
Genome Re-Annotation and Transcriptome Analyses of Sanghuangporus sanghuang

Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Material
2.2. Transcriptome Sequencing
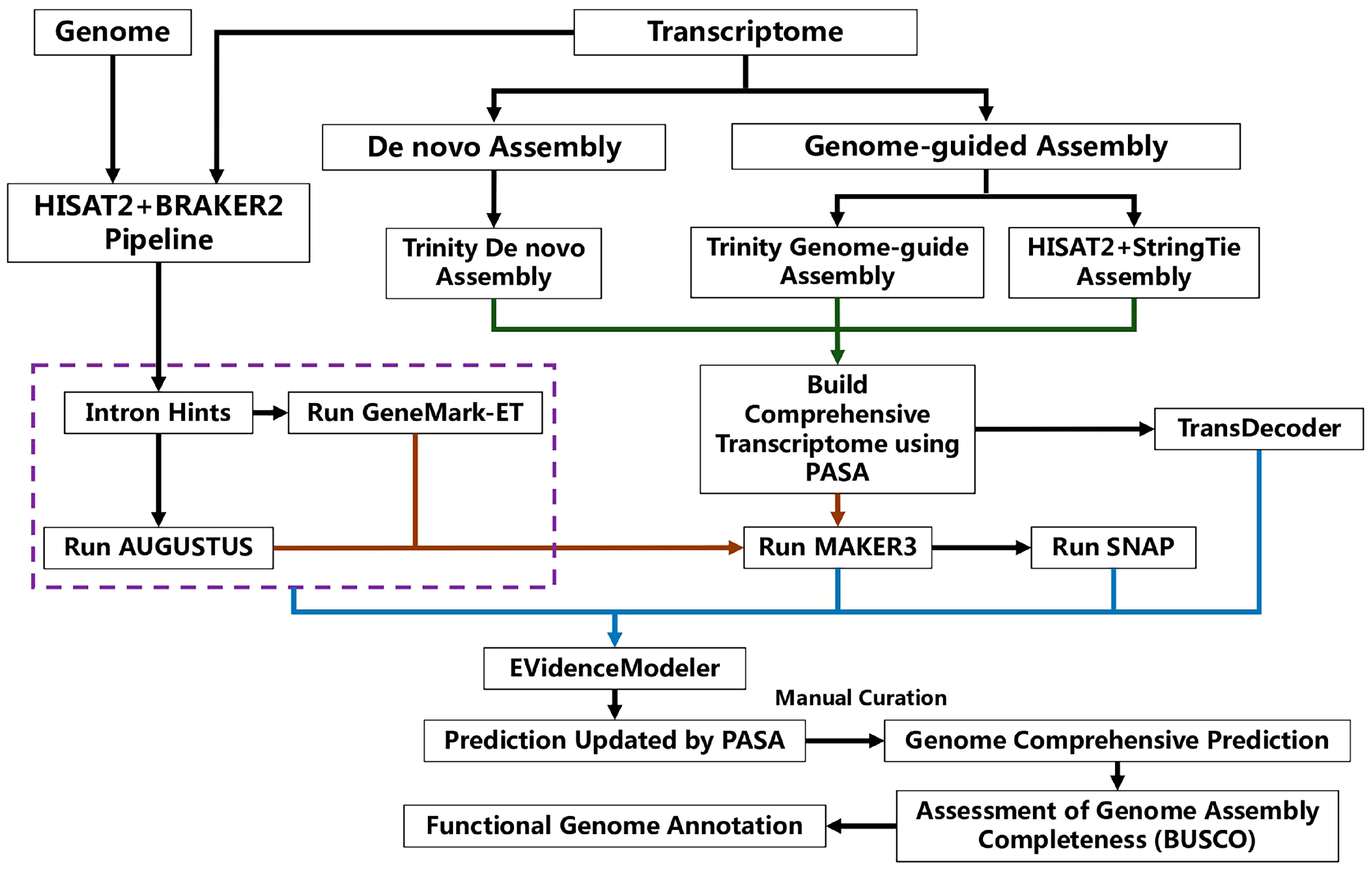
2.3. Genome Assembly and Prediction
2.4. Transcriptome Assembly
2.5. Gene Annotation
2.6. Phylogenetic Analysis of Cytochrome P450 (CYP)
3. Results
3.1. Transcriptome Sequencing and Updated Genome Assembly
3.2. Transcriptome Assembly
3.3. Comparison of MS2_V1 and MS2_V2 Assemblies
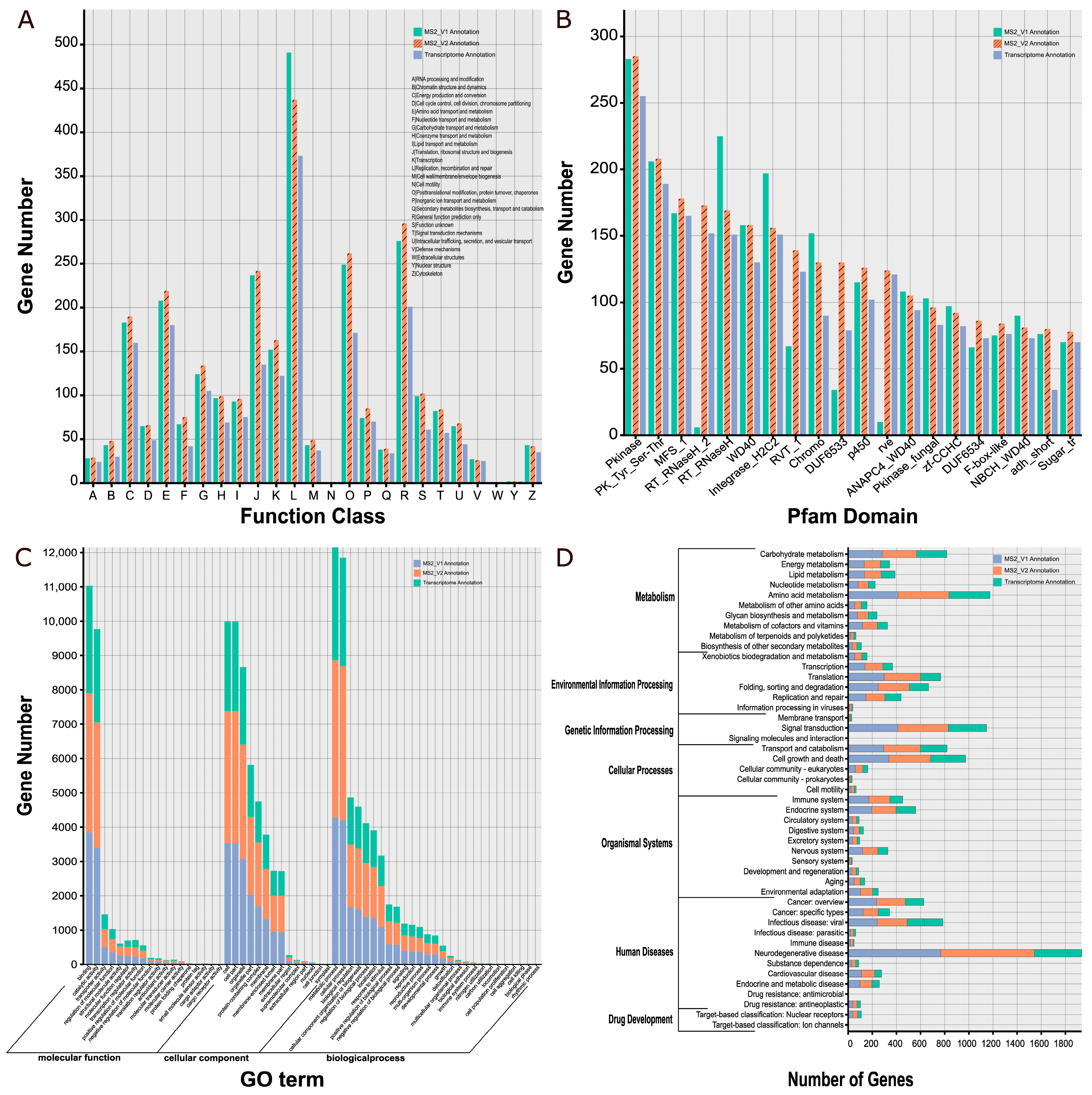
3.4. Functional Annotation of Protein-Coding Genes
3.5. Identification of Genes Involved in Synthesis of Secondary Metabolites
3.5.1. Terpenoid Biosynthesis
3.5.2. Polysaccharide Biosynthesis
3.5.3. Ubiquinone and Other Terpenoid Quinone Biosynthesis
3.5.4. Steroid Biosynthesis
3.5.5. Flavonoid Biosynthesis
3.6. CAZyme
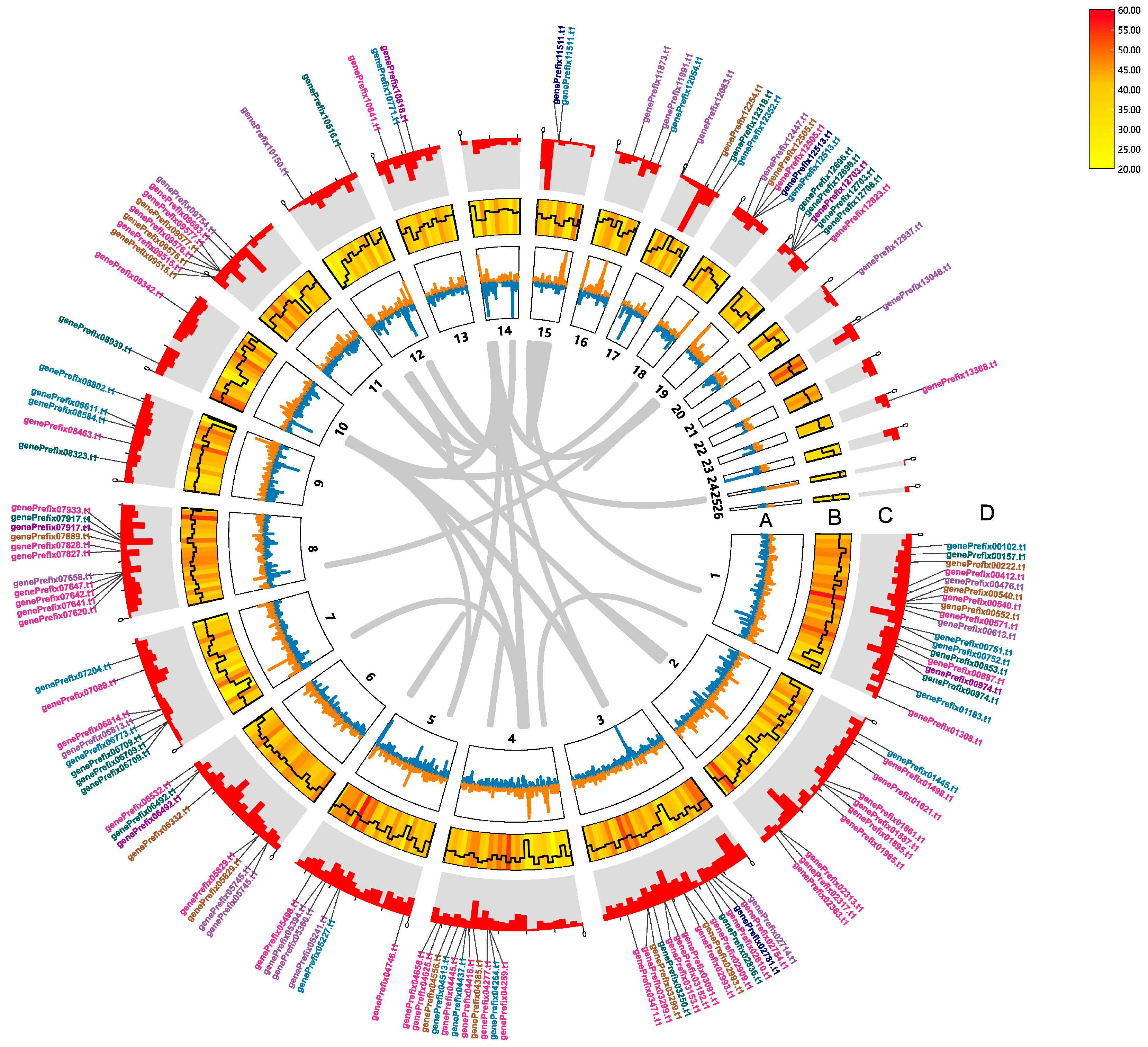
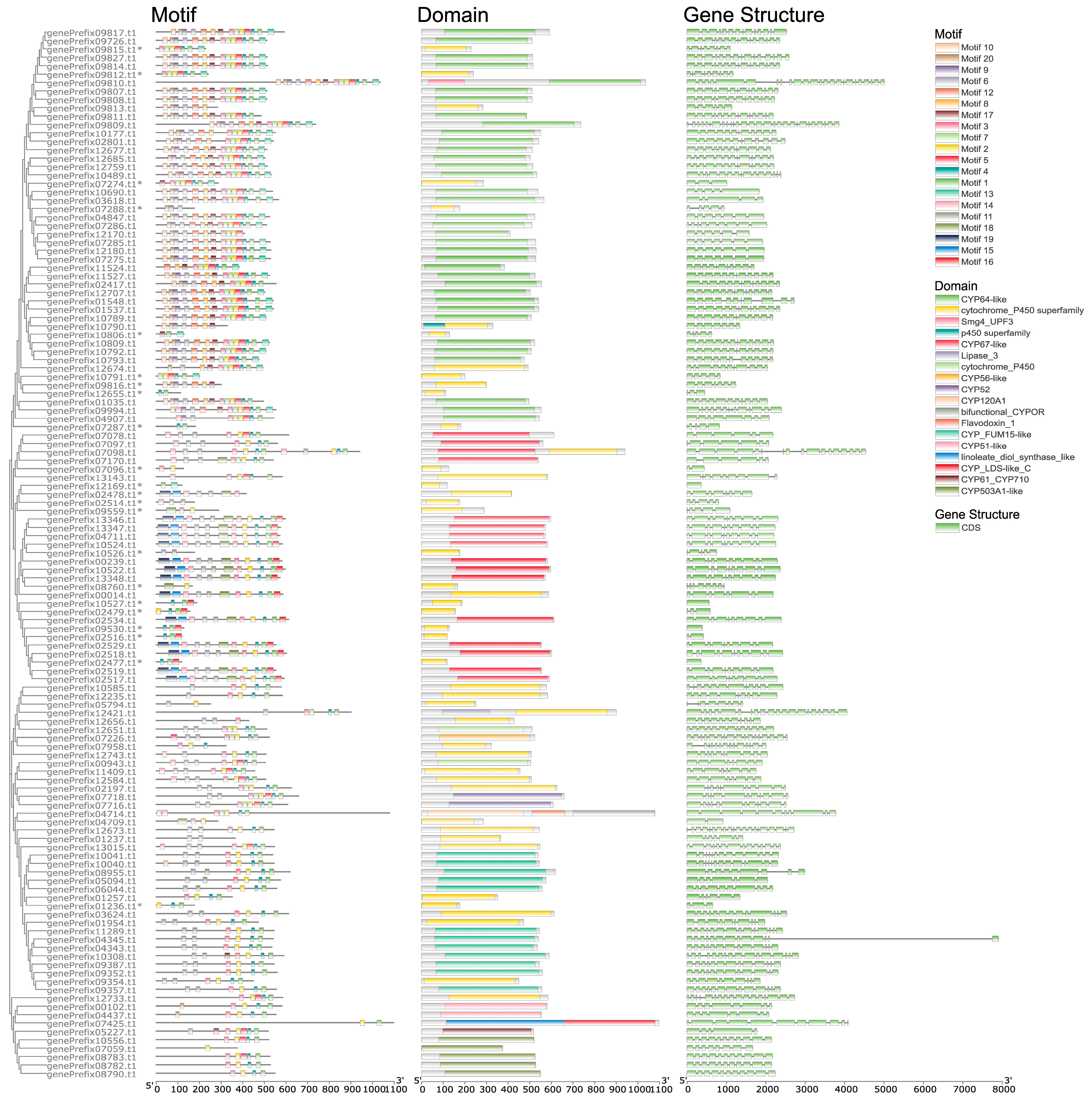
3.7. CYP
3.8. Gene Cluster of Secondary Metabolites
4. Discussion
4.1. Methodology of Genome Re-Annotation
4.2. Substantial Transcription of Genes Related to Secondary Metabolite Biosynthesis
4.3. Identification of Genes Related to Flavonoid Synthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wu, F.; Zhou, L.W.; Yang, Z.L.; Bau, T.; Li, T.H.; Dai, Y.C. Resource diversity of Chinese macrofungi: Edible, medicinal and poisonous species. Fungal Divers. 2019, 98, 1–76. [Google Scholar] [CrossRef]
- Zhou, L.W.; Vlasák, J.; Decock, C.; Assefa, A.; Stenlid, J.; Abate, D.; Wu, S.H.; Dai, Y.C. Global diversity and taxonomy of the Inonotus linteus complex (Hymenochaetales, Basidiomycota): Sanghuangporus gen. nov., Tropicoporus excentrodendri and T. guanacastensis gen. et spp. nov., and 17 new combinations. Fungal Divers. 2016, 77, 335–347. [Google Scholar] [CrossRef]
- Chen, J.H.; Shen, S.; Zhou, L.W. Modeling current geographic distribution and future range shifts of Sanghuangporus under multiple climate change scenarios in China. Front. Microbiol. 2022, 13, 1064451. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Liu, S.L.; Jiang, J.H.; Zhou, L.W. Addressing widespread misidentifications of traditional medicinal mushrooms in Sanghuangporus (Basidiomycota) through ITS barcoding and designation of reference sequences. IMA Fungus 2021, 12, 10. [Google Scholar] [CrossRef]
- Wu, F.; Zhou, L.W.; Vlasák, J.; Dai, Y.C. Global diversity and systematics of Hymenochaetaceae with poroid hymenophore. Fungal Divers. 2022, 113, 1–192. [Google Scholar] [CrossRef]
- Wu, S.H.; Wei, C.L.; Chang, C.C. Sanghuangporus vitexicola sp. nov. (Hymenochaetales, Basidiomycota) from tropical Taiwan. Phytotaxa 2020, 475, 43–51. [Google Scholar] [CrossRef]
- Zhou, L.W.; Ghobad-Nejhad, M.; Tian, X.M.; Wang, Y.F.; Wu, F. Current status of ‘Sanghuang’ as a group of medicinal mushrooms and their perspective in industry development. Food Rev. Int. 2022, 38, 589–607. [Google Scholar] [CrossRef]
- Liu, X.; Hou, R.; Xu, K.; Chen, L.; Wu, X.; Lin, W.; Zheng, M.; Fu, J. Extraction, characterization and antioxidant activity analysis of the polysaccharide from the solid-state fermentation substrate of Inonotus hispidus. Int. J. Biol. Macromol. 2019, 123, 468–476. [Google Scholar] [CrossRef]
- Gründemann, C.; Arnhold, M.; Meier, S.; Bäcker, C.; Garcia-Käufer, M.; Grunewald, F.; Steinborn, C.; Klemd, A.M.; Wille, R.; Huber, R.; et al. Effects of Inonotus hispidus extracts and compounds on human immunocompetent cells. Planta Med. 2016, 82, 1359–1367. [Google Scholar] [CrossRef]
- Angelini, P.; Girometta, C.; Tirillini, B.; Moretti, S.; Covino, S.; Cipriani, M.; D’Ellena, E.; Angeles, G.; Federici, E.; Savino, E.; et al. A comparative study of the antimicrobial and antioxidant activities of Inonotus hispidus fruit and their mycelia extracts. Int. J. Food Prop. 2019, 22, 768–783. [Google Scholar] [CrossRef]
- Zhou, L.W. Systematics is crucial for the traditional Chinese medicinal studies and industry of macrofungi. Fungal Biol. Rev. 2020, 34, 10–12. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, X.H.; Dai, Y.C.; Zhou, L.W.; Cai, W.M.; Guo, L.D.; Cui, B.K.; Li, N.; Lei, P.; Li, C.T.; et al. Sanghuang industry in China: Current status, challenges and perspectives—The Qiandao Lake declaration for sanghuang industry development. Mycosystema 2023, 42, 855–873. [Google Scholar]
- Mancheron, A.; Uricaru, R.; Rivals, E. An alternative approach to multiple genome comparison. Nucleic Acids Res. 2011, 39, e101. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.H.; Wu, S.H.; Zhou, L.W. The first whole genome sequencing of Sanghuangporus sanghuang provides insights into its medicinal application and evolution. J. Fungi 2021, 7, 787. [Google Scholar] [CrossRef]
- Schrimpe-Rutledge, A.C.; Jones, M.B.; Chauhan, S.; Purvine, S.O.; Sanford, J.A.; Monroe, M.E.; Brewer, H.M.; Payne, S.H.; Ansong, C.; Frank, B.C.; et al. Comparative omics-driven genome annotation refinement: Application across Yersiniae. PLoS ONE 2012, 7, e33903. [Google Scholar] [CrossRef]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef]
- Lu, M.Y.J.; Fan, W.L.; Wang, W.F.; Chen, T.C.; Tang, Y.C.; Chu, F.H.; Chang, T.T.; Wang, S.Y.; Li, M.Y.; Chen, Y.H.; et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA 2014, 111, 4743–4752. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, X.; Yang, Y.L.; Xing, Y.M.; Zhang, Q.; Li, J.M.; Ma, K.; Liu, H.W.; Guo, S.X. Genomic and transcriptomic analyses reveal differential regulation of diverse terpenoid and polyketides secondary metabolites in Hericium erinaceus. Sci. Rep. 2017, 7, 10151. [Google Scholar] [CrossRef]
- Yang, L.; Guan, R.; Shi, Y.; Ding, J.; Dai, R.; Ye, W.; Xu, K.; Chen, Y.; Shen, L.; Liu, Y.; et al. Comparative genome and transcriptome analysis reveal the medicinal basis and environmental adaptation of artificially cultivated Taiwanofungus camphoratus. Mycol. Prog. 2018, 17, 871–883. [Google Scholar] [CrossRef]
- Shao, Y.; Guo, H.; Zhang, J.; Liu, H.; Wang, K.; Zuo, S.; Xu, P.; Xia, Z.; Zhou, Q.; Zhang, H.; et al. The genome of the medicinal macrofungus Sanghuang provides insights into the synthesis of diverse secondary metabolites. Front. Microbiol. 2020, 10, 3035. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, J.; Jiang, H.; Wang, G.; Wang, Y. Deep sequencing of the Sanghuangporus vaninii transcriptome reveals dynamic landscapes of candidate genes involved in the biosynthesis of active compounds. Arch. Microbiol. 2021, 203, 2315–2324. [Google Scholar] [CrossRef] [PubMed]
- Kimes, N.E.; Callaghan, A.V.; Aktas, D.F.; Smith, W.L.; Sunner, J.; Golding, B.T.; Drozdowska, M.; Hazen, T.C.; Suflita, J.M.; Morris, P.J. Metagenomic analysis and metabolite profiling of deep-sea sediments from the Gulf of Mexico following the Deepwater Horizon oil spill. Front. Microbiol. 2013, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Hoff, K.J.; Lomsadze, A.; Borodovsky, M.; Stanke, M. Whole-genome annotation with BRAKER. Methods Mol. Biol. 2019, 1962, 65–95. [Google Scholar] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 5–60. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, J.; Sun, L.; Bonito, G.; Wang, J.; Cui, W.; Fu, Y.; Li, Y. Genome sequencing illustrates the genetic basis of the pharmacological properties of Gloeostereum incarnatum. Genes 2019, 10, 188. [Google Scholar] [CrossRef]
- Duan, Y.; Han, H.; Qi, J.; Gao, J.-m.; Xu, Z.; Wang, P.; Zhang, J.; Liu, C. Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genom. 2022, 23, 314. [Google Scholar] [CrossRef]
- Yu, F.; Song, J.; Liang, J.; Wang, S.; Lu, J. Whole genome sequencing and genome annotation of the wild edible mushroom, Russula griseocarnosa. Genomics 2020, 112, 603–614. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, L.; Hu, H.; Cai, M.; Liang, X.; Li, X.; Zhang, Z.; Xie, Y.; Xiao, C.; Chen, S.; et al. Whole-genome assembly of Ganoderma leucocontextum (Ganodermataceae, Fungi) discovered from the Tibetan Plateau of China. G3-Genes Genom. Genet. 2021, 11, jkab337. [Google Scholar] [CrossRef]
- Min, B.; Park, H.; Jang, Y.; Kim, J.-J.; Kim, K.H.; Pangilinan, J.; Lipzen, A.; Riley, R.; Grigoriev, I.V.; Spatafora, J.W.; et al. Genome sequence of a white rot fungus Schizopora paradoxa KUC8140 for wood decay and mycoremediation. J. Biotechnol. 2015, 211, 42–43. [Google Scholar] [CrossRef]
- Miyauchi, S.; Kiss, E.; Kuo, A.; Drula, E.; Kohler, A.; Sanchez-Garcia, M.; Morin, E.; Andreopoulos, B.; Barry, K.W.; Bonito, G.; et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat. Commun. 2020, 11, 5125. [Google Scholar] [CrossRef]
- Caballero, J.R.I.; Ata, J.P.; Leddy, K.A.; Glenn, T.C.; Kieran, T.J.; Klopfenstein, N.B.; Kim, M.-S.; Stewart, J.E. Genome comparison and transcriptome analysis of the invasive brown root rot pathogen, Phellinus noxius, from different geographic regions reveals potential enzymes associated with degradation of different wood substrates. Fungal Biol. 2020, 124, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Ren, J.; Yu, M.S.; Kim, H.; Kim, W.Y.; Shen, J.; Yoo, S.M.; Eyun, S.I.; Na, D. Construction of a tunable promoter library to optimize gene expression in Methylomonas sp. Dh-1, a methanotroph, and its application to cadaverine production. Biotechnol. Biofuels 2021, 14, 228. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kim, J.-E.; Lee, Y.-W.; Son, H. Fungal cytochrome P450s and the P450 complement (CYPome) of Fusarium graminearum. Toxins 2018, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Xiang, W.; Wen, L.; Lu, W.; Shi, Y.; Liu, Y.; Li, Z. Genome-wide identification, characterization and expression analysis of bes1 gene family in tomato. BMC Plant Biol. 2021, 21, 161. [Google Scholar] [CrossRef]
- Liu, K.; Xiao, X.; Wang, J.; Chen, C.Y.O.; Hu, H. Polyphenolic composition and antioxidant, antiproliferative, and antimicrobial activities of mushroom Inonotus sanghuang. LWT-Food Sci. Technol. 2017, 82, 154–161. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | MS2_V1 | MS2_V2 |
|---|---|---|
| Number of protein-coding genes | 10,913 | 13,531 |
| Total length of protein-coding genes (bp) | 21,053,817 | 21,409,391 |
| Average length of protein-coding genes (bp) | 1929.24 | 1582.25 |
| Average exon length (bp) | 267.00 | 236.43 |
| Average CDS length (bp) | 1559.82 | 1266.30 |
| Complete BUSCOs (C) | 11.7% (207) | 92.8% (1637) |
| Complete and single-copy BUSCOs (S) | 11.7% (207) | 92.5% (1632) |
| Complete and duplicated BUSCOs (D) | 0% (0) | 0.3% (5) |
| Total BUSCO groups searched | 1764 | 1764 |
| Database | MS2_V1 | MS2_V2 | Transcriptome |
|---|---|---|---|
| NR | 6033 (55.38%) | 6404 (47.33%) | 4480 (33.11%) |
| Swiss-Prot | 5378 (49.28%) | 5681 (41.99%) | 4103 (30.32%) |
| KOG | 2381 (21.82%) | 2444 (18.06%) | 1770 (13.08%) |
| eggNOG | 8362 (76.63%) | 9215 (68.10%) | 6140 (45.38%) |
| Interproscan | 8055 (73.83%) | 8828 (65.24%) | 6083 (44.96%) |
| Pfam | 7968 (73.01%) | 7968 (58.89%) | 5583 (41.26%) |
| GO | 6387 (58.53%) | 6910 (51.07%) | 4939 (36.50%) |
| KEGG | 3743 (34.30%) | 3972 (29.35%) | 2840 (20.99%) |
| CAZymes | 401 (3.67%) | 448 (3.31%) | 335 (2.48%) |
| FTFD | 321 (2.94%) | 358 (2.65%) | 296 (2.19%) |
| Total number | 10,913 | 13,531 | 13,531 |
| Characteristic | Item | MS2_V1 | MS2_V2 | Transcriptome |
|---|---|---|---|---|
| Gene involved in the pathway of medicinal metabolites | Terpenoid backbone biosynthesis | 16 | 18 | 14 |
| Ubiquinone and other terpenoid-quinone biosynthesis | 13 | 17 | 12 | |
| Sesquiterpenoid and triterpenoid biosynthesis | 3 | 3 | 3 | |
| Polysaccharide biosynthesis | 53 | 52 | 35 | |
| Steroid biosynthesis | 19 | 19 | 16 | |
| Uridine diphosphate glucose biosynthesis | 14 | 15 | 14 | |
| Phenylpropanoid biosynthesis | 5 | 5 | 5 | |
| Gene encoding CAZymes | CBM | 22 | 30 | 19 |
| CE | 30 | 49 | 21 | |
| GH | 187 | 193 | 156 | |
| GT | 55 | 57 | 56 | |
| PL | 27 | 29 | 21 | |
| AA | 79 | 89 | 61 | |
| Sum | 400 | 447 | 334 | |
| Gene encoding cytochromes P450 | Pisatin demethylase-like | 7 | 6 | 5 |
| P450, CYP52 | 8 | 7 | 7 | |
| E-class P450, group IV | 6 | 7 | 4 | |
| E-class P450, group I | 71 | 75 | 64 | |
| Undetermined | 21 | 32 | 23 | |
| Sum | 119 | 127 | 103 | |
| Gene cluster of secondary metabolites | terpene | 12 | 11 | -- |
| T1PKS | 4 | 4 | -- | |
| NRPS | 1 | 0 | -- | |
| NRPS-like | 4 | 5 | -- | |
| Sum | 21 | 20 | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Z.-Q.; Jiang, J.-H.; Li, C.-T.; Li, Y.; Zhou, L.-W. Genome Re-Annotation and Transcriptome Analyses of Sanghuangporus sanghuang. J. Fungi 2023, 9, 505. https://doi.org/10.3390/jof9050505
Shen Z-Q, Jiang J-H, Li C-T, Li Y, Zhou L-W. Genome Re-Annotation and Transcriptome Analyses of Sanghuangporus sanghuang. Journal of Fungi. 2023; 9(5):505. https://doi.org/10.3390/jof9050505
Chicago/Turabian StyleShen, Zi-Qi, Ji-Hang Jiang, Chang-Tian Li, Yu Li, and Li-Wei Zhou. 2023. "Genome Re-Annotation and Transcriptome Analyses of Sanghuangporus sanghuang" Journal of Fungi 9, no. 5: 505. https://doi.org/10.3390/jof9050505