

Complete Chloroplast Genome Sequence of Dahlia imperialis (Asteraceae): Comparative Analysis and Phylogenetic Relationships

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction, Genome Sequencing, and Annotation
2.3. Codon Usage Analysis
2.4. Interspersed Repeats and SSRs
2.5. Comparison of the Complete Chloroplast Genomes
2.6. Phylogenetic Analysis
3. Results
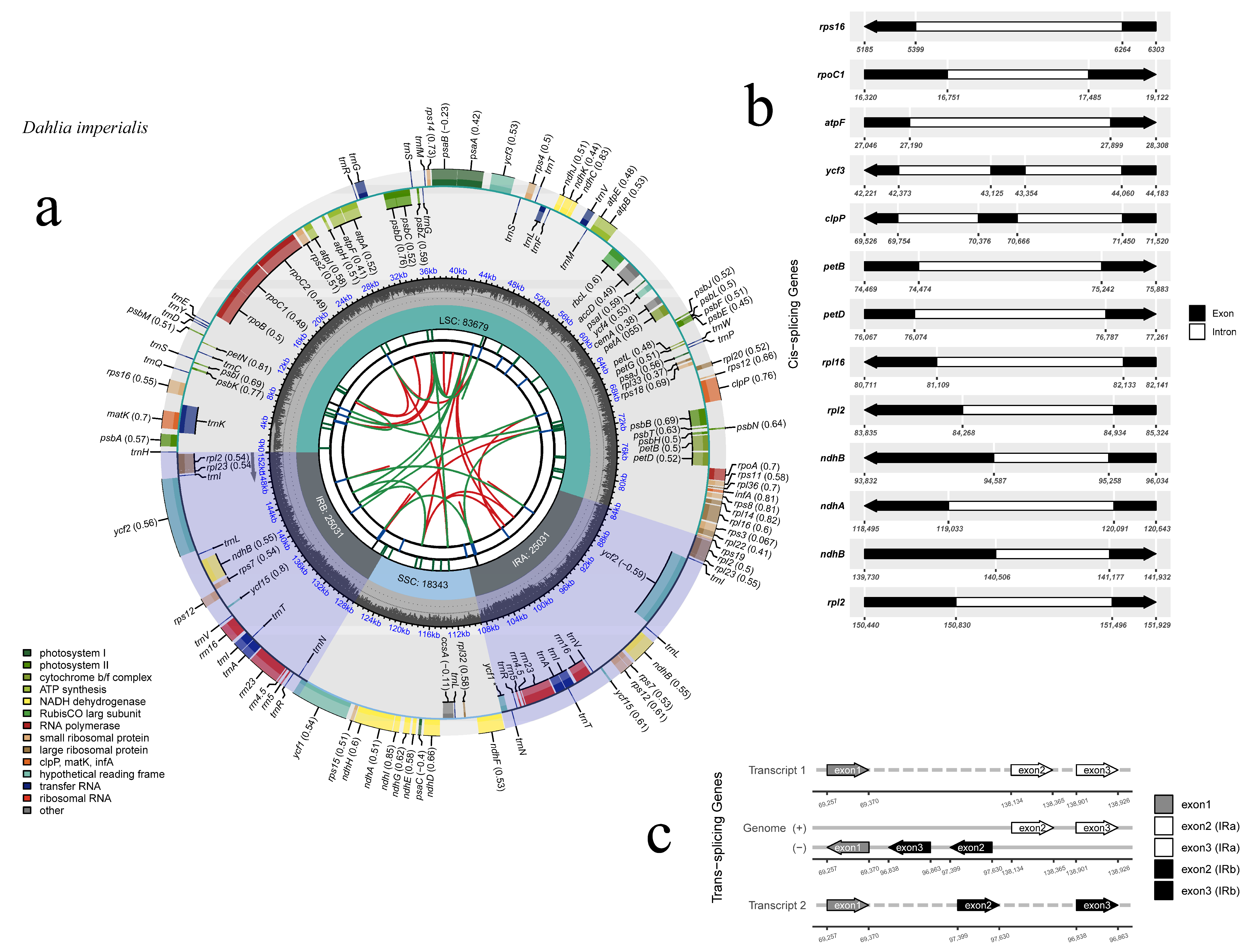
3.1. Chloroplast Genome Features
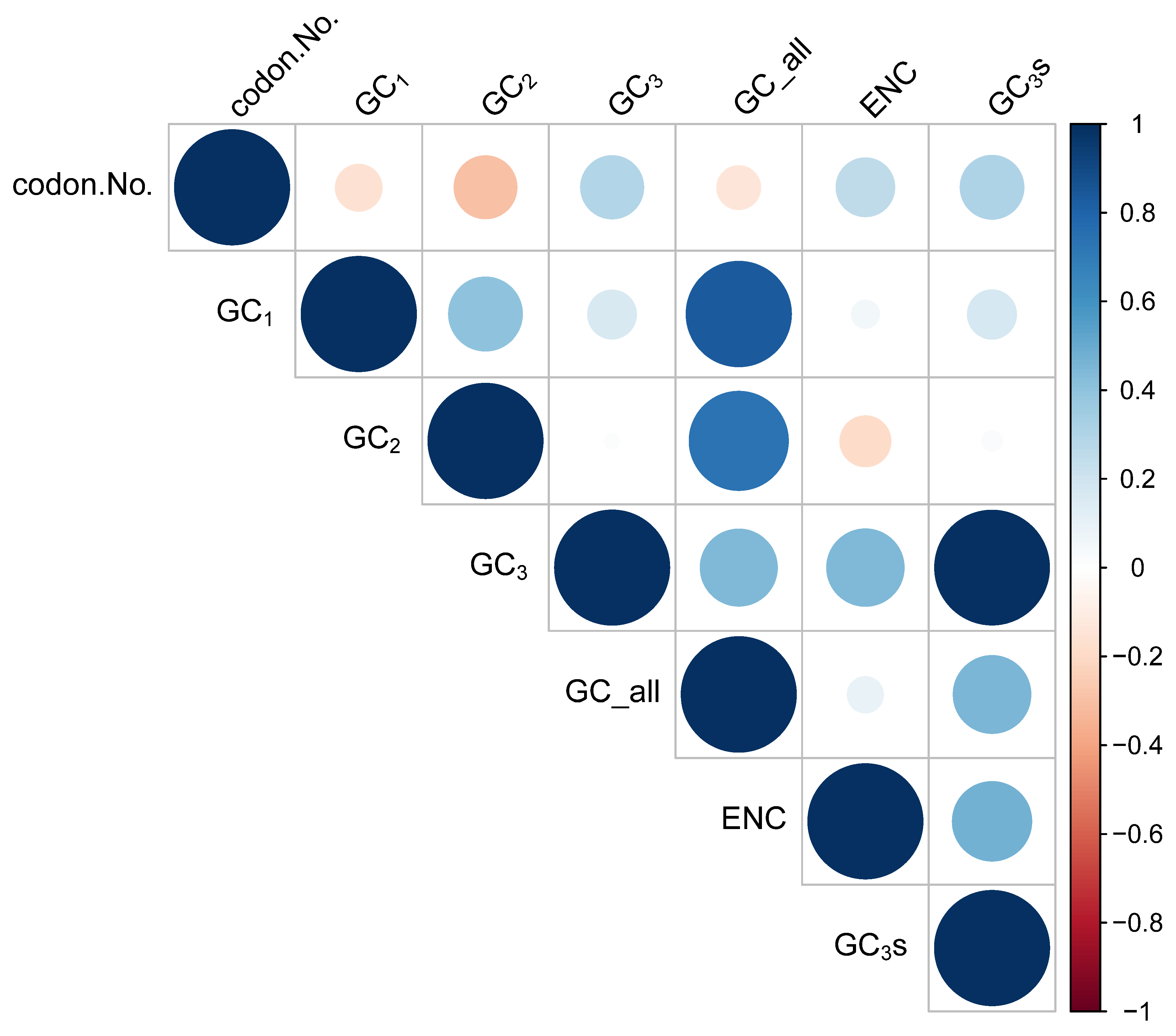
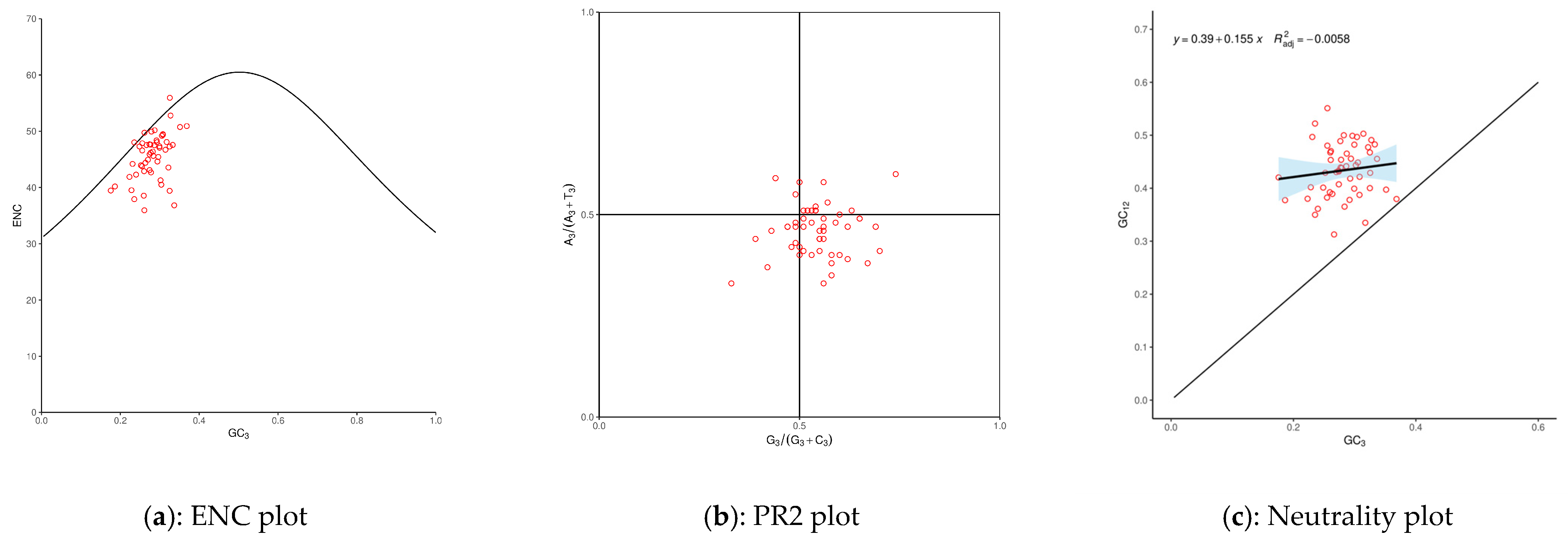
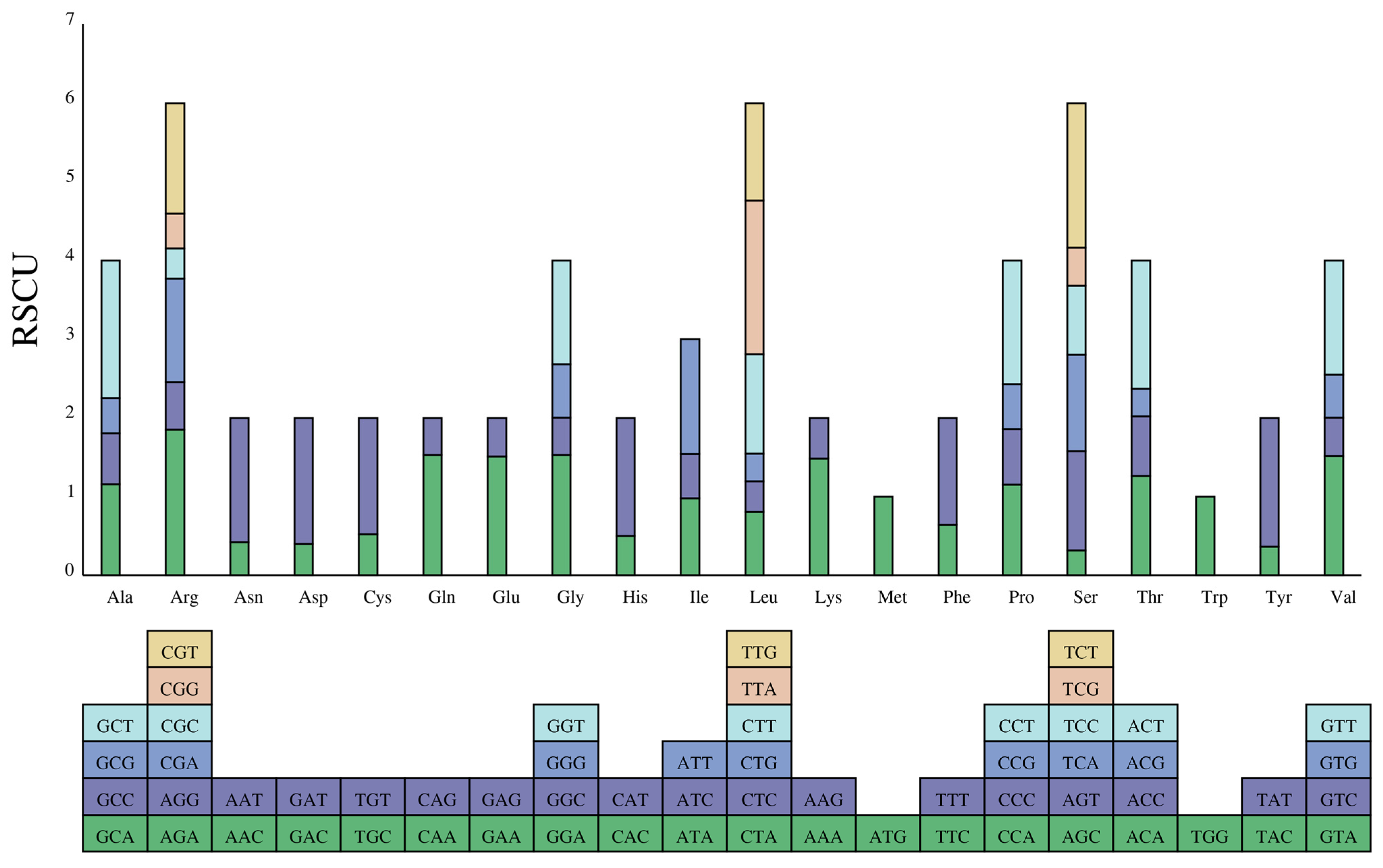
3.2. Codon Usage Bias (CUB)
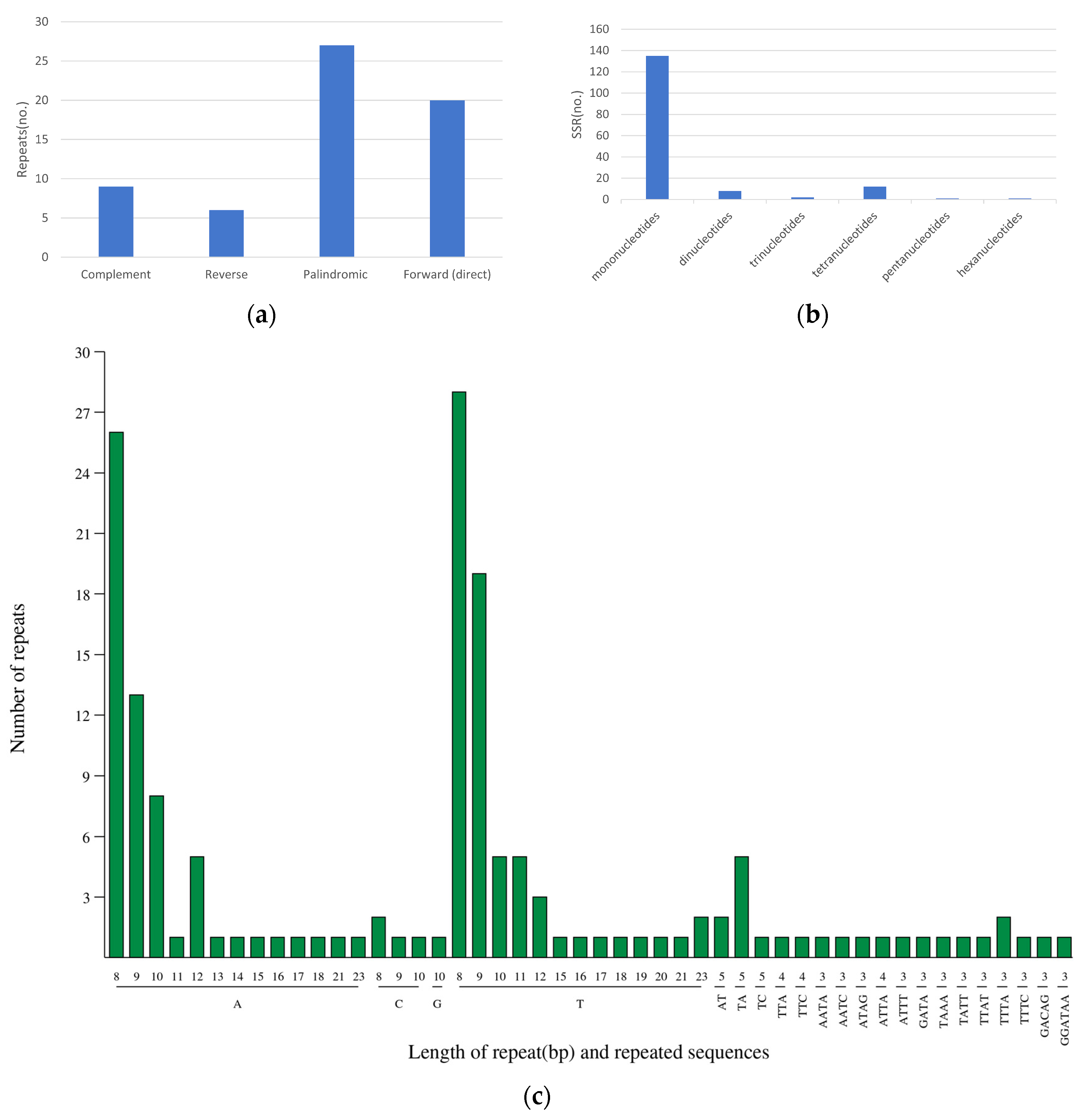
3.3. Interspersed Repeats and Simple Sequence Repeats (SSRs)
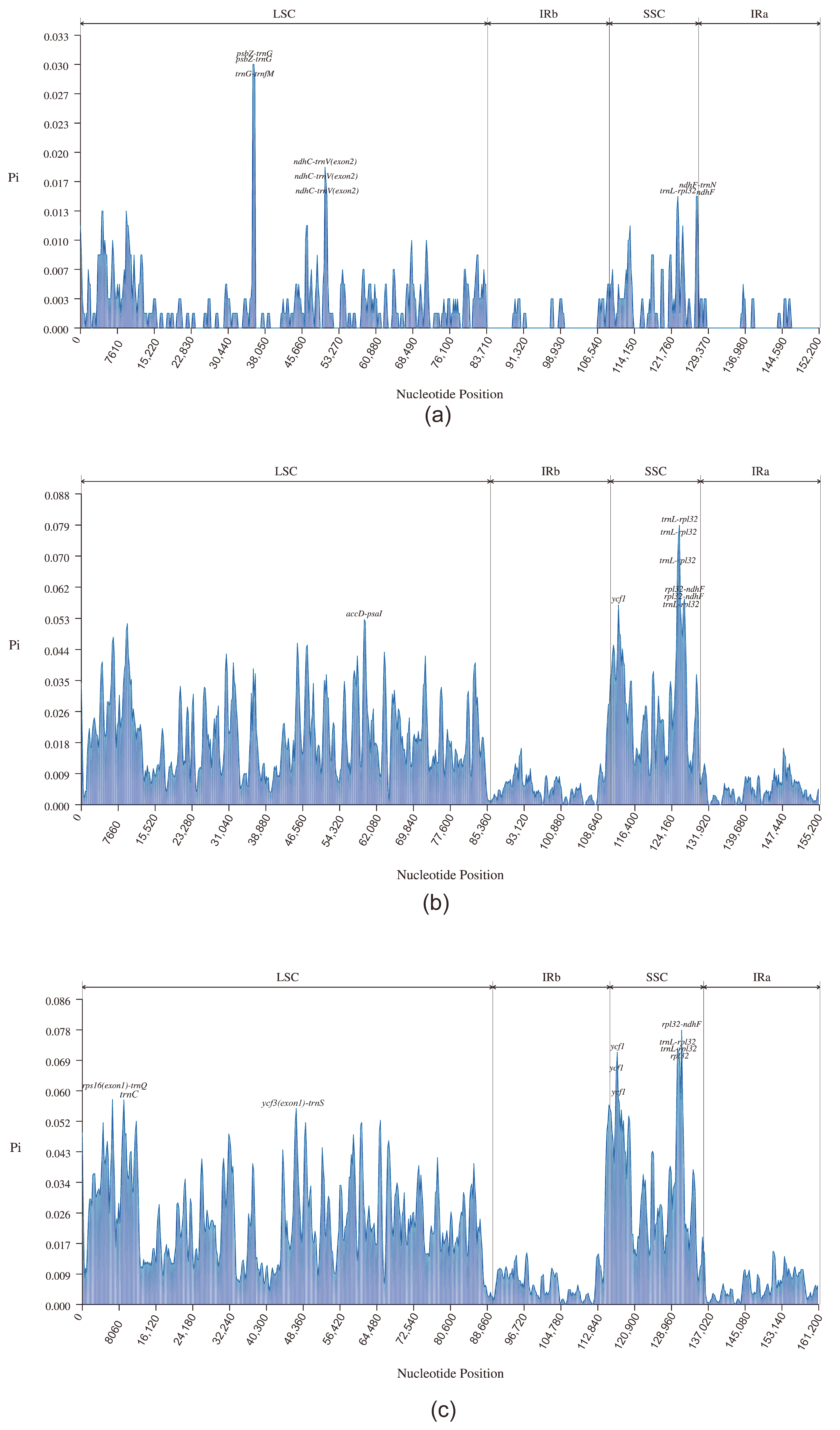
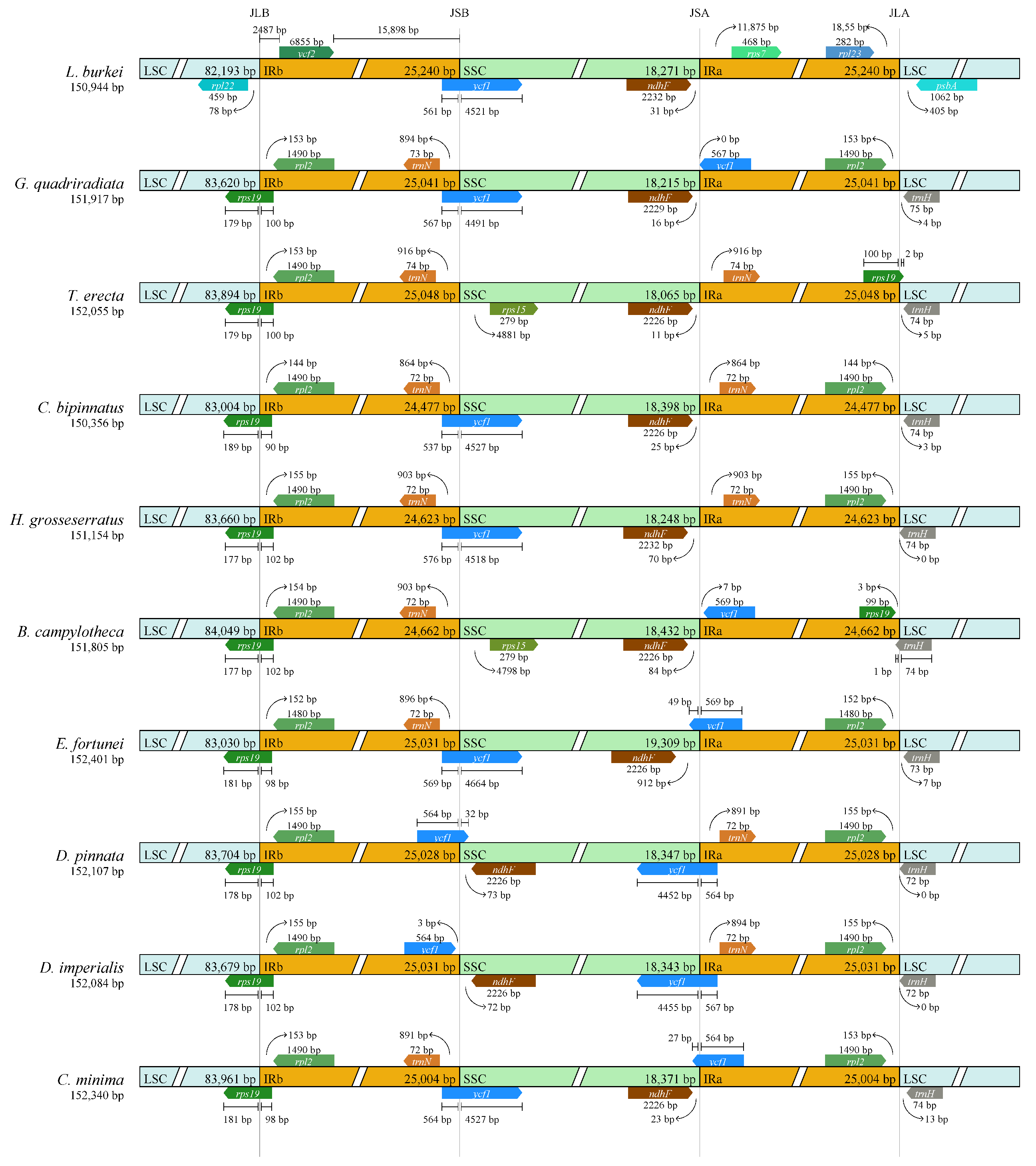
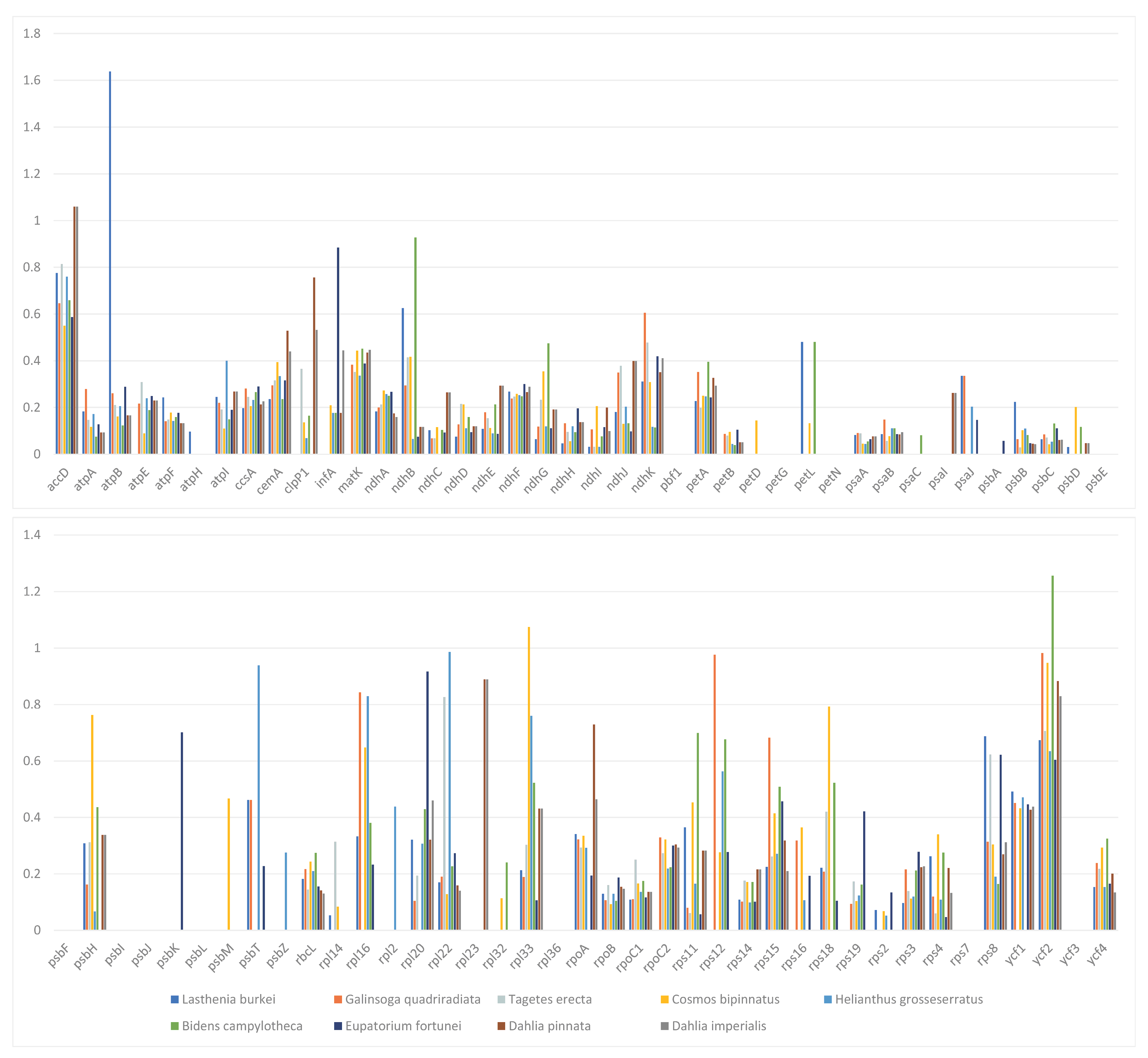
3.4. Comparative Analysis of the Chloroplast Genomes of D. imperialis and Its Related Species
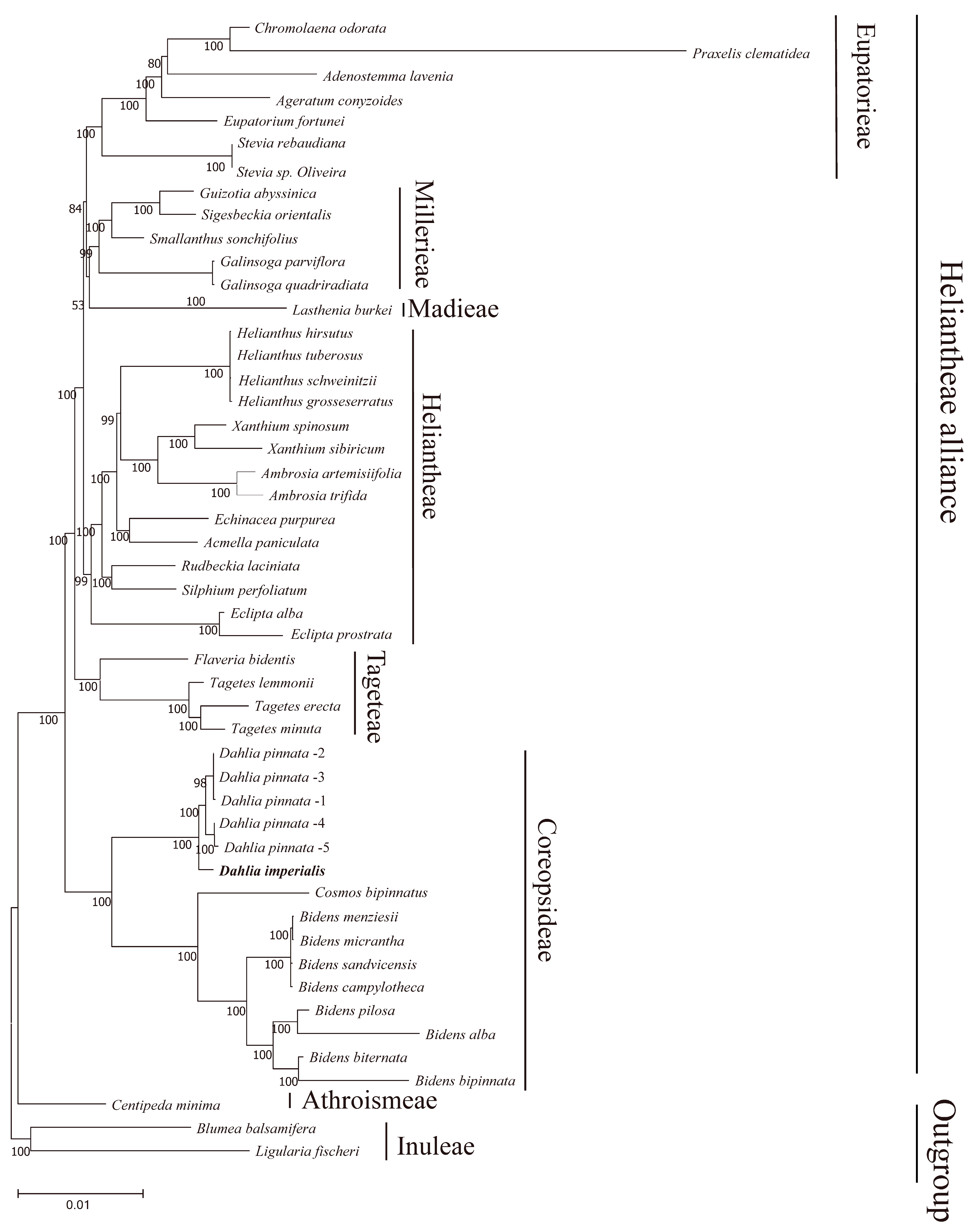
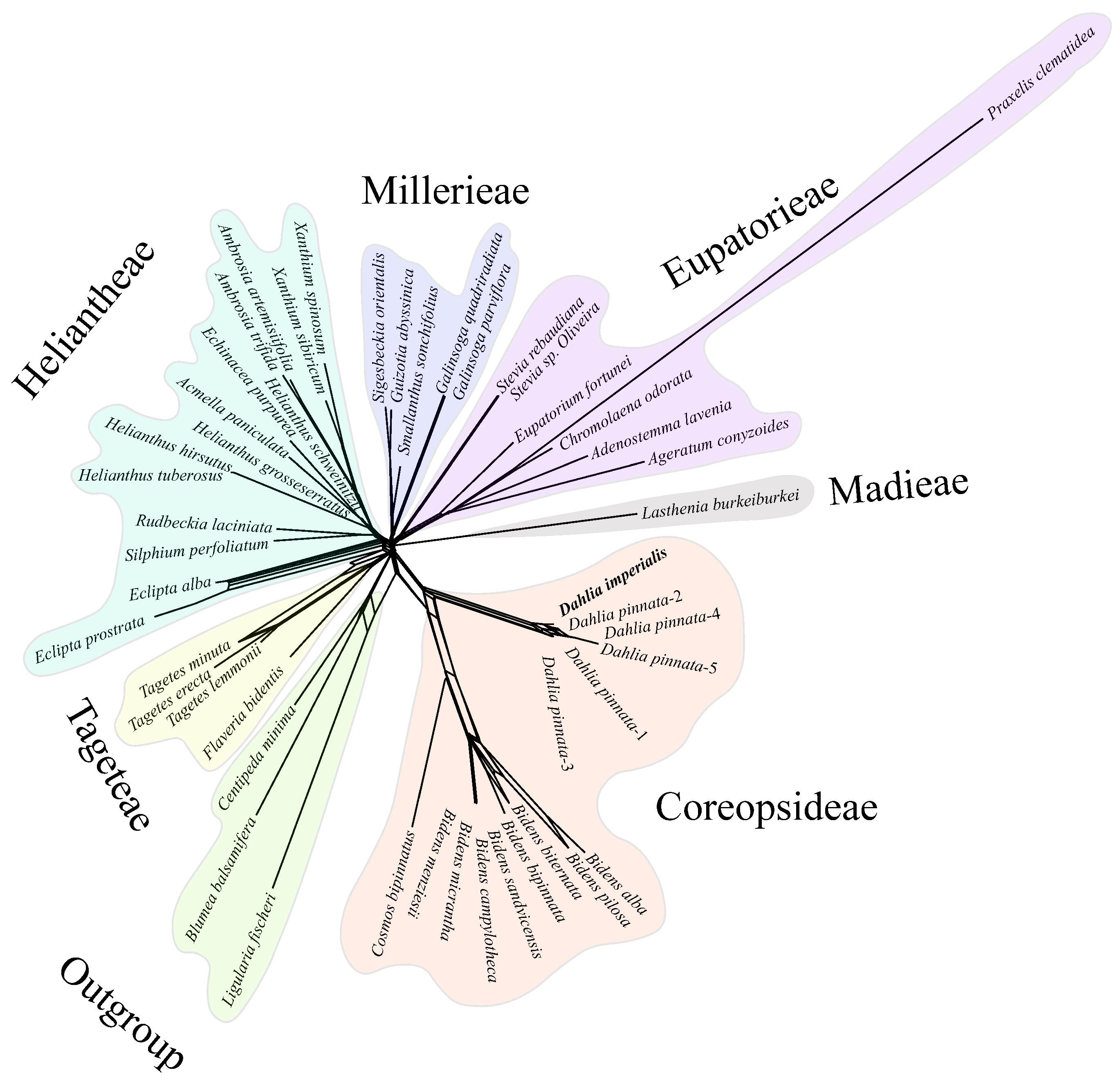
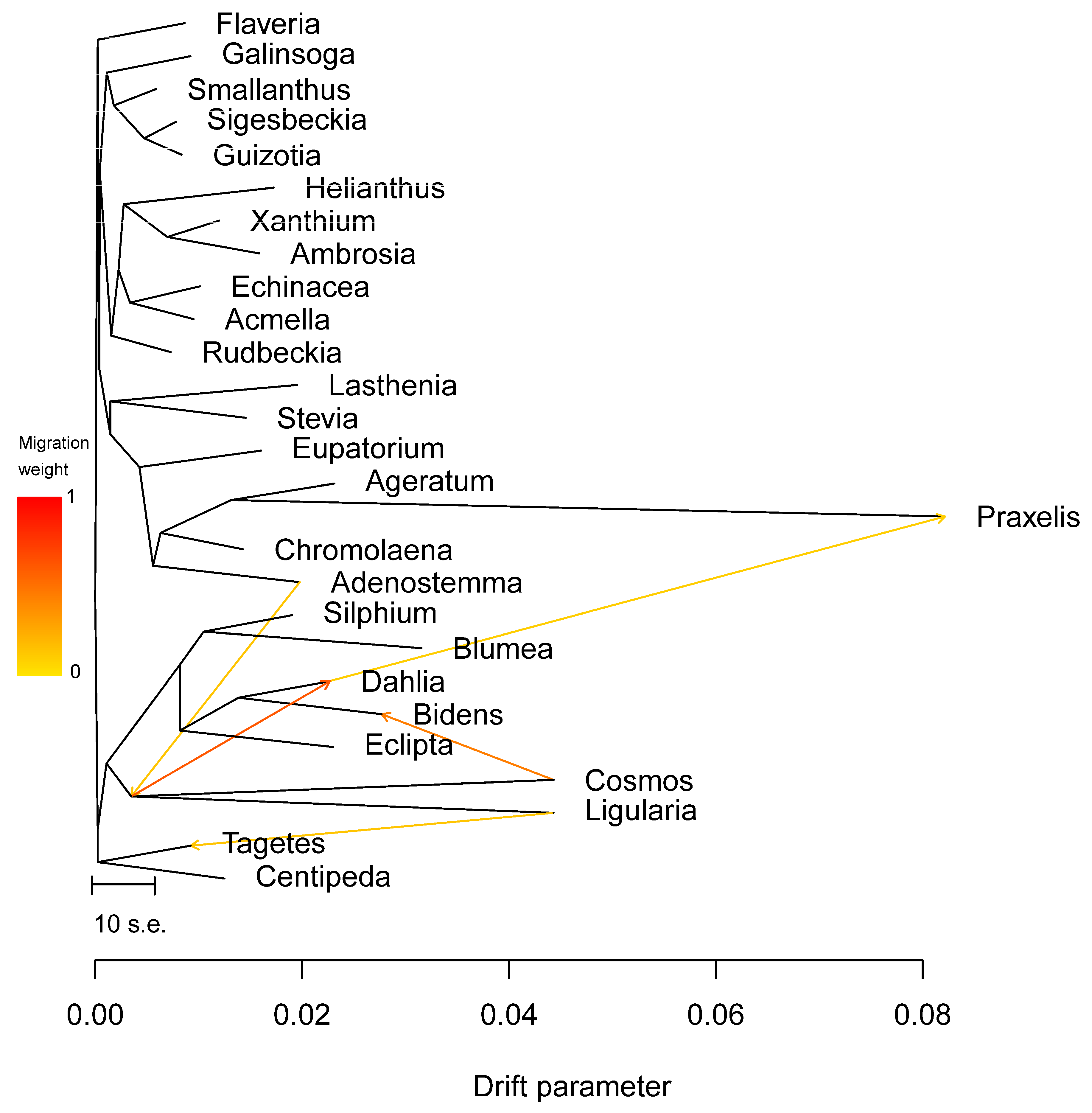
3.5. Phylogenetic Inference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Carrasco-Ortiz, M.; Munguía-Lino, G.; Castro-Castro, A.; Vargas-Amado, G.; Harker, M.; Rodríguez, A. Riqueza, distribución geográfica y estado de conservación del género Dahlia (Asteraceae) en México. Acta Bot. Mex. 2019, 126, 25. [Google Scholar] [CrossRef]
- Villaseñor, J.L.; Redonda-Martinez, R. A new species of Dahlia (Asteraceae, Coreopsideae) from the state of Oaxaca, Mexico. Phytotaxa 2018, 362, 239–243. [Google Scholar] [CrossRef]
- Yan, G.X. Overview and enlightenment of Dahlia industry development. Anhui Agric. Sci. Bull. 2020, 26, 38–39. (In Chinese) [Google Scholar]
- Whitley, G.R. The medicinal and nutritional properties of Dahlia spp. J. Ethnopharmacol. 1985, 14, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, Y.; Devaraj, B.S.; Velusamy, N.K.; Soundirarajan, P.S.; Kandaswamy, K. Phytochemical extracts of Leucas aspera and Dahlia pinnata exhibit antimicrobial properties in Escherichia coli and Enterococcus faecalis. Curr. Biotechnol. 2020, 9, 297–303. [Google Scholar] [CrossRef]
- Brighenti, F.; Casiraghi, M.C.; Canzi, E.; Ferrari, A. Effect of consumption of a ready-to-eat breakfast cereal containing inulin on the intestinal milieu and blood lipids in healthy male volunteers. Eur. J. Clin. Nutr. 1999, 53, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Hilman, A.; Harmayani, E.; Cahyanto, M.N. The potential of Gembili (Dioscorea esculenta L.) and Dahlia (Dahlia spp L.) from Indonesia as prebiotic compound. In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2021; Volume 782, p. 032109. [Google Scholar]
- Pal, S.; Kumar, A.; Chaturvedi, P.; Srivastava, R.; Tripathi, S. Determination of lethal dose for gamma rays induced mutagenesis in different cultivars of Dahlia. J. Hill Agric. 2017, 8, 279–282. [Google Scholar] [CrossRef]
- Ohno, S.; Hori, W.; Hosokawa, M.; Tatsuzawa, F.; Doi, M. Petal color is associated with leaf flavonoid accumulation in a labile bicolor flowering Dahlia (Dahlia variabilis)‘Yuino’. Hort. J. 2016, 85, 177–186. [Google Scholar] [CrossRef]
- Saar, D.E.; Polans, N.O.; Sørensen, P.D. A phylogenetic analysis of the genus Dahlia (Asteraceae) based on internal and external transcribed spacer regions of nuclear ribosomal DNA. Syst. Bot. 2003, 28, 627–639. [Google Scholar]
- Panero, J.L.; Funk, V.A. The value of sampling anomalous taxa in phylogenetic studies: Major clades of the Asteraceae revealed. Mol. Phylogenet. Evol. 2008, 47, 757–782. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, C.F.; Huang, J.X.; Ma, H. Reconstructing phylogenetic relationships among Asterinae subfamilies of Compositae using low-copy nuclear genes. Acta Bot. 2015, 50, 549. [Google Scholar]
- Zhang, C.F.; Huang, C.H.; Liu, M.; Hu, Y.; Panero, J.L.; Luebert, F.; Gao, T.G.; Ma, H. Phylotranscriptomic insights into Asteraceae diversity, polyploidy, and morphological innovation. J. Integr. Plant Biol. 2021, 63, 1273–1293. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M. The Chloroplast Genome; Springer: Dodrecht, The Netherlands, 1992; Volume 19, pp. 149–168. [Google Scholar]
- Lin, C.P.; Wu, C.S.; Huang, Y.Y.; Chaw, S.M. The complete chloroplast genome of Ginkgo biloba reveals the mechanism of inverted repeat contraction. Genome Biol. Evol. 2012, 4, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.I.; Lee, H.O.; Lee, I.H.; Kim, I.S.; Lee, S.W.; Yang, T.J.; Shim, D. Complete chloroplast genome of Pinus densiflora Siebold & Zucc. and comparative analysis with five pine trees. Forests 2019, 10, 600. [Google Scholar]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Boore, J.L. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.J.; Zhang, X.; Landis, J.B.; Sun, Y.X.; Sun, J.; Kuang, T.H.; Li, L.J.; Bashir, B.T.; Deng, T.; Sun, H.; et al. Phylogenomic and comparative analyses of Rheum (Polygonaceae, Polygonoideae). J. Integr. Plant Biol. 2022, 60, 1229–1240. [Google Scholar] [CrossRef]
- Guo, Y.Y.; Luo, Y.B.; Liu, Z.J.; Wang, X.Q. Reticulate evolution and sea-level fluctuations together drove species diversification of slipper orchids (P aphiopedilum) in South-East Asia. Mol. Ecol. 2015, 24, 2838–2855. [Google Scholar] [CrossRef]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.F.; Su, K.L.; Zhao, Y.R.; Song, Z.; Sun, J. Analysis of codon usage in the chloroplast genome of Medicago truncatula. Cao Ye Xue Bao 2015, 24, 171–179. [Google Scholar]
- Wei, L.; He, J.; Jia, X.; Qi, Q.; Liang, Z.S.; Zheng, H.; Ping, Y.; Liu, S.Y.; Sun, J.C. Analysis of codon usage bias of mitochondrial genome in Bombyx mori and its relation to evolution. BMC Evol. Biol. 2014, 14, 262. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, Q.; Xu, L.; Gao, H.; Liu, L.; Zhou, X. CPJSdraw: Analysis and visualization of junction sites of chloroplast genomes. PeerJ 2023, 11, e15326. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Estimating Phylogenetic Trees and Networks Using SplitsTree 4. Manuscript in Preparation, Software. 2005. Available online: https://www.splitstree.org (accessed on 20 September 2023).
- Pickrell, J.; Pritchard, J. Inference of population splits and mixtures from genome-wide allele frequency data. Nature Preced. 2012, 8, e1002967. [Google Scholar]
- Qian, W.; Yang, J.R.; Pearson, N.M.; Maclean, C.; Zhang, J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012, 8, e1002603. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Kress, W.J.; García-Robledo, C.; Uriarte, M.; Erickson, D.L. DNA barcodes for ecology, evolution, and conservation. Trends Ecol. Evol. 2015, 30, 25–35. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Shahzadi, I.; Mehmood, F.; Ali, Z.; Ahmed, I.; Mirza, B. Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 2020, 112, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, L.; Qi, J.; Zhang, L. Complete chloroplast genome sequence of Hibiscus cannabinus and comparative analysis of the Malvaceae Family. Front. Genet. 2020, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Mehmood, F.; Ali, Z.; Malik, M.S.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Comparative analyses of chloroplast genomes among three Firmiana species: Identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 2019, 19, 100199. [Google Scholar]
- Qin, Z.; Wang, Y.; Wang, Q.; Li, A.; Hou, F.; Zhang, L. Evolution analysis of simple sequence repeats in plant genome. PLoS ONE 2015, 10, e0144108. [Google Scholar] [CrossRef] [PubMed]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef]
- Gichira, A.W.; Avoga, S.; Li, Z.; Hu, G.; Wang, Q.; Chen, J. Comparative genomics of 11 complete chloroplast genomes of Senecioneae (Asteraceae) species: DNA barcodes and phylogenetics. Bot. Stud. 2019, 60, 17. [Google Scholar] [CrossRef]
- Wang, C.; Liu, J.; Su, Y.; Li, M.; Xie, X.; Su, J. Complete chloroplast genome sequence of sonchus brachyotus helps to elucidate evolutionary relationships with related species of asteraceae. BioMed Res. Int. 2021, 2021, 9410496. [Google Scholar] [CrossRef]
- Huang, C.; Liu, D.; Li, Z.A.; Molloy, D.P.; Luo, Z.F.; Su, Y.; Li, H.O.; Liu, Q.; Wang, R.Z.; Xiao, L.T. The PPR protein RARE1-mediated editing of chloroplast accD transcripts is required for fatty acid biosynthesis and heat tolerance in Arabidopsis. Plant Commun. 2023, 4, 100461. [Google Scholar] [CrossRef]
- Rogalski, M.; Schottler, M.A.; Thiele, W.; Schulze, W.X.; Bock, R. Rpl33, a nonessential plastid-encoded ribosomal protein in Tobacco, is required under cold stress conditions. Plant Cell 2008, 20, 2221–2237. [Google Scholar] [CrossRef]
- Rogalski, M.; Schottler, M.A.; Thiele, W.; Schulze, W.X.; Bock, R. Plastid phylogenomic analysis of green plants: A billion years of evolutionary history. Am. J. Bot. 2018, 105, 291–301. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Item | Describe |
|---|---|---|
| Cp genome structure | Cp genome/bp | 152,084 |
| LSC/bp | 83,679 | |
| SSC/bp | 18,343 | |
| IRa/IRb/bp | 25,031 | |
| Gene composition | Cp gene | 134 |
| tRNA | 38 | |
| rRNA | 8 | |
| Protein coding | 86 | |
| Pseudo | 2 | |
| GC content (%) | Cp gene | 38.45 |
| LSC | 35.63% | |
| SSC | 31.15% | |
| IRa/IRb | 43.01% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, S.-D.; Liu, Y.; Hao, L.-H.; Xiang, D.-Y.; Yu, W.-B.; Liang, J.; Chen, D.-F.; Niu, S.-C. Complete Chloroplast Genome Sequence of Dahlia imperialis (Asteraceae): Comparative Analysis and Phylogenetic Relationships. Horticulturae 2024, 10, 7. https://doi.org/10.3390/horticulturae10010007
Duan S-D, Liu Y, Hao L-H, Xiang D-Y, Yu W-B, Liang J, Chen D-F, Niu S-C. Complete Chloroplast Genome Sequence of Dahlia imperialis (Asteraceae): Comparative Analysis and Phylogenetic Relationships. Horticulturae. 2024; 10(1):7. https://doi.org/10.3390/horticulturae10010007
Chicago/Turabian StyleDuan, Shan-De, Yang Liu, Li-Hong Hao, Di-Ying Xiang, Wen-Bin Yu, Juan Liang, Duan-Fen Chen, and Shan-Ce Niu. 2024. "Complete Chloroplast Genome Sequence of Dahlia imperialis (Asteraceae): Comparative Analysis and Phylogenetic Relationships" Horticulturae 10, no. 1: 7. https://doi.org/10.3390/horticulturae10010007