

High-Throughput Virtual Screening of Quinones for Aqueous Redox Flow Batteries: Status and Perspectives

Abstract
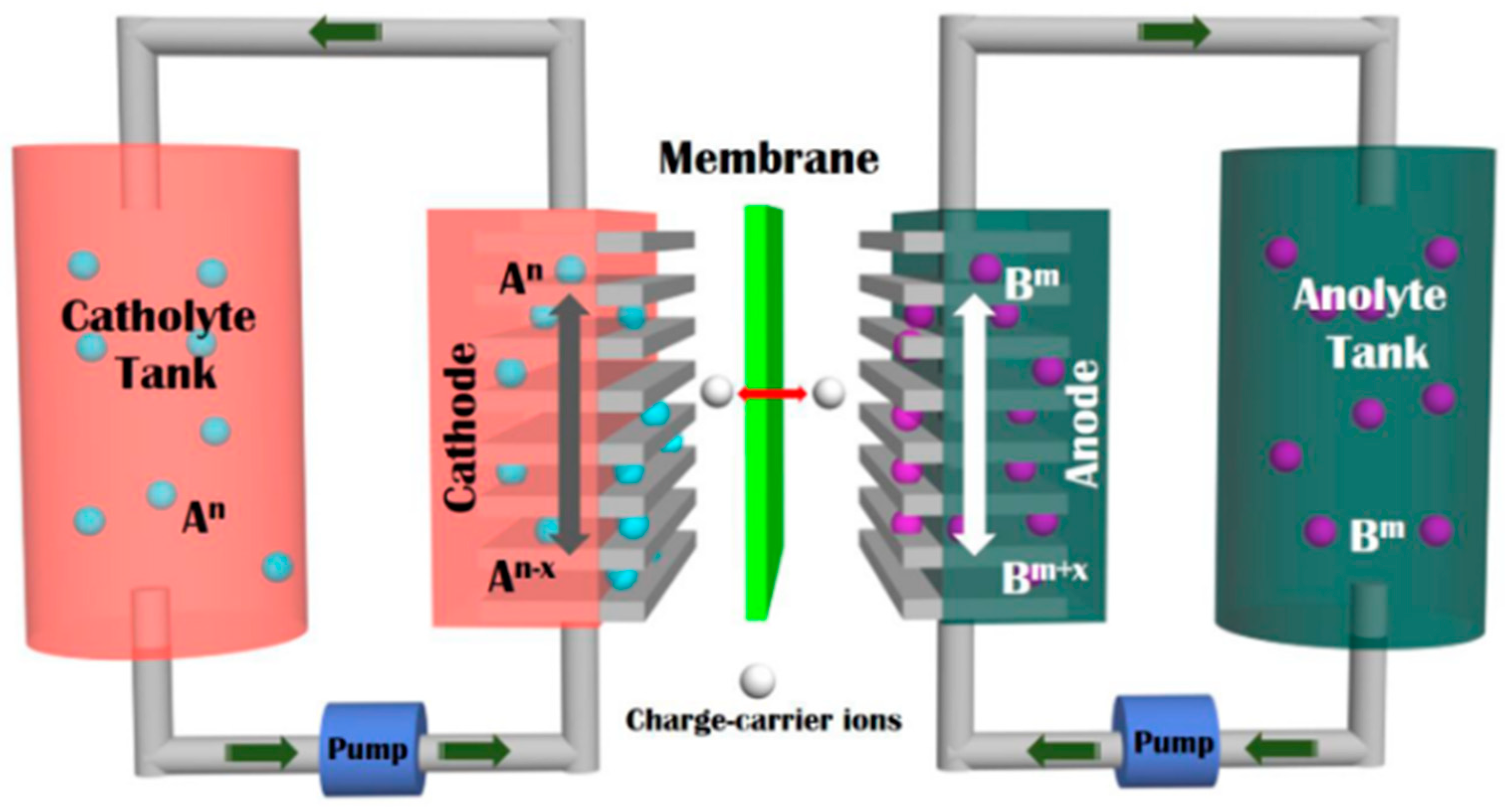
:1. Introduction
2. Redox Potential
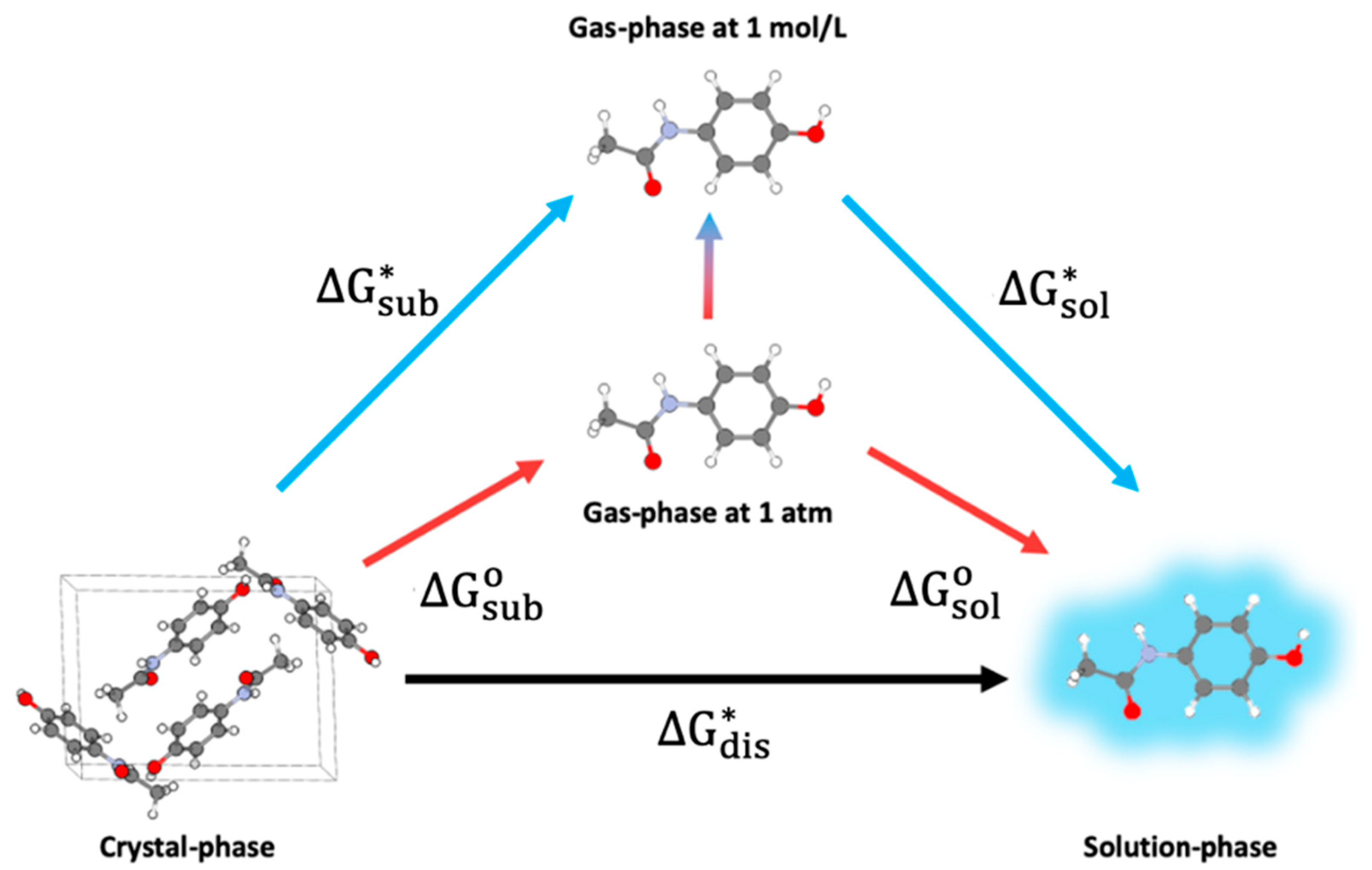
3. Aqueous Solubility
4. High-Throughput Virtual Screening: Status and Challenges
5. Summary and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Z.; Lu, Y.-C. Material Design of Aqueous Redox Flow Batteries: Fundamental Challenges and Mitigation Strategies. Adv. Mater. 2020, 32, 2002132. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Díez, E.; Ventosa, E.; Guarnieri, M.; Trovò, A.; Flox, C.; Marcilla, R.; Soavi, F.; Mazur, P.; Aranzabe, E.; Ferret, R. Redox flow batteries: Status and perspective towards sustainable stationary energy storage. J. Power Sources 2021, 481, 228804. [Google Scholar] [CrossRef]
- Narayan, S.R.; Nirmalchandar, A.; Murali, A.; Yang, B.; Hoober-Burkhardt, L.; Krishnamoorthy, S.; Prakash, G.K.S. Next-generation aqueous flow battery chemistries. Curr. Opin. Electrochem. 2019, 18, 72–80. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, T.; Ali, M.; Wu, C.; Chen, W. Recent Progress in Organic Species for Redox Flow Batteries. Energy Storage Mater. 2022, 50, 105–138. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, C. Cost-effective iron-based aqueous redox flow batteries for large-scale energy storage application: A review. J. Power Sources 2021, 493, 229445. [Google Scholar] [CrossRef]
- Wang, G.; Zou, H.; Zhu, X.; Ding, M.; Jia, C. Recent progress in zinc-based redox flow batteries: A review. J. Phys. D Appl. Phys. 2021, 55, 163001. [Google Scholar] [CrossRef]
- Khor, A.; Leung, P.; Mohamed, M.R.; Flox, C.; Xu, Q.; An, L.; Wills, R.G.A.; Morante, J.R.; Shah, A.A. Review of zinc-based hybrid flow batteries: From fundamentals to applications. Mater. Today Energy 2018, 8, 80–108. [Google Scholar] [CrossRef]
- Fischer, P.; Mazúr, P.; Krakowiak, J. Family Tree for Aqueous Organic Redox Couples for Redox Flow Battery Electrolytes: A Conceptual Review. Molecules 2022, 27, 560. [Google Scholar] [CrossRef]
- Zhong, F.; Yang, M.; Ding, M.; Jia, C. Organic Electroactive Molecule-Based Electrolytes for Redox Flow Batteries: Status and Challenges of Molecular Design. Front. Chem. 2020, 8. Available online: https://www.frontiersin.org/articles/10.3389/fchem.2020.00451 (accessed on 4 November 2022). [CrossRef]
- Zhao, Z.; Zhang, C.; Li, X. Opportunities and challenges of organic flow battery for electrochemical energy storage technology. J. Energy Chem. 2022, 67, 621–639. [Google Scholar] [CrossRef]
- Fang, X.; Li, Z.; Zhao, Y.; Yue, D.; Zhang, L.; Wei, X. Multielectron Organic Redoxmers for Energy-Dense Redox Flow Batteries. ACS Mater. Lett. 2022, 4, 277–306. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, C.; Zhang, L.; Zhou, Y.; Yu, G. Molecular engineering of organic electroactive materials for redox flow batteries. Chem. Soc. Rev. 2018, 47, 69–103. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Tian, J.; Xu, J.; Wang, Y. Organic Flow Batteries: Recent Progress and Perspectives. Energy Fuels 2020, 34, 13384–13411. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Q.; Zhang, X.; Ran, J.; Han, X.; Yang, Z.; Xu, T. Degradation of electrochemical active compounds in aqueous organic redox flow batteries. Curr. Opin. Electrochem. 2022, 32, 100895. [Google Scholar] [CrossRef]
- Kwabi, D.G.; Ji, Y.; Aziz, M.J. Electrolyte Lifetime in Aqueous Organic Redox Flow Batteries: A Critical Review. Chem. Rev. 2020, 120, 6467–6489. [Google Scholar] [CrossRef]
- Brushett, F.R.; Aziz, M.J.; Rodby, K.E. On Lifetime and Cost of Redox-Active Organics for Aqueous Flow Batteries. ACS Energy Lett. 2020, 5, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Huskinson, B.; Marshak, M.P.; Suh, C.; Er, S.; Gerhardt, M.R.; Galvin, C.J.; Chen, X.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J. A metal-free organic–inorganic aqueous flow battery. Nature 2014, 505, 195–198. [Google Scholar] [CrossRef]
- Wu, M.; Jing, Y.; Wong, A.A.; Fell, E.M.; Jin, S.; Tang, Z.; Gordon, R.G.; Aziz, M.J. Extremely Stable Anthraquinone Negolytes Synthesized from Common Precursors. Chem 2020, 6, 1432–1442. [Google Scholar] [CrossRef]
- Guiheneuf, S.; Godet-Bar, T.; Fontmorin, J.-M.; Jourdin, C.; Floner, D.; Geneste, F. A new hydroxyanthraquinone derivative with a low and reversible capacity fading process as negolyte in alkaline aqueous redox flow batteries. J. Power Sources 2022, 539, 231600. [Google Scholar] [CrossRef]
- Kwabi, D.G.; Lin, K.; Ji, Y.; Kerr, E.F.; Goulet, M.-A.; De Porcellinis, D.; Tabor, D.P.; Pollack, D.A.; Aspuru-Guzik, A.; Gordon, R.G.; et al. Alkaline Quinone Flow Battery with Long Lifetime at pH 12. Joule 2018, 2, 1894–1906. [Google Scholar] [CrossRef]
- Wedege, K.; Dražević, E.; Konya, D.; Bentien, A. Organic Redox Species in Aqueous Flow Batteries: Redox Potentials, Chemical Stability and Solubility. Sci Rep 2016, 6, 39101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoober-Burkhardt, L.; Krishnamoorthy, S.; Yang, B.; Murali, A.; Nirmalchandar, A.; Prakash, G.K.S.; Narayanan, S.R. A New Michael-Reaction-Resistant Benzoquinone for Aqueous Organic Redox Flow Batteries. J. Electrochem. Soc. 2017, 164, A600. [Google Scholar] [CrossRef]
- Ji, Y.; Goulet, M.-A.; Pollack, D.A.; Kwabi, D.G.; Jin, S.; De Porcellinis, D.; Kerr, E.F.; Gordon, R.G.; Aziz, M.J. A Phosphonate-Functionalized Quinone Redox Flow Battery at Near-Neutral pH with Record Capacity Retention Rate. Adv. Energy Mater. 2019, 9, 1900039. [Google Scholar] [CrossRef]
- Feng, R.; Zhang, X.; Murugesan, V.; Hollas, A.; Chen, Y.; Shao, Y.; Walter, E.; Wellala, N.P.N.; Yan, L.; Rosso, K.M.; et al. Reversible ketone hydrogenation and dehydrogenation for aqueous organic redox flow batteries. Science 2021, 372, 836–840. [Google Scholar] [CrossRef]
- Yang, X.; Garcia, S.N.; Janoschka, T.; Kónya, D.; Hager, M.D.; Schubert, U.S. Novel, Stable Catholyte for Aqueous Organic Redox Flow Batteries: Symmetric Cell Study of Hydroquinones with High Accessible Capacity. Molecules 2021, 26, 3823. [Google Scholar] [CrossRef]
- Wang, C.; Yu, B.; Liu, Y.; Wang, H.; Zhang, Z.; Xie, C.; Li, X.; Zhang, H.; Jin, Z. N-alkyl-carboxylate-functionalized anthraquinone for long-cycling aqueous redox flow batteries. Energy Storage Mater. 2021, 36, 417–426. [Google Scholar] [CrossRef]
- Hollas, A.; Wei, X.; Murugesan, V.; Nie, Z.; Li, B.; Reed, D.; Liu, J.; Sprenkle, V.; Wang, W. A biomimetic high-capacity phenazine-based anolyte for aqueous organic redox flow batteries. Nat Energy 2018, 3, 508–514. [Google Scholar] [CrossRef]
- Pang, S.; Wang, X.; Wang, P.; Ji, Y. Biomimetic Amino Acid Functionalized Phenazine Flow Batteries with Long Lifetime at Near-Neutral pH. Angew. Chem. Int. Ed. 2021, 60, 5289–5298. [Google Scholar] [CrossRef]
- Lin, K.; Gómez-Bombarelli, R.; Beh, E.S.; Tong, L.; Chen, Q.; Valle, A.; Aspuru-Guzik, A.; Aziz, M.J.; Gordon, R.G. A redox-flow battery with an alloxazine-based organic electrolyte. Nat. Energy 2016, 1, 16102. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-L.; Fu, Y. Theoretical study of reduction potentials of substituted flavins. J. Mol. Struct. THEOCHEM 2008, 856, 112–118. [Google Scholar] [CrossRef]
- Orita, A.; Verde, M.G.; Sakai, M.; Meng, Y.S. A biomimetic redox flow battery based on flavin mononucleotide. Nat. Commun. 2016, 7, 13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Pang, S.; Wang, X.; Wang, P.; Ji, Y. Ultrastable aqueous phenazine flow batteries with high capacity operated at elevated temperatures. Joule 2021, 5, 2437–2449. [Google Scholar] [CrossRef]
- Symons, P. Quinones for redox flow batteries. Curr. Opin. Electrochem. 2021, 29, 100759. [Google Scholar] [CrossRef]
- Pyzer-Knapp, E.O.; Suh, C.; Gómez-Bombarelli, R.; Aguilera-Iparraguirre, J.; Aspuru-Guzik, A. What Is High-Throughput Virtual Screening? A Perspective from Organic Materials Discovery. Annu. Rev. Mater. Res. 2015, 45, 195–216. Available online: https://www.annualreviews.org/doi/10.1146/annurev-matsci-070214-020823 (accessed on 20 September 2022). [CrossRef] [Green Version]
- Er, S.; Suh, C.; Marshak, M.P.; Aspuru-Guzik, A. Computational design of molecules for an all-quinone redox flow battery. Chem. Sci. 2015, 6, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Pineda Flores, S.D.; Martin-Noble, G.C.; Phillips, R.L.; Schrier, J. Bio-Inspired Electroactive Organic Molecules for Aqueous Redox Flow Batteries. 1. Thiophenoquinones. J. Phys. Chem. C 2015, 119, 21800–21809. [Google Scholar] [CrossRef]
- Huynh, M.T.; Anson, C.W.; Cavell, A.C.; Stahl, S.S.; Hammes-Schiffer, S. Quinone 1 e− and 2 e−/2 H+ Reduction Potentials: Identification and Analysis of Deviations from Systematic Scaling Relationships. J. Am. Chem. Soc. 2016, 138, 15903–15910. [Google Scholar] [CrossRef]
- Han, Y.-K.; Jin, C.-S. Computational screening of electroactive indolequinone derivatives as high-performance active materials for aqueous redox flow batteries. Curr. Appl. Phys. 2018, 18, 1507–1512. [Google Scholar] [CrossRef]
- Tabor, D.P.; Gómez-Bombarelli, R.; Tong, L.; Gordon, R.G.; Aziz, M.J.; Aspuru-Guzik, A. Mapping the frontiers of quinone stability in aqueous media: Implications for organic aqueous redox flow batteries. J. Mater. Chem. A 2019, 7, 12833–12841. [Google Scholar] [CrossRef]
- Fornari, R.P.; Mesta, M.; Hjelm, J.; Vegge, T.; de Silva, P. Molecular Engineering Strategies for Symmetric Aqueous Organic Redox Flow Batteries. ACS Mater. Lett. 2020, 2, 239–246. [Google Scholar] [CrossRef]
- Kristensen, S.B.; van Mourik, T.; Pedersen, T.B.; Sørensen, J.L.; Muff, J. Simulation of electrochemical properties of naturally occurring quinones. Sci. Rep. 2020, 10, 13571. [Google Scholar] [CrossRef] [PubMed]
- Schwan, S.; Schröder, D.; Wegner, H.A.; Janek, J.; Mollenhauer, D. Substituent Pattern Effects on the Redox Potentials of Quinone-Based Active Materials for Aqueous Redox Flow Batteries. ChemSusChem 2020, 13, 5480–5488. [Google Scholar] [CrossRef]
- Zhang, Q.; Khetan, A.; Sorkun, E.; Niu, F.; Loss, A.; Pucher, I.; Er, S. Data-driven discovery of small electroactive molecules for energy storage in aqueous redox flow batteries. Energy Storage Mater. 2022, 47, 167–177. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Li, X. Machine learning for flow batteries: Opportunities and challenges. Chem. Sci. 2022, 13, 4740–4752. [Google Scholar] [CrossRef] [PubMed]
- Frey, D.; Shin, J.H.; Musco, C.; Modestino, M.A. Chemically-informed data-driven optimization (ChIDDO): Leveraging physical models and Bayesian learning to accelerate chemical research. React. Chem. Eng. 2022, 7, 855–865. [Google Scholar] [CrossRef]
- Wang, F.; Cheng, J. Automated workflow for computation of redox potentials, acidity constants, and solvation free energies accelerated by machine learning. J. Chem. Phys. 2022, 157, 024103. [Google Scholar] [CrossRef] [PubMed]
- Boobier, S.; Hose, D.R.J.; Blacker, A.J.; Nguyen, B.N. Machine learning with physicochemical relationships: Solubility prediction in organic solvents and water. Nat. Commun. 2020, 11, 5753. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.; Dandu, N.; Blaiszik, B.; Narayanan, B.; Assary, R.S.; Redfern, P.C.; Foster, I.; Curtiss, L.A. Graph-Based Approaches for Predicting Solvation Energy in Multiple Solvents: Open Datasets and Machine Learning Models. J. Phys. Chem. A 2021, 125, 5990–5998. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Yang, X.; Tang, Y.-H.; Zheng, M.; Andersen, A.; Murugesan, V.; Hollas, A.; Wang, W. Graphical Gaussian process regression model for aqueous solvation free energy prediction of organic molecules in redox flow batteries. Phys. Chem. Chem. Phys. 2021, 23, 24892–24904. [Google Scholar] [CrossRef]
- Jinich, A.; Sanchez-Lengeling, B.; Ren, H.; Harman, R.; Aspuru-Guzik, A. A Mixed Quantum Chemistry/Machine Learning Approach for the Fast and Accurate Prediction of Biochemical Redox Potentials and Its Large-Scale Application to 315 000 Redox Reactions. ACS Cent. Sci. 2019, 5, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jinich, A.; Aspuru-Guzik, A. MultiDK: A Multiple Descriptor Multiple Kernel Approach for Molecular Discovery and Its Application to Organic Flow Battery Electrolytes. J. Chem. Inf. Model. 2017, 57, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, J.; Lu, X.; He, M.; Wang, R. Redox potentials of aryl derivatives from hybrid functional based first principles molecular dynamics. Phys. Chem. Chem. Phys. 2016, 18, 14911–14917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Goodson, T.; Zimmerman, P.M. Achieving Accurate Reduction Potential Predictions for Anthraquinones in Water and Aprotic Solvents: Effects of Inter- and Intramolecular H-Bonding and Ion Pairing. J. Phys. Chem. C 2016, 120, 22235–22247. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, T.-S.; Pan, D. Tuning the Performance of Aqueous Organic Redox Flow Batteries via First-Principles Calculations. J. Phys. Chem. Lett. 2020, 11, 10433–10438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Khetan, A.; Er, S. Comparison of computational chemistry methods for the discovery of quinone-based electroactive compounds for energy storage. Sci. Rep. 2020, 10, 22149. [Google Scholar] [CrossRef] [PubMed]
- Fornari, R.P.; de Silva, P. A Computational Protocol Combining DFT and Cheminformatics for Prediction of pH-Dependent Redox Potentials. Molecules 2021, 26, 3978. [Google Scholar] [CrossRef]
- Gaudin, T.; Aubry, J.-M. Prediction of Pourbaix diagrams of quinones for redox flow battery by COSMO-RS. J. Energy Storage 2022, 49, 104152. [Google Scholar] [CrossRef]
- Guo, C.; Wang, W.; Feng, W.; Li, P. Insights into the one-electron reduction behavior of tetrachloro-o-benzoquinone: A DFT and molecular dynamics study. RSC Adv. 2017, 7, 12775–12782. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Assary, R.S.; Qu, X.; Jain, A.; Ong, S.P.; Rajput, N.N.; Persson, K.; Curtiss, L.A. Accelerating Electrolyte Discovery for Energy Storage with High-Throughput Screening. J. Phys. Chem. Lett. 2015, 6, 283–291. [Google Scholar] [CrossRef]
- Assary, R.S.; Zhang, L.; Huang, J.; Curtiss, L.A. Molecular Level Understanding of the Factors Affecting the Stability of Dimethoxy Benzene Catholyte Candidates from First-Principles Investigations. J. Phys. Chem. C 2016, 120, 14531–14538. [Google Scholar] [CrossRef]
- Ding, Y.; Li, Y.; Yu, G. Exploring Bio-inspired Quinone-Based Organic Redox Flow Batteries: A Combined Experimental and Computational Study. Chem 2016, 1, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, K.M.; Cheng, L.; Curtiss, L.A. Effects of Functional Groups in Redox-Active Organic Molecules: A High-Throughput Screening Approach. J. Phys. Chem. C 2017, 121, 237–245. [Google Scholar] [CrossRef]
- Lambert, F.; Danten, Y.; Gatti, C.; Frayret, C. A tool for deciphering the redox potential ranking of organic compounds: A case study of biomass-extracted quinones for sustainable energy. Phys. Chem. Chem. Phys. 2020, 22, 20212–20226. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.R.; Bodman, S.E.; Burney, A.M.; Hughes, C.D.; Crittenden, D.L. Experimental Validation of a Computational Screening Approach to Predict Redox Potentials for a Diverse Variety of Redox-Active Organic Molecules. J. Phys. Chem. C 2020, 124, 24105–24114. [Google Scholar] [CrossRef]
- Zhou, X.; Khetan, A.; Er, S. Evaluation of Computational Chemistry Methods for Predicting Redox Potentials of Quinone-Based Cathodes for Li-Ion Batteries. Batteries 2021, 7, 71. [Google Scholar] [CrossRef]
- Fenini, F.; Drazevic, E.; Bentien, A. Impact of pH management on utilization and performance of anthraquinone/ ferrocyanide flow batteries. J. Power Sources 2022, 540, 231641. [Google Scholar] [CrossRef]
- Marenich, A.V.; Ho, J.; Coote, M.L.; Cramer, C.J.; Truhlar, D.G. Computational electrochemistry: Prediction of liquid-phase reduction potentials. Phys. Chem. Chem. Phys. 2014, 16, 15068–15106. [Google Scholar] [CrossRef]
- Méndez-Hernández, D.D.; Tarakeshwar, P.; Gust, D.; Moore, T.A.; Moore, A.L.; Mujica, V. Simple and accurate correlation of experimental redox potentials and DFT-calculated HOMO/LUMO energies of polycyclic aromatic hydrocarbons. J. Mol. Model. 2013, 19, 2845–2848. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Persson, K.A. Toward Accurate Modeling of the Effect of Ion-Pair Formation on Solute Redox Potential. J. Chem. Theory Comput. 2016, 12, 4501–4508. [Google Scholar] [CrossRef]
- Costanzo, F.; Sulpizi, M.; Valle, R.G.D.; Sprik, M. The oxidation of tyrosine and tryptophan studied by a molecular dynamics normal hydrogen electrode. J. Chem. Phys. 2011, 134, 244508. [Google Scholar] [CrossRef]
- Ghosh, D.; Roy, A.; Seidel, R.; Winter, B.; Bradforth, S.; Krylov, A.I. First-Principle Protocol for Calculating Ionization Energies and Redox Potentials of Solvated Molecules and Ions: Theory and Application to Aqueous Phenol and Phenolate. J. Phys. Chem. B 2012, 116, 7269–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiki, R.; Matsui, T.; Shigeta, Y.; Nakano, H.; Yoshida, N. Recent Developments of Computational Methods for pKa Prediction Based on Electronic Structure Theory with Solvation Models. J 2021, 4, 849–864. [Google Scholar] [CrossRef]
- Ho, J. Are thermodynamic cycles necessary for continuum solvent calculation of pKas and reduction potentials? Phys. Chem. Chem. Phys. 2014, 17, 2859–2868. [Google Scholar] [CrossRef]
- Navo, C.D.; Jiménez-Osés, G. Computer Prediction of pKa Values in Small Molecules and Proteins. ACS Med. Chem. Lett. 2021, 12, 1624–1628. [Google Scholar] [CrossRef]
- Bauer, S.; Namyslo, J.C.; Kaufmann, D.E.; Turek, T. Evaluation of Options and Limits of Aqueous All-Quinone-Based Organic Redox Flow Batteries. J. Electrochem. Soc. 2020, 167, 110522. [Google Scholar] [CrossRef]
- Mao, J.; Ruan, W.; Chen, Q. Understanding the Aqueous Solubility of Anthraquinone Sulfonate Salts: The Quest for High Capacity Electrolytes of Redox Flow Batteries. J. Electrochem. Soc. 2020, 167, 070522. [Google Scholar] [CrossRef]
- Huang, Z.; Lee, J.; Henkensmeier, D.; Hempelmann, R.; Kim, S.; Chen, R. Effect of Molecular Structure and Coordinating Ions on the Solubility and Electrochemical Behavior of Quinone Derivatives for Aqueous Redox Flow Batteries. J. Electrochem. Soc. 2020, 167, 160502. [Google Scholar] [CrossRef]
- Raevsky, O.A.; Grigorev, V.Y.; Polianczyk, D.E.; Raevskaja, O.E.; Dearden, J.C. Aqueous Drug Solubility: What Do We Measure, Calculate and QSPR Predict? Mini-Rev. Med. Chem. 2019, 19, 362–372. Available online: https://www.eurekaselect.com/article/92000 (accessed on 4 November 2022). [CrossRef] [PubMed]
- Duarte Ramos Matos, G.; Mobley, D.L. Challenges in the use of atomistic simulations to predict solubilities of drug-like molecules. F1000Research 2019, 7, 686. [Google Scholar] [CrossRef] [Green Version]
- Bergström, C.A.S.; Larsson, P. Computational prediction of drug solubility in water-based systems: Qualitative and quantitative approaches used in the current drug discovery and development setting. Int. J. Pharm. 2018, 540, 185–193. [Google Scholar] [CrossRef]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.R.; van Mourik, T.; Mitchell, J.B.O. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the General Solubility Equation: In Silico Prediction of Aqueous Solubility Incorporating the Effect of Topographical Polar Surface Area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, F.; Lagorce, D.; Reynès, C.; Villoutreix, B.O.; Vayer, P.; Miteva, M.A. In Silico Prediction of Aqueous Solubility: A Multimodel Protocol Based on Chemical Similarity. Mol. Pharm. 2012, 9, 3127–3135. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, J.L.; Nath, N.; De Ferrari, L.; van Mourik, T.; Mitchell, J.B.O. Uniting Cheminformatics and Chemical Theory To Predict the Intrinsic Aqueous Solubility of Crystalline Druglike Molecules. J. Chem. Inf. Model. 2014, 54, 844–856. [Google Scholar] [CrossRef] [Green Version]
- Boothroyd, S.; Kerridge, A.; Broo, A.; Buttar, D.; Anwar, J. Solubility prediction from first principles: A density of states approach. Phys. Chem. Chem. Phys. 2018, 20, 20981–20987. [Google Scholar] [CrossRef] [Green Version]
- Fowles, D.J.; Palmer, D.S.; Guo, R.; Price, S.L.; Mitchell, J.B.O. Toward Physics-Based Solubility Computation for Pharmaceuticals to Rival Informatics. J. Chem. Theory Comput. 2021, 17, 3700–3709. [Google Scholar] [CrossRef]
- Sorkun, M.C.; Koelman, J.M.V.A.; Er, S. Pushing the limits of solubility prediction via quality-oriented data selection. iScience 2021, 24, 101961. [Google Scholar] [CrossRef]
- Panapitiya, G.; Girard, M.; Hollas, A.; Sepulveda, J.; Murugesan, V.; Wang, W.; Saldanha, E. Evaluation of Deep Learning Architectures for Aqueous Solubility Prediction. ACS Omega 2022, 7, 15695–15710. [Google Scholar] [CrossRef]
- Oja, M.; Sild, S.; Piir, G.; Maran, U. Intrinsic Aqueous Solubility: Mechanistically Transparent Data-Driven Modeling of Drug Substances. Pharmaceutics 2022, 14, 2248. [Google Scholar] [CrossRef]
- Llinas, A.; Avdeef, A. Solubility Challenge Revisited after Ten Years, with Multilab Shake-Flask Data, Using Tight (SD ∼ 0.17 log) and Loose (SD ∼ 0.62 log) Test Sets. J. Chem. Inf. Model. 2019, 59, 3036–3040. [Google Scholar] [CrossRef]
- Palmer, D.S.; Mitchell, J.B.O. Is Experimental Data Quality the Limiting Factor in Predicting the Aqueous Solubility of Druglike Molecules? Mol. Pharm. 2014, 11, 2962–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llinàs, A.; Glen, R.C.; Goodman, J.M. Solubility Challenge: Can You Predict Solubilities of 32 Molecules Using a Database of 100 Reliable Measurements? J. Chem. Inf. Model. 2008, 48, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Henkensmeier, D.; Chen, R. Imidazolium cation enabled reversibility of a hydroquinone derivative for designing aqueous redox electrolytes. Sustain. Energy Fuels 2020, 4, 2998–3005. [Google Scholar] [CrossRef]
- McDonagh, J.L.; Palmer, D.S.; van Mourik, T.; Mitchell, J.B.O. Are the Sublimation Thermodynamics of Organic Molecules Predictable? J. Chem. Inf. Model. 2016, 56, 2162–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emami, S.; Jouyban, A.; Valizadeh, H.; Shayanfar, A. Are Crystallinity Parameters Critical for Drug Solubility Prediction? J. Solut. Chem. 2015, 44, 2297–2315. [Google Scholar] [CrossRef]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the sixth blind test of organic crystal structure prediction methods. Acta Cryst. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 439–459. [Google Scholar] [CrossRef]
- Li, J.; Xu, H.; Wang, J.; Wang, Y.; Lu, D.; Liu, J.; Wu, J. Theoretical insights on the hydration of quinones as catholytes in aqueous redox flow batteries. Chin. J. Chem. Eng. 2021, 37, 72–78. [Google Scholar] [CrossRef]
- Ringe, S.; Hormann, N.G.; Oberhofer, H.; Reuter, K. Implicit Solvation Methods for Catalysis at Electrified Interfaces. Chem. Rev. 2022, 122, 10777–10820. Available online: https://pubs.acs.org/doi/10.1021/acs.chemrev.1c00675 (accessed on 4 November 2022). [CrossRef]
- Zhang, J.; Zhang, H.; Wu, T.; Wang, Q.; van der Spoel, D. Comparison of Implicit and Explicit Solvent Models for the Calculation of Solvation Free Energy in Organic Solvents. J. Chem. Theory Comput. 2017, 13, 1034–1043. [Google Scholar] [CrossRef]
- Sadowsky, D.; Arey, J.S. Prediction of aqueous free energies of solvation using coupled QM and MM explicit solvent simulations. Phys. Chem. Chem. Phys. 2020, 22, 8021–8034. [Google Scholar] [CrossRef]
- Sorkun, M.C.; Khetan, A.; Er, S. AqSolDB, a curated reference set of aqueous solubility and 2D descriptors for a diverse set of compounds. Sci. Data 2019, 6, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhigalko, M.V.; Shishkin, O.V.; Gorb, L.; Leszczynski, J. Out-of-plane deformability of aromatic systems in naphthalene, anthracene and phenanthrene. J. Mol. Struct. 2004, 693, 153–159. [Google Scholar] [CrossRef]
- de la Cruz, C.; Molina, A.; Patil, N.; Ventosa, E.; Marcilla, R.; Mavrandonakis, A. New insights into phenazine-based organic redox flow batteries by using high-throughput DFT modelling. Sustain. Energy Fuels 2020, 4, 5513–5521. [Google Scholar] [CrossRef]
- Zhang, Q.; Khetan, A.; Er, S. A quantitative evaluation of computational methods to accelerate the study of alloxazine-derived electroactive compounds for energy storage. Sci. Rep. 2021, 11, 4089. [Google Scholar] [CrossRef] [PubMed]
- Carretero-González, J.; Castillo-Martínez, E.; Armand, M. Highly water-soluble three-redox state organic dyes as bifunctional analytes. Energy Environ. Sci. 2016, 9, 3521–3530. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. Available online: https://pubs.acs.org/doi/10.1021/ci049714%2B (accessed on 3 November 2022). [CrossRef] [Green Version]
- Wu, M.; Bahari, M.; Jing, Y.; Amini, K.; Fell, E.M.; George, T.Y.; Gordon, R.G.; Aziz, M.J. Highly Stable, Low Redox Potential Quinone for Aqueous Flow Batteries. Batter. Supercaps 2022, 5, e202200009. [Google Scholar] [CrossRef]
- Murali, A.; Nirmalchandar, A.; Krishnamoorthy, S.; Hoober-Burkhardt, L.; Yang, B.; Soloveichik, G.; Prakash, G.K.S.; Narayanan, S.R. Understanding and Mitigating Capacity Fade in Aqueous Organic Redox Flow Batteries. J. Electrochem. Soc. 2018, 165, A1193. [Google Scholar] [CrossRef]
- Jing, Y.; Zhao, E.W.; Goulet, M.-A.; Bahari, M.; Fell, E.M.; Jin, S.; Davoodi, A.; Jónsson, E.; Wu, M.; Grey, C.P.; et al. In situ electrochemical recomposition of decomposed redox-active species in aqueous organic flow batteries. Nat. Chem. 2022, 14, 1103–1109. [Google Scholar] [CrossRef]
- Zhao, E.W.; Liu, T.; Jónsson, E.; Lee, J.; Temprano, I.; Jethwa, R.B.; Wang, A.; Smith, H.; Carretero-González, J.; Song, Q.; et al. In situ NMR metrology reveals reaction mechanisms in redox flow batteries. Nature 2020, 579, 224–228. [Google Scholar] [CrossRef]
- Carney, T.J.; Collins, S.J.; Moore, J.S.; Brushett, F.R. Concentration-Dependent Dimerization of Anthraquinone Disulfonic Acid and Its Impact on Charge Storage. Chem. Mater. 2017, 29, 4801–4810. [Google Scholar] [CrossRef]
- Deb, K.; Gupta, H. Searching for Robust Pareto-Optimal Solutions in Multi-objective Optimization. In Evolutionary Multi-Criterion Optimization; Springer: Berlin/Heidelberg, Germany, 2005; pp. 150–164. [Google Scholar]
- Wang, Z.; Rangaiah, G.P. Application and Analysis of Methods for Selecting an Optimal Solution from the Pareto-Optimal Front obtained by Multiobjective Optimization. Ind. Eng. Chem. Res. 2017, 56, 560–574. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, T.; Abbasi, M.; Ribeiro, B.; Arrais, J.P. Diversity oriented Deep Reinforcement Learning for targeted molecule generation. J. Cheminformatics 2021, 13, 21. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Battery Performance Metric |
|---|---|
| redox potential | energy and power density, cost |
| aqueous solubility | energy and power density, energy and charge capacity, cost |
| rate kinetics | power density, efficiency |
| stability (chemical/electrochemical) | efficiency, lifetime |
| stability (calendar-life) | lifetime, cost |
| ionic conductivity | power density, efficiency |
| dynamic viscosity | power density, efficiency |
| permeation across the membrane | energy and power density, efficiency, and lifetime |
| toxicity | environmental impact |
| synthesizability | cost |
| Abbreviation | Meaning |
|---|---|
| HTVS | High-throughput virtual screening |
| ARFB | Aqueous redox flow battery |
| RAM | Redox active material |
| EDG | Electron-donating group |
| EWG | Electron-withdrawing group |
| PCET | Proton-coupled-electron-transfer |
| SHE | Standard hydrogen electrode |
| DFT | Density functional theory |
| DFTB | Density functional tight binding |
| SEQM | Semi-empirical quantum mechanics |
| MD | Molecular dynamics |
| QM/MM | Quantum mechanics/molecular mechanics |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| RMSE | Root mean squared error |
| MAD/MAE | Mean absolute deviation/Mean absolute error |
| PCM | Polarizable continuum model |
| PBF | Poisson Boltzmann formalism |
| COSMO | Conductor-like screening model |
| GSE | Generalized solubility equation |
| QSPR | Quantitative structure-property relationships |
| ML | Machine learning |
| Equilibrium redox potential with respect to SHE | |
| Acid dissociation constant | |
| Aqueous solubility | |
| Equilibrium constant for hydration | |
| Gibbs free energy of sublimation | |
| Gibbs free energy of aqueous solvation |
| Total Cores/[Ref] | Substituents | Library |
|---|---|---|
| Total: 17 (benzoquinone, naphthoquinone, anthraquinone)/[35] | 18 (−N(CH3)2, −NH2, −OCH3, −OH, −SH, −CH3, −SiH3, −F, −Cl, −C2H3, −CHO, −COOCH3, −CF3, −CN, −COOH, −PO3H2, −SO3H, −NO2) | 1710 |
| Total: 3 (caldariella, sulfolobus, benzodithiophenoquinone)/[36] | 8 (−NH2, −OH, −CH3, −F, −COOH, −PO3H2, −SO3H, −NO2) | 10,611 |
| Total: 1 (benzoquinone)/[37] | 22 (−COCH3, −N(CH3)2, −NH2, −C6H5, −CH3, −COOCH3, −C(CH3)3, −OCH3, −CH2CH3, −OH, −OCH2CH3, −F, −Cl, −Br, −SH, −SiH3, −CHO, −CF3, −CN, −COOH, −SO3–, −NO2) | 134 |
| Total: 3 (indolequinone)/[38] | 18 (−N(CH3)2, −NH2, −OCH3, −OH, −SH, −CH3, −SiH3, −F, −Cl, −C2H3, −CHO, −COOCH3, −CF3, −CN, −COOH, −PO3H2, −SO3H, −NO2) | 180 |
| Total: 16 (benzoquinone, naphthoquinone, anthraquinone, phenanthrene)/[39] | 4 (−OH, −COOH, −PO3H2, −SO3H) | 146,857 |
| Total: 35 (fused variations of quinones and pyrazines)/[40] | 2 (−OCH3, −SO3H) | 78 |
| Total: n.a (various cores derived from bacteria, fungi, yeast, algae, etc.)/[41] | n.a. | 990 |
| Total: 3 (benzoquinone, naphthoquinone, anthraquinone)/[42] | 7 (−CH3, −OCH3, −OH, −F, −CN, −SO3–, −NO2) | 592 |
| Total: 29 (benzoquinone, various cyclopentenediones)/[43] | 5 (−NH2, −OH, −F, −COOH, −SO3H) | 3257 |
| Ref | Redox Potential | Solubility | Candidates |
|---|---|---|---|
| [35] | Model: Equation (8) with no thermochemical corrections Method: PBE/Planewave and PBE/6-31G ** with Poisson–Boltzmann implicit solvation (PBF) Accuracy: (32 points) R2 = 0.974 | - 31 molecules with < 0.2 V and < −81.5 kJ/mol - 28 molecules with > 1.0 V and < −81.5 kJ/mol | |
| [36] | Model: Equation (8) with unspecified free energy corrections at T = 298.15 K, p = 1 atm Method: B3LYP/6-311 + G(d,p) with SMD implicit solvation Accuracy: (12 points) R2 = 0.981, MAD = 0.034 V | ChemAxon | - 36 molecules with < 0.25 V and > 2 mol/L - 15 molecules with > 0.95 V and > 2 mol/L |
| [37] | Model: Equation (7) with zero-point energy and entropic corrections at T = 298.15 K, p = 1 atm Method: B3LYP/6-31++G ** with a conductor, such as polarizable continuum model (C-PCM) Accuracy: (25 points) R2 = 0.934 | ||
| [38] | Model: Equation (7) with vibrational corrections included no solvation correction Method: B3LYP/6-311 + G(d,p) Accuracy: (5 points) R2 = 0.954 | ChemAxon | - 10 molecules with < 0.2 V - 9 molecules with > 0.9 V - 3 molecules with > 2 mol/L |
| [39] | Model: Equation (8) with no thermochemical corrections Method: PM7 (semi-empirical) with COSMO solvation model Accuracy: (28 points) R2 = 0.938, MAD = 0.067 V Stability model: where Q is the oxidized form of the quinone and QOH2 is the Michael product | ChemAxon | - 46 molecules with < 1.0, < 0.03 eV and > 0.95 (no catechol, no phenanthrene) - 377 molecules with < 1.0, < 0.03 eV and > 0.95 (with catechol, no phenanthrene) - 1545 molecules with < 1.0 and > 0.95 V of (only phenanthrenes lacking Michael sites) |
| [40] | Model: Equation (7) with zero-point, vibrational energy and entropic corrections at T = 298 K Method: B3LYP/6-311 + G(d,p) with Conductor, such as Polarizable Continuum model (C-PCM) Accuracy: n.a. | ChemAxon | |
| [41] | Model: Equation (8) with no thermochemical or solvation corrections Method: PBE/6-31G ** Accuracy: (6 points) R2 = 0.983 | (with PCM) | |
| [42] | Model: Equation (7) with zero-point, vibrational energy, and entropic corrections at T = 298 K Method: PBE-D3/aug-ccpVDZ with Polarizable Continuum model (PCM) Accuracy: (19 points) MAD = 0.071 V | ||
| [43] | Model: Equation (8) with no thermochemical corrections Method: PBE/LACVP **++ with Poisson–Boltzmann implicit solvation (PBF) Accuracy: (43 points) R2 = 0.977, RMSE = 0.051 V | AqSolPred | - 205 molecules with > 0.06 mol/L and −0.1 V < > 0.294 V |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khetan, A. High-Throughput Virtual Screening of Quinones for Aqueous Redox Flow Batteries: Status and Perspectives. Batteries 2023, 9, 24. https://doi.org/10.3390/batteries9010024
Khetan A. High-Throughput Virtual Screening of Quinones for Aqueous Redox Flow Batteries: Status and Perspectives. Batteries. 2023; 9(1):24. https://doi.org/10.3390/batteries9010024
Chicago/Turabian StyleKhetan, Abhishek. 2023. "High-Throughput Virtual Screening of Quinones for Aqueous Redox Flow Batteries: Status and Perspectives" Batteries 9, no. 1: 24. https://doi.org/10.3390/batteries9010024