II)alamin base-on cofactor and homocysteine ion joint model [141]: (a) HOMO2; (b) HOMO3; (c)LUMO1; (d) LUMO2.
Figure 13.
The MCSCF frontier molecular orbital surfaces of the imidazole-methylcob(II)alamin base-on cofactor and homocysteine ion joint model [141]: (a) HOMO2; (b) HOMO3; (c)LUMO1; (d) LUMO2.

Figure 14.
The mechanism of the base-on histidine-methylcob(II)alamin cofactor species interaction with the homocysteine dissociate ion [141].
Figure 14.
The mechanism of the base-on histidine-methylcob(II)alamin cofactor species interaction with the homocysteine dissociate ion [141].

Figure 15.
The cob(I)alamin cofactor species mechanism interaction with a 5-methyltetrahydrofolate protonated ion [141].
Figure 15.
The cob(I)alamin cofactor species mechanism interaction with a 5-methyltetrahydrofolate protonated ion [141].

Figure 16.
The Methionine Synthase process for HOMO2 and LUMO surfaces at the beginning of the CASSCF geometry optimization [143]: (RCo-N = 2.08 A; RCo-N = 2.35 A).
Figure 16.
The Methionine Synthase process for HOMO2 and LUMO surfaces at the beginning of the CASSCF geometry optimization [143]: (RCo-N = 2.08 A; RCo-N = 2.35 A).

Figure 17.
The Co-N bond cleavage mechanism in methylcobalamin cofactor in the presence of the homocysteine and histidine substrates [143]: (a) the charge transfer from the substrate to methylcobalamin; (b) the products.
Figure 17.
The Co-N bond cleavage mechanism in methylcobalamin cofactor in the presence of the homocysteine and histidine substrates [143]: (a) the charge transfer from the substrate to methylcobalamin; (b) the products.

Figure 18.
The schematic structures of an AdoMet positive ion without and with imidazole-cob(II)alamin [143]: (a) AdoMet positive ion; (b) base-on imidazole-cob(II)alamin (the side hydrogens that replaced the side chains are not shown) with an AdoMet positive ion common model used in CASSCF calculations.
Figure 18.
The schematic structures of an AdoMet positive ion without and with imidazole-cob(II)alamin [143]: (a) AdoMet positive ion; (b) base-on imidazole-cob(II)alamin (the side hydrogens that replaced the side chains are not shown) with an AdoMet positive ion common model used in CASSCF calculations.

Figure 19.
The AdoMet positive-ion and cob(II)alamin inactive-particle model HOMO (a) and LUMO (b) surfaces during the CASSCF geometry optimization process [143].
Figure 19.
The AdoMet positive-ion and cob(II)alamin inactive-particle model HOMO (a) and LUMO (b) surfaces during the CASSCF geometry optimization process [143].

Figure 20.
The –CH3 group mechanism transfer from the AdoMet positive particle to the inactive cob(II)alamin cofactor compound [143]: (a) the starting model movement; (b) the transfer of the methyl radical; (c) the final products.
Figure 20.
The –CH3 group mechanism transfer from the AdoMet positive particle to the inactive cob(II)alamin cofactor compound [143]: (a) the starting model movement; (b) the transfer of the methyl radical; (c) the final products.

Figure 21.
The CASSCF without a total-energy-barrier-determined mechanism of the Methionine Synthase process [143]. Hcy, His, MeTHF, THF, Met, and AdoMet indicate homocysteine, histidine, 5-methyltetrahydrofolate, tetrahydrofolate, methionine, and S-adenosylmethionine, respectively. The initial stage is black, and the regular turnover process stages are blue.
Figure 21.
The CASSCF without a total-energy-barrier-determined mechanism of the Methionine Synthase process [143]. Hcy, His, MeTHF, THF, Met, and AdoMet indicate homocysteine, histidine, 5-methyltetrahydrofolate, tetrahydrofolate, methionine, and S-adenosylmethionine, respectively. The initial stage is black, and the regular turnover process stages are blue.

Figure 22.
The Isobutyryl-CoA Mutase process’s substrate activation by a valine radical after its catalytic activation by the adenosylcobalamin cofactor [87,142].

Figure 23.
Models of adenosylcobalamin used in CASSCF electronic structure calculations [142]: (a) with the dimethylbenzimidazol ligand substituted with histidine; (b) with side chains substituted with hydrogens and CH3COO−; (c) with side chains substituted with hydrogens and CH3O−.
Figure 23.
Models of adenosylcobalamin used in CASSCF electronic structure calculations [142]: (a) with the dimethylbenzimidazol ligand substituted with histidine; (b) with side chains substituted with hydrogens and CH3COO−; (c) with side chains substituted with hydrogens and CH3O−.

Figure 24.
Adenosylcob(III)alamin and CH3COO− common model CASSCF HOMO-LUMO surfaces, energy levels, and the electron density population therein [142]: (a) Co-C bond distance of 2.00 Å; (b) Co-C bond distance of 3.17 Å. The data inside the rectangle refer to LUMO2, LUMO3, and LUMO4.
Figure 24.
Adenosylcob(III)alamin and CH3COO− common model CASSCF HOMO-LUMO surfaces, energy levels, and the electron density population therein [142]: (a) Co-C bond distance of 2.00 Å; (b) Co-C bond distance of 3.17 Å. The data inside the rectangle refer to LUMO2, LUMO3, and LUMO4.

Figure 25.
Adenosylcob(III)alamin and CH3COO− common model CASSCF HOMO-LUMO surfaces, energy levels, and the electron density population therein [142]: (left side) Co-C bond distance of 2.50 Å; (right side) Co-C bond distance of 2.65 Å.
Figure 25.
Adenosylcob(III)alamin and CH3COO− common model CASSCF HOMO-LUMO surfaces, energy levels, and the electron density population therein [142]: (left side) Co-C bond distance of 2.50 Å; (right side) Co-C bond distance of 2.65 Å.

Figure 26.
The CASSCF without a total energy barrier determined by the combined Co-C bond breaking mechanism and the substrate (SHCOO− model) transferring hydrogen to the 5′-deoxy-5′-adenosyl radical during the adenosylcobalamin cofactor-catalyzed reactions [142].
Figure 26.
The CASSCF without a total energy barrier determined by the combined Co-C bond breaking mechanism and the substrate (SHCOO− model) transferring hydrogen to the 5′-deoxy-5′-adenosyl radical during the adenosylcobalamin cofactor-catalyzed reactions [142].

Figure 27.
Adenosylcob(III)alamin and CH3O− common model CASSCF HOMO-LUMO surfaces, energy levels, and the electron density population therein [142]: (a) Co-C bond distance of 2.00 Å; (b) Co-C bond distance of 3.05 Å. The data inside the rectangle refer to LUMO2, LUMO3, and LUMO4.
Figure 27.
Adenosylcob(III)alamin and CH3O− common model CASSCF HOMO-LUMO surfaces, energy levels, and the electron density population therein [142]: (a) Co-C bond distance of 2.00 Å; (b) Co-C bond distance of 3.05 Å. The data inside the rectangle refer to LUMO2, LUMO3, and LUMO4.

Figure 28.
The CASSCF without a total energy barrier determines the combined Co-C bond-breaking mechanism and the substrate (RHO− model) transferring hydrogen to the 5′-deoxy-5′-adenosyl-radical during the adenosylcobalamin cofactor-catalyzed reactions [142].
Figure 28.
The CASSCF without a total energy barrier determines the combined Co-C bond-breaking mechanism and the substrate (RHO− model) transferring hydrogen to the 5′-deoxy-5′-adenosyl-radical during the adenosylcobalamin cofactor-catalyzed reactions [142].

Figure 29.
The 1,2-propanediol and 5′deoxy-5′-adenosyl radical joint model HOMO-LUMO surface profiles at the starting stage of the direct hydrogen transfer process [142].
Figure 29.
The 1,2-propanediol and 5′deoxy-5′-adenosyl radical joint model HOMO-LUMO surface profiles at the starting stage of the direct hydrogen transfer process [142].

Figure 30.
The de-hydrogenated 2-methylene-glutarate and 5′deoxy-5′-adenosyl radical joint model HOMO-LUMO surface profiles at the starting stage of the forbidden reverse hydrogen transfer process [142].
Figure 30.
The de-hydrogenated 2-methylene-glutarate and 5′deoxy-5′-adenosyl radical joint model HOMO-LUMO surface profiles at the starting stage of the forbidden reverse hydrogen transfer process [142].

Figure 31.
Two adenosylcobalamin base-on forms [144]: (a) base-on form with an axial dimethylbenzimidazole ligand; (b) base-on form with the dimethylbenzimidazole ligand substituted with an axial histidine ligand.
Figure 31.
Two adenosylcobalamin base-on forms [144]: (a) base-on form with an axial dimethylbenzimidazole ligand; (b) base-on form with the dimethylbenzimidazole ligand substituted with an axial histidine ligand.

Figure 32.
The catalyzed-by-adenosylcobalamin cofactor compound: Glutamate Mutase and Methylmalonyl-CoA Mutase bioprocesses [144].
Figure 32.
The catalyzed-by-adenosylcobalamin cofactor compound: Glutamate Mutase and Methylmalonyl-CoA Mutase bioprocesses [144].

Figure 33.
The model for CASSCF electronic structure calculations [144].
Figure 33.
The model for CASSCF electronic structure calculations [144].

Figure 34.
The CASSCF histidine, adenosylcobalamin, and CH3COO− model frontier molecular orbital surfaces [144].
Figure 34.
The CASSCF histidine, adenosylcobalamin, and CH3COO− model frontier molecular orbital surfaces [144].

Figure 35.
A Co-N bond cleavage in the adenosylcobalamin cofactor under the influence of the proposed mechanism of the histidine and CH3COO− substrates [144]: (a) charge transfer from the substrate to adenosylcobalamin; (b) the products.
Figure 35.
A Co-N bond cleavage in the adenosylcobalamin cofactor under the influence of the proposed mechanism of the histidine and CH3COO− substrates [144]: (a) charge transfer from the substrate to adenosylcobalamin; (b) the products.

Figure 36.
Common models for the cob(I)alamin cofactor with organic halides for the CASSCF geometry-optimization procedures: (a) with 3,5-dibrom-4-hidroxobenzoic acid; (b) with 3-bromo-4-hidroxobenzoic acid; (c) with 1,2-dichloroethene [211].
Figure 36.
Common models for the cob(I)alamin cofactor with organic halides for the CASSCF geometry-optimization procedures: (a) with 3,5-dibrom-4-hidroxobenzoic acid; (b) with 3-bromo-4-hidroxobenzoic acid; (c) with 1,2-dichloroethene [211].

Figure 37.
The cob(I)alamin cofactor with organic halide compound model surfaces of the intermolecular molecular orbitals and their electron density populations (P): (a) with 3,5-dibromo-4-hidroxobenzoic acid; (b) with 3-bromo-4-hidroxobenzoic acid; (c) with 1,2-dichloroethene [211].
Figure 37.
The cob(I)alamin cofactor with organic halide compound model surfaces of the intermolecular molecular orbitals and their electron density populations (P): (a) with 3,5-dibromo-4-hidroxobenzoic acid; (b) with 3-bromo-4-hidroxobenzoic acid; (c) with 1,2-dichloroethene [211].

Figure 38.
The cob(I)alamin cofactor interaction with the 3,5-dibromo-4-hydroxybenzoic acid mechanism: (a) charge transfer; (b) halogen transfer [211].
Figure 38.
The cob(I)alamin cofactor interaction with the 3,5-dibromo-4-hydroxybenzoic acid mechanism: (a) charge transfer; (b) halogen transfer [211].

Figure 39.
The cob(I)alamin cofactor interaction with the 3-bromo-4-hydroxybenzoic acid mechanism: (a) charge transfer; (b) halogen transfer [211].
Figure 39.
The cob(I)alamin cofactor interaction with the 3-bromo-4-hydroxybenzoic acid mechanism: (a) charge transfer; (b) halogen transfer [211].

Figure 40.
The cob(I)alamin cofactor interaction with the 1,2-dichloroethene mechanism: (a) charge transfer; (b) halogen transfer [211].
Figure 40.
The cob(I)alamin cofactor interaction with the 1,2-dichloroethene mechanism: (a) charge transfer; (b) halogen transfer [211].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Spataru, T. The Miracle of Vitamin B12 Biochemistry. Reactions 2024, 5, 20-76. https://doi.org/10.3390/reactions5010002
AMA Style
Spataru T. The Miracle of Vitamin B12 Biochemistry. Reactions. 2024; 5(1):20-76. https://doi.org/10.3390/reactions5010002
Chicago/Turabian StyleSpataru, Tudor. 2024. "The Miracle of Vitamin B12 Biochemistry" Reactions 5, no. 1: 20-76. https://doi.org/10.3390/reactions5010002