
The First Whole Genome Sequencing of Agaricus bitorquis and Its Metabolite Profiling

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strain and Strain Culture
2.2. Genome Sequencing, De Novo Assembly, and Annotation
2.2.1. Extraction of Genome DNA
2.2.2. Sequencing and De Novo Assembly
2.2.3. Gene Prediction and Annotation
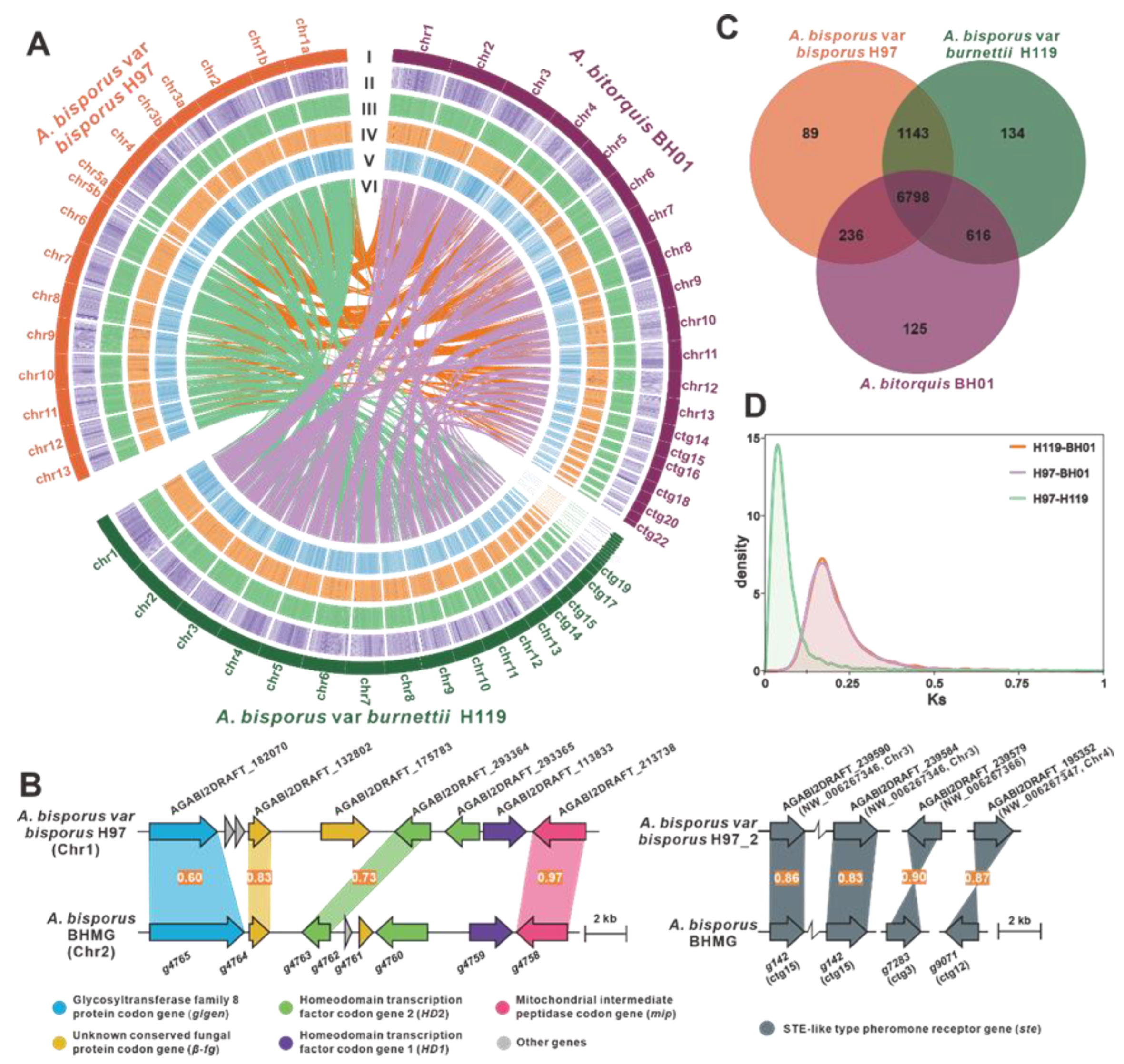
2.3. Comparative Genomics Analysis
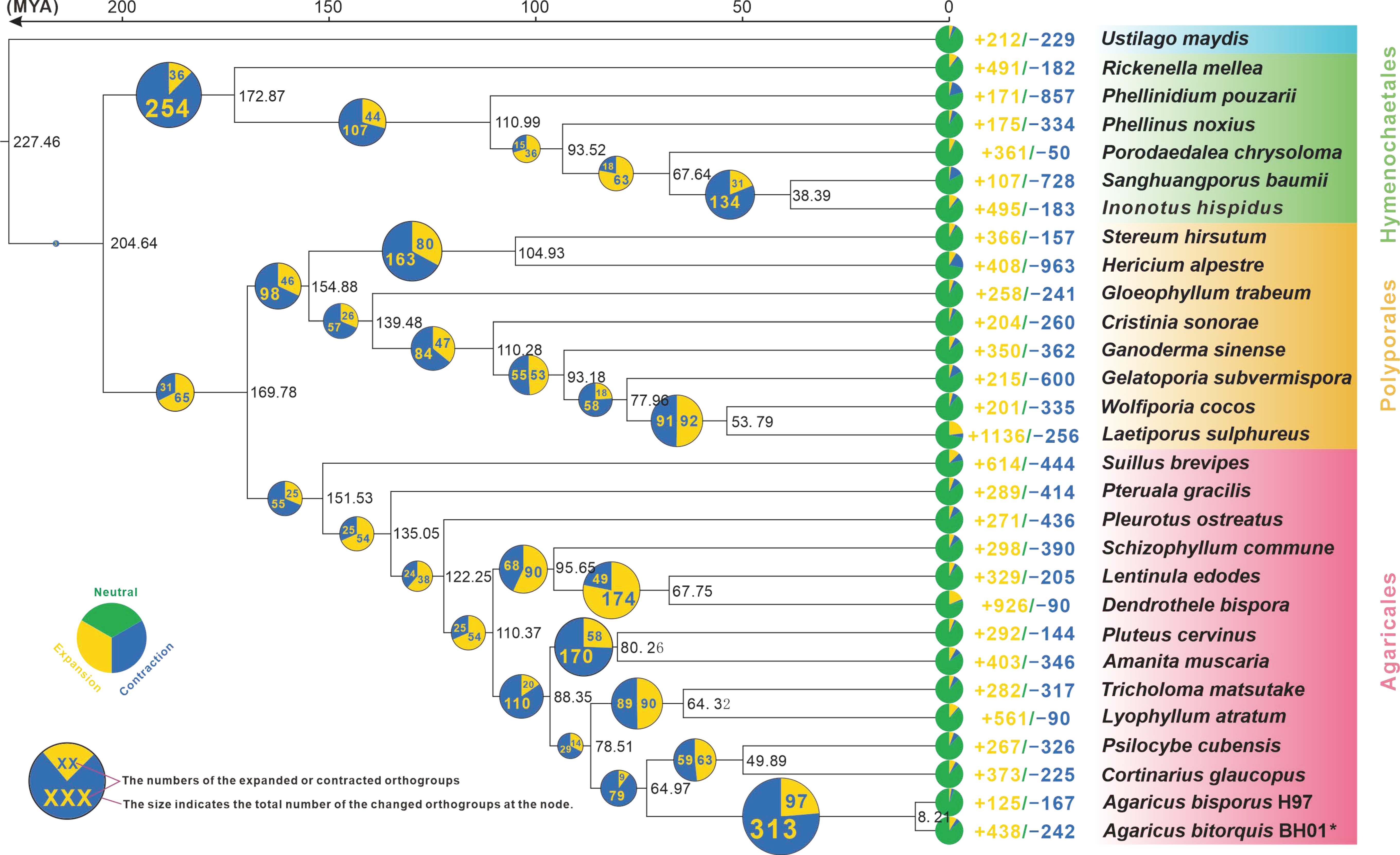
2.4. Phylogenomic Analysis
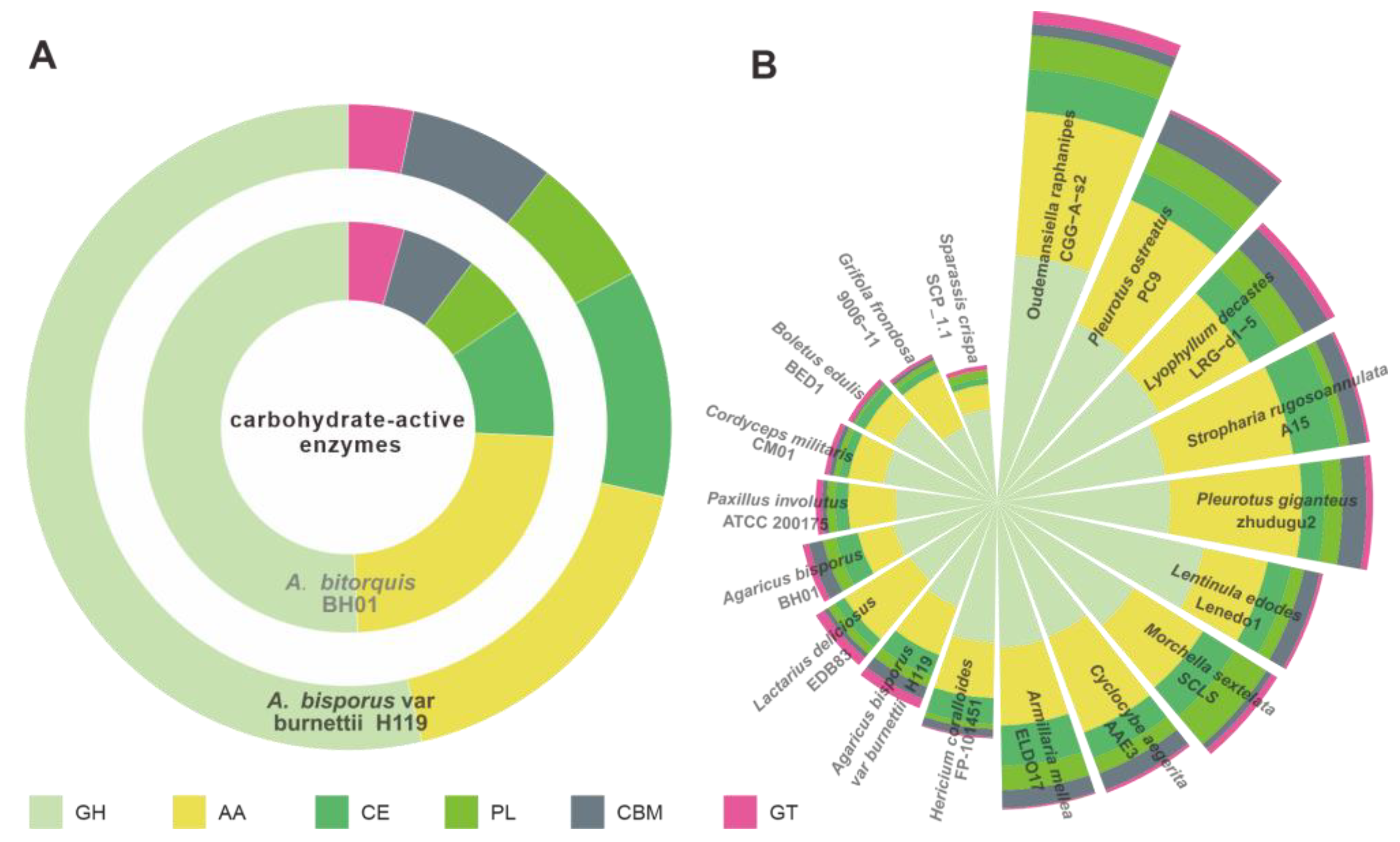
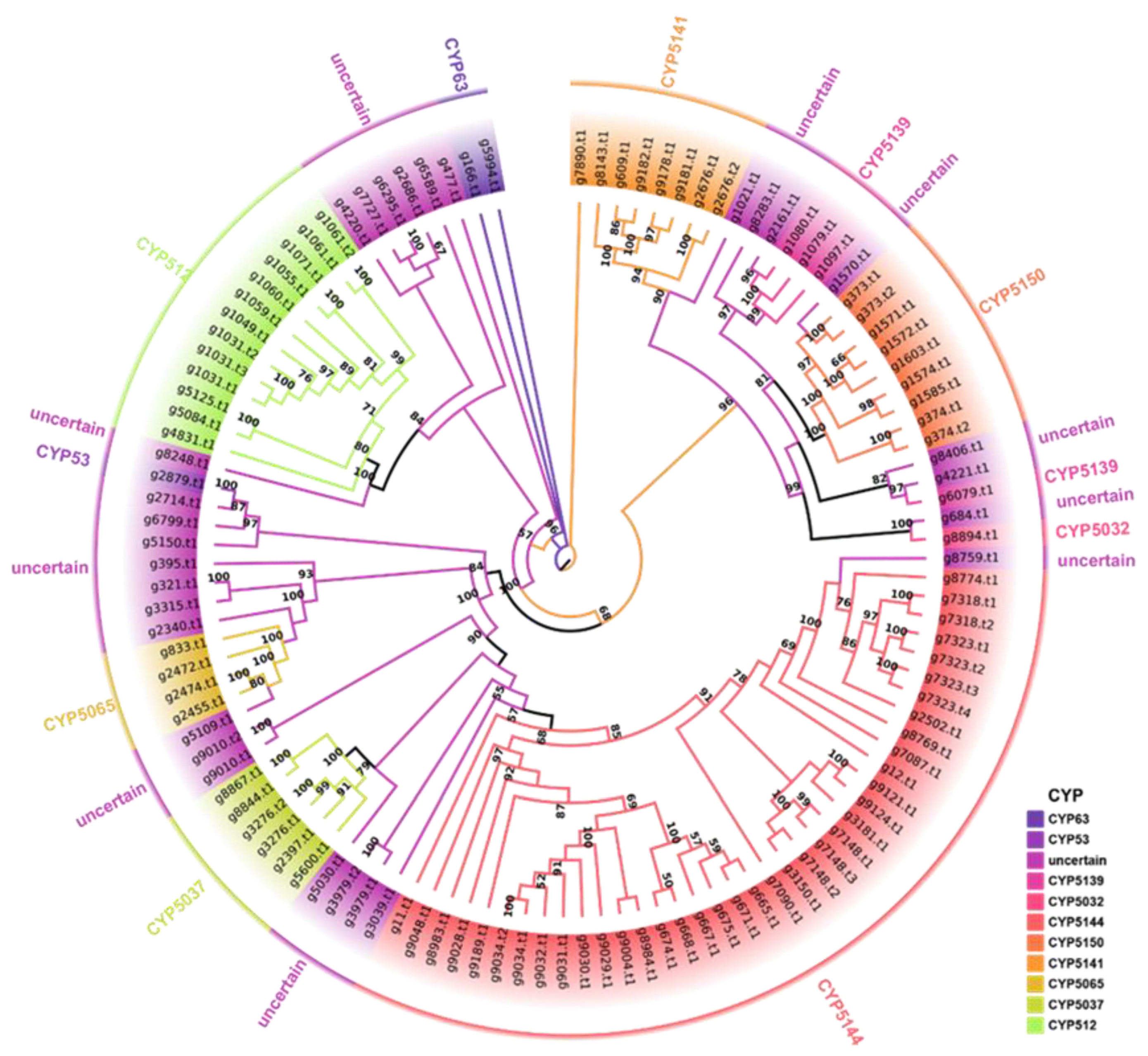
2.5. CAZy Family and Cytochrome P450 Analyses
2.6. Sequencing, Assembly, Annotation, and Comparative Analysis of Mitogenome
2.7. Metabolite Profiling Comparison
2.8. Data Availability
3. Results
3.1. Genome Sequence Assembly and Annotation of A. bitorquis BH01
3.2. Identification of the Mating Genes
3.3. Phylogenomic and Evolutionary Analysis
3.4. CAZymes Analysis
3.5. Cytochrome P450 Family Analysis
3.6. Comparative Genomic Analysis within Agaricus Species
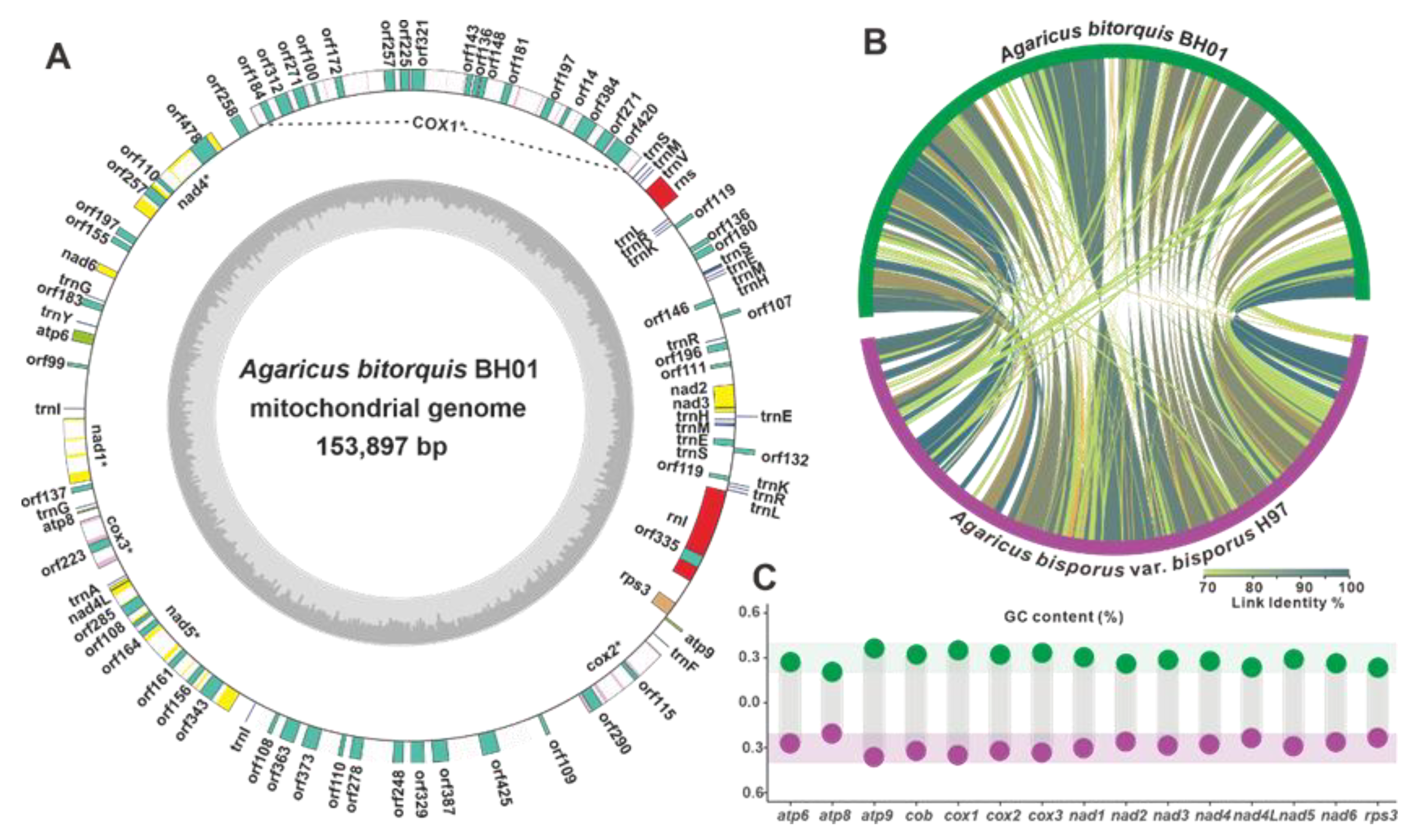
3.7. The Mitogenome of A. bitorquis and Comparative Mitogenomic Analysis within Agaricus Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | A. bitorquis BH01 | A. bisporus var. bisporus H97 |
|---|---|---|
| Total length (bp) | 153,897 | 135,055 |
| GC content (%) | 29.01% | 29.07% |
| AT-skew | 0.019 | 0.011 |
| GC-skew | 0.0304 | 0.0088 |
| No. of tRNA | 27 | 35 |
| No. of un_ORFs | 58 | 29 |
| No. of introns | 47 | 46 |
| Length of the cox1 gene (bp) | 30,178 | 29,908 |
| References | This study | [35] |
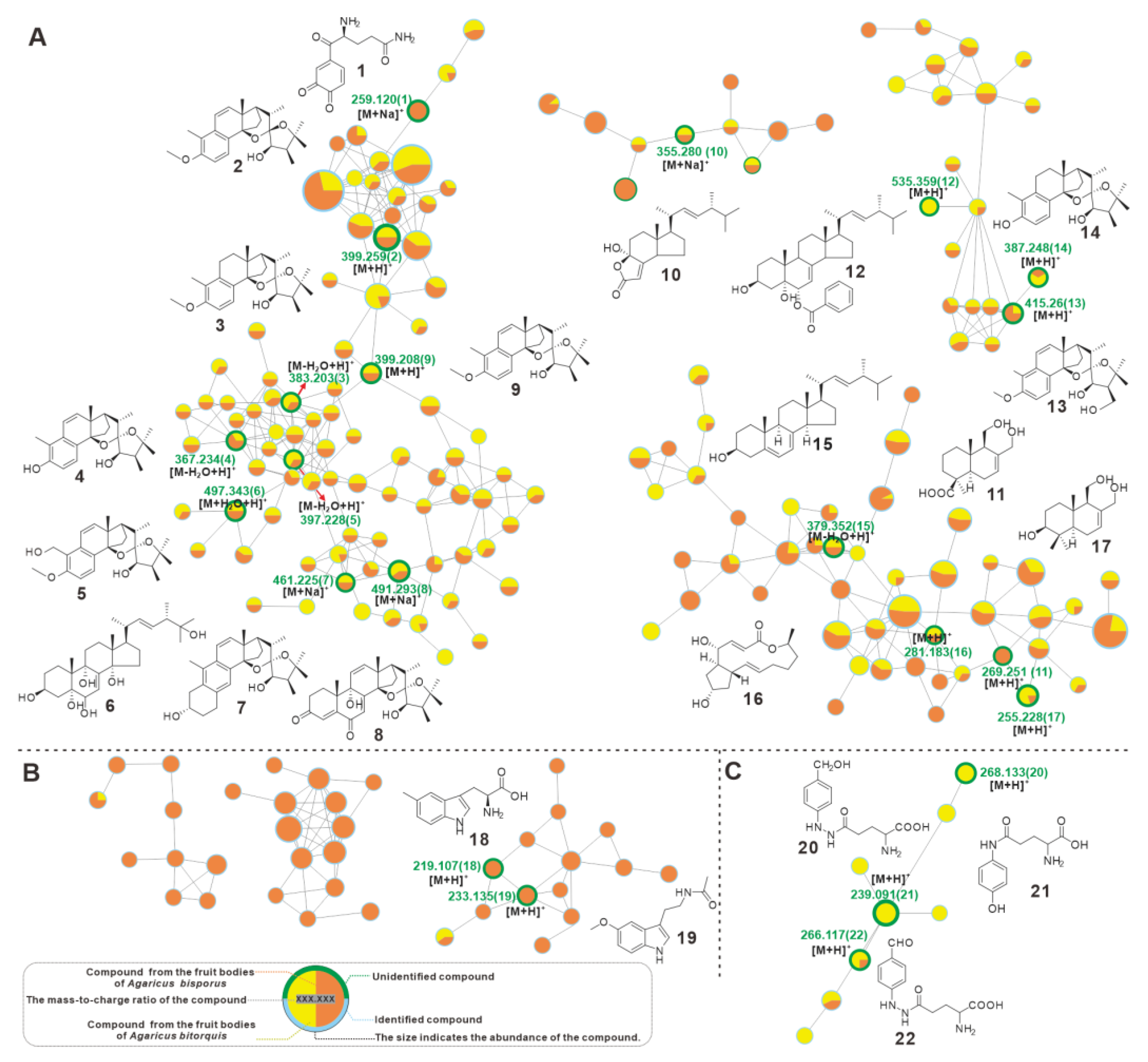
3.8. Metabolic Profiling Comparison between A. bitorquis and A. bisporus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cappelli, A. Fungi Europaei. Agaricus L Ex Fr. Ss Karsten (Psaliota Fr.); Edizioni Candusso: Origgio, Italy, 1984; pp. 123–125. [Google Scholar]
- Flegg, P.; Spencer, D.M.; Wood, D.A. The Biology and Technology of the Cultivated Mushroom; Wiley: Hoboken, NJ, USA, 1985; pp. 295–336. [Google Scholar]
- Ozturk, M.; Duru, M.E.; Kivrak, S.; Mercan-Dogan, N.; Turkoglu, A.; Ozler, M.A. In vitro antioxidant, anticholinesterase and antimicrobial activity studies on three Agaricus species with fatty acid compositions and iron contents: A comparative study on the three most edible mushrooms. Food Chem. Toxicol. 2011, 49, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Glamočlija, J.; Stojković, D.; Nikolić, M.; Ćirić, A.; Reis, F.S.; Barros, L.; Ferreira, I.C.F.R.; Soković, M. A comparative study on edible Agaricus mushrooms as functional foods. Food Funct. 2015, 6, 1900–1910. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Jiao, Y.; Wang, W.; Wang, F.; Chen, Q. Characterization and antioxidant activities of intracellular polysaccharides from Agaricus bitorquis (QuéL.) Sacc. Chaidam ZJU-CDMA-12. Int. J. Biol. Macromol. 2020, 156, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, W.; Yuen, H.; Yuen, M.; Peng, Q. Immunomodulatory effect of intracellular polysaccharide from mycelia of Agaricus bitorquis (QuéL.) Sacc. Chaidam by TLR4-mediated MyD88 dependent signaling pathway. Int. J. Biol. Macromol. 2021, 183, 79–89. [Google Scholar] [CrossRef]
- Nandan, C.K.; Patra, P.; Bhanja, S.K.; Adhikari, B.; Sarkar, R.; Mandal, S.; Islam, S.S. Structural characterization of a water-soluble β-(1→6)-linked D-glucan isolated from the hot water extract of an edible mushroom, Agaricus bitorquis. Carbohydr. Res. 2008, 343, 3120–3122. [Google Scholar] [CrossRef]
- Beattie, K.D.; Ulrich, R.; Grice, I.D.; Uddin, S.J.; Blake, T.B.; Wood, K.A.; Steele, J.; Iu, F.; May, T.W.; Tiralongo, E. Ethanolic and aqueous extracts derived from Australian fungi inhibit cancer cell growth in vitro. Mycologia 2011, 103, 458–465. [Google Scholar] [CrossRef]
- Falandysz, J. Selenium in Edible Mushrooms. J. Environ. Sci. Health Pt. C-Environ. Carcinog. Ecotoxicol. Rev. 2008, 26, 256–299. [Google Scholar] [CrossRef]
- Distribution of Agaricus bitorquis (Quél.) Sacc. Available online: https://www.gbif.org/species/5243374 (accessed on 17 March 2023).
- Mingguang, L.; Weili, C.; Fengjun, G. Research progress of the biological characteristics of Agaricus bitorquis and its development. Edible and Medicinal Mushrooms 2022, 30, 32–35. [Google Scholar]
- Navarro, M.J.; López-Serrano, F.R.; Escudero-Colomar, L.A.; Gea, F.J. Cultivation of Agaricus bitorquis mushroom as an strategy for the Integrated Pest Management of the myceliophagous mite Microdispus lambi. Pest Manag. Sci. 2020, 76, 2953–2958. [Google Scholar] [CrossRef]
- Colauto, N.B.; Fermor, T.R.; Eira, A.F.; Linde, G.A. Pseudomonas putida stimulates primordia on Agaricus bitorquis. Curr. Microbiol. 2016, 72, 482–488. [Google Scholar] [CrossRef]
- Duan, Y.; Han, H.; Qi, J.; Gao, J.-M.; Xu, Z.; Wang, P.; Zhang, J.; Liu, C. Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genom. 2022, 23, 314. [Google Scholar] [CrossRef]
- Gong, W.; Wang, Y.; Xie, C.; Zhou, Y.; Zhu, Z.; Peng, Y. Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 2020, 112, 2393–2399. [Google Scholar] [CrossRef]
- Dong, W.G.; Wang, Z.X.; Feng, X.L.; Zhang, R.Q.; Shen, D.Y.; Du, S.; Gao, J.M.; Qi, J. Chromosome-Level Genome Sequences, Comparative Genomic Analyses, and Secondary-Metabolite Biosynthesis Evaluation of the Medicinal Edible Mushroom Laetiporus sulphureus. Microbiol. Spectr. 2022, 10, e0243922. [Google Scholar] [CrossRef]
- Lu, M.Y.; Fan, W.L.; Wang, W.F.; Chen, T.; Tang, Y.C.; Chu, F.H.; Chang, T.T.; Wang, S.Y.; Li, M.Y.; Chen, Y.H.; et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA 2014, 111, E4743–E4752. [Google Scholar] [CrossRef]
- Zhang, R.-Q.; Feng, X.-L.; Wang, Z.-X.; Xie, T.-C.; Duan, Y.; Liu, C.; Gao, J.-M.; Qi, J. Genomic and Metabolomic Analyses of the Medicinal Fungus Inonotus hispidus for Its Metabolite’s Biosynthesis and Medicinal Application. J. Fungi 2022, 8, 1245. [Google Scholar] [CrossRef]
- Zhu, L.; Gao, X.; Zhang, M.; Hu, C.; Yang, W.; Guo, L.; Yang, S.; Yu, H.; Yu, H. Whole Genome Sequence of an Edible Mushroom Oudemansiella raphanipes (Changgengu). J. Fungi 2023, 9, 266. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y.; Jiang, H.; Yang, K.; Wang, S.; Wang, R.; Li, S.; Lei, P.; Xu, H.; Qiu, Y.; et al. Whole Genome Sequencing and Annotation of Naematelia aurantialba (Basidiomycota, Edible-Medicinal Fungi). J. Fungi 2022, 8, 6. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, M.; Sun, Y.; Li, Q.; Liu, J.; Song, C.; Shang, X.; Tan, Q.; Zhang, L.; Yu, H. Whole-genome sequence of a high-temperature edible mushroom Pleurotus giganteus (zhudugu). Front. Microbiol. 2022, 13, 941889. [Google Scholar] [CrossRef]
- Sun, L.; Fu, Y.; Yang, Y.; Wang, X.; Cui, W.; Li, D.; Yuan, X.; Zhang, Z.; Fu, Y.; Li, Y. Genomic Analyses Reveal Evidence of Independent Evolution, Demographic History, and Extreme Environment Adaptation of Tibetan Plateau Agaricus bisporus. Front. Microbiol. 2019, 10, 1786. [Google Scholar] [CrossRef]
- Morin, E.; Kohler, A.; Baker, A.R.; Foulongne-Oriol, M.; Lombard, V.; Nagye, L.G.; Ohm, R.A.; Patyshakuliyeva, A.; Brun, A.; Aerts, A.L.; et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA 2012, 109, 17501–17506. [Google Scholar] [CrossRef]
- Hei, Y.; Zhang, H.; Tan, N.; Zhou, Y.; Wei, X.; Hu, C.; Liu, Y.; Wang, L.; Qi, J.; Gao, J.M. Antimicrobial activity and biosynthetic potential of cultivable actinomycetes associated with Lichen symbiosis from Qinghai-Tibet Plateau. Microbiol. Res. 2021, 244, 126652. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, A.S.M.; Sedaghat-Telgerd, N.; Lavrijssen, B.; Ohm, R.A.; Hendrickx, P.M.; Scholtmeijer, K.; Baars, J.J.P.; van Peer, A. Telomere-to-telomere assembled and centromere annotated genomes of the two main subspecies of the button mushroom Agaricus bisporus reveal especially polymorphic chromosome ends. Sci. Rep. 2020, 10, 14653. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Casselton, L.A. Mating in mushrooms: Increasing the chances but prolonging the affair. Trends Genet. 2001, 17, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Bistis, G.N. Genetics of Sexuality in Higher Fungi. Mycologia 1967, 59, 384. [Google Scholar] [CrossRef]
- Kües, U. From two to many: Multiple mating types in Basidiomycetes. Fungal Biol. Rev. 2015, 29, 126–166. [Google Scholar] [CrossRef]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; De Vries, R.P.; Mäkelä, M.R. Plant-Polysaccharide-Degrading Enzymes from Basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef]
- Vanden Bossche, H.; Marichal, P.; Gorrens, J.; Coene, M.C. Biochemical basis for the activity and selectivity of oral antifungal drugs. Br. J. Clin. Pract. 1990, 71, 41–46. [Google Scholar]
- Li, Q.; Yang, L.; Xiang, D.; Wan, Y.; Wu, Q.; Huang, W.; Zhao, G. The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): Features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 2020, 145, 974–984. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Yao, W.; Shen, J.; Chen, M.; Gao, M.; Ren, J.; Li, Q.; Liu, N. The 256 kb mitochondrial genome of Clavaria fumosa is the largest among phylum Basidiomycota and is rich in introns and intronic ORFs. IMA Fungus 2020, 11, 26. [Google Scholar] [CrossRef]
- Singer, G.A.C.; Hickey, D.A. Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol. Biol. Evol. 2000, 17, 1581–1588. [Google Scholar] [CrossRef]
- Férandon, C.; Xu, J.; Barroso, G. The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet. Biol. 2013, 55, 85–91. [Google Scholar] [CrossRef]
- Da Silva de Souza, A.C.; Correa, V.G.; Goncalves, G.A.; Soares, A.A.; Bracht, A.; Peralta, R.M. Agaricus blazei Bioactive Compounds and their Effects on Human Health: Benefits and Controversies. Curr. Pharm. Des. 2017, 23, 2807–2834. [Google Scholar] [CrossRef]
- Tomatsu, M.; Shimakage, A.; Shinbo, M.; Yamada, S.; Takahashi, S. Novel angiotensin I-converting enzyme inhibitory peptides derived from soya milk. Food Chem. 2013, 136, 612–616. [Google Scholar] [CrossRef]
- Weaver, R.F.; Rajagopalan, K.V.; Handler, P.; Rosenthal, D.; Jeffs, P.W. Isolation from the Mushroom Agaricus bisporus and Chemical Synthesis of γ-l-Glutaminyl-4-hydroxybenzene. J. Biol. Chem. 1971, 246, 2010–2014. [Google Scholar] [CrossRef]
- Hirotani, M.; Sai, K.; Hirotani, S.; Yoshikawa, T. Blazeispirols B, C, E and F, des-A-ergostane-type compounds, from the cultured mycelia of the fungus Agaricus Blazei. Phytochem. 2001, 59, 571–577. [Google Scholar] [CrossRef]
- Rao, J.S.; Kumar, K.R. Bioactive molecules and their antioxidant activity of Agaricus bisporus. JCBPS 2013, 3, 1222–1228. [Google Scholar]
- Ueguchi, Y.; Matsunami, K.; Otsuka, H.; Kondo, K. Constituents of cultivated Agaricus blazei. J. Nat. Med. 2011, 65, 307–312. [Google Scholar] [CrossRef]
- Zhao, J.Y.; Ding, J.H.; Li, Z.H.; Dong, Z.J.; Feng, T.; Zhang, H.B.; Liu, J.K. Two new sesquiterpenes from cultures of the basidiomycete Agaricus arvensis. J. Asian Nat. Prod. Res. 2013, 15, 305–309. [Google Scholar] [CrossRef]
- Hirotani, M.; Sai, K.; Nagai, R.; Hirotani, S.; Takayanagi, H.; Yoshikawa, T. Blazeispirane and protoblazeispirane derivatives from the cultured mycelia of the fungus Agaricus blazei. Phytochemistry 2002, 61, 589–595. [Google Scholar] [CrossRef]
- Takaku, T.; Kimura, Y.; Okuda, H. Isolation of an Antitumor Compound from Agaricus blazei Murill and Its Mechanism of Action. J. Nutr. 2001, 131, 1409–1413. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Furutani, Y.; Kimura, S.; Zhu, Y.; Kawabata, K.; Furutani, M.; Nishikawa, T.; Tanaka, T.; Masaki, T.; Matsuoka, R.; et al. Brefeldin A is an estrogenic, Erk1/2-activating component in the extract of Agaricus blazei mycelia. J. Agric. Food Chem. 2013, 61, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Muszyńska, B.; Sułkowska-Ziaja, K.; Ekiert, H. Indole compounds in fruiting bodies of some edible Basidiomycota species. Food Chem. 2011, 125, 1306–1308. [Google Scholar] [CrossRef]
- Tsuji, H.; Ogawa, T.; Bando, N.; Sasaoka, K. Incorporation of chorismic acid and 4-aminobenzoic acid into the 4-hydroxyaniline moiety of N-(gamma-L-glutamyl)-4-hydroxyaniline in Agaricus bisporus. Biochim. Et Biophys. Acta BBA-Gen. Subj. 1985, 840, 287–290. [Google Scholar] [CrossRef]
- Chulia, A.J.; Bernillon, J.; Favre-Bonvin, J.; Kaouadji, M.; Arpin, N. Isolation of β-N-(γ-glutamyl)-4-formylphenylhydrazine (agaritinal) from Agaricus campestris. Phytochemistry 1988, 27, 929–930. [Google Scholar] [CrossRef]
- De Groot, P.W.J.; Schaap, P.J.; Van Griensven, L.J.L.D.; Visser, J. Isolation of developmentally regulated genes from the edible mushroom Agaricus bisporus. Microbiology 1997, 143, 1993–2001. [Google Scholar] [CrossRef]
- Wagemaker, M.J.M.; Eastwood, D.C.; van der Drift, C.; Jetten, M.S.M.; Burton, K.; Van Griensven, L.J.L.D.; Op den Camp, H.J.M. Expression of the urease gene of Agaricus bisporus: A tool for studying fruit body formation and post-harvest development. Appl. Microbiol. Biotechnol. 2006, 71, 486–492. [Google Scholar] [CrossRef]
- Li, N.-Y.; Cai, W.-M.; Jin, Q.-L.; Qin, Q.-P.; Ran, F.-L. Molecular Cloning and Expression of Polyphenoloxidase Genes from the Mushroom, Agaricus bisporus. Agric. Sci. China 2011, 10, 185–194. [Google Scholar] [CrossRef]
- Yuan, S.; Qiuhui, H.; Anxiang, S.; Fei, P.; Gaoxing, M.; Ning, M.; Wenjian, Y. Transcriptomic Analysis of the Mechanism Underlying the Effect of Methyl Jasmonate on the Size of Agaricus bisporus. Shipin Kexue/Food Sci. 2021, 42, 130–137. [Google Scholar]
- Bishop, K.S.; Kao, C.H.; Xu, Y.; Glucina, M.P.; Paterson, R.R.; Ferguson, L.R. From 2000 years of Ganoderma lucidum to recent developments in nutraceuticals. Phytochemistry 2015, 114, 56–65. [Google Scholar] [CrossRef]
- Wang, Z.X.; Feng, X.L.; Liu, C.; Gao, J.M.; Qi, J. Diverse Metabolites and Pharmacological Effects from the Basidiomycetes Inonotus hispidus. Antibiotics 2022, 11, 1097. [Google Scholar] [CrossRef]
- Lee, I.K.; Yun, B.S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 2011, 64, 349–359. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]







| Entry | A. bitorquis BH01 | A. bisporus var. bisporus H97 | A. bisporus var. burnettii H119 |
|---|---|---|---|
| Sequencing technology | lumina NovaSeq 6000 Nanopore PromethION | Sanger dideoxy | PacBio RSII |
| Sequencing depth | 145× | 8.29× | 70× |
| No. of contigs | 22 | 70 | 16 |
| Total length (bp) | 32,345,193 | 30,417,844 | 30,702,502 |
| Largest length (bp) | 3,210,751 | 3,550,205 | 3,639,502 |
| Scaffold N50 (bp) | 2,052,213 | 2,550,681 | 1,931,181 |
| Scaffold L50 | 10 | 5 | 6 |
| GC content (%) | 46.05 | 46.5 | 46.6 |
| No. of protein-coding genes | 10,028 | 10,438 | 10,421 |
| GenBank accession No. | PRJNA946023 | GCA_000300575.1 | GCA_014872705.1 |
| References | This study | [23] | [25] |
| No | Putative Metabolite | Molecular Formula | Adduct | m/z | Source | Reference | |
|---|---|---|---|---|---|---|---|
| A. bisporus | A. bitorquis | ||||||
| 1 | γ-L-Glutaminyl-3,4-benzoquinone | C11H12N2O4 | [M + Na]+ | 259.12 | √ | NA | Weaver, et al. [38] |
| 2 | blazeispirol B | C25H34O4 | [M + H]+ | 399.259 | √ | √ | Masao, et al. [39] |
| 3 | blazeispirol C | C25H36O4 | [M-H2O + H]+ | 383.203 | √ | √ | Masao, et al. [39] |
| 4 | blazeispirol D | C24H32O4 | [M-H2O + H]+ | 367.234 | √ | √ | Masao, et al. [39] |
| 5 | blazeispirol E | C25H34O5 | [M-H2O + H]+ | 397.228 | √ | √ | Masao, et al. [39] |
| 6 | (22E,24R)-3β,5α,6β,9α,14α,25-hexhydroxyergosta-7,22-diene | C28H46O6 | [M + H2O + H]+ | 497.343 | √ | √ | Rao, et al. [40] |
| 7 | blazeispirol X | C28H38O4 | [M + Na]+ | 461.225 | √ | √ | Masao, et al. [39] |
| 8 | blazeispirol Y | C28H36O6 | [M + Na]+ | 491.293 | √ | √ | Masao, et al. [39] |
| 9 | blazeispirol A | C25H34O4 | [M + H]+ | 399.208 | √ | √ | Masao, et al. [39] |
| 10 | demethyl-incisterol A3 | C21H32O3 | [M + Na]+ | 355.28 | √ | √ | Yumi, et al. [41] |
| 11 | 11,12-dihydroxy-15-drimeneoic acid | C15H24O4 | [M + H]+ | 269.251 | √ | NA | Zhao, et al. [42] |
| 12 | benzoyl ergostane | C35H50O4 | [M + H]+ | 535.359 | NA | √ | Yumi, et al. [41] |
| 13 | blazeispirol I | C25H34O5 | [M + H]+ | 415.26 | √ | √ | Masao, et al. [43] |
| 14 | blazeispirol F | C24H35O4 | [M + H]+ | 387.248 | √ | √ | Masao, et al. [43] |
| 15 | ergosterol | C28H44O | [M-H2O + H]+ | 379.352 | √ | √ | Takeshi, et al. [44] |
| 16 | brefeldin A | C16H24O4 | [M + H]+ | 281.183 | √ | √ | Dong, et al. [45] |
| 17 | 3β,11,12-trihydroxydrimene | C15H26O3 | [M + H]+ | 255.228 | √ | √ | Zhao, et al. [42] |
| 18 | 5-CH3-Tryptophan 5-methyltryptamine | C12H14N2O2 | [M + H]+ | 219.107 | √ | NA | Muszyńska, et al. [46] |
| 19 | melatonin | C13H16N2O2 | [M + H]+ | 233.135 | √ | NA | Muszyńska, et al. [46] |
| 20 | agaritine | C12H17N3O4 | [M + H]+ | 268.133 | NA | √ | Levenberg |
| 21 | N-(γ-L-glutamyl)-4-hydroxyaniline | C11H14N2O4 | [M + H]+ | 239.091 | NA | √ | Tsuji, et al. [47] |
| 22 | β-N-(γ-glutamyl)-4-formylphenyl-hydrazine | C12H15N3O4 | [M + H]+ | 266.117 | NA | √ | Albert J., et al. [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, C.; Feng, X.-l.; Wang, Z.-x.; Qi, J. The First Whole Genome Sequencing of Agaricus bitorquis and Its Metabolite Profiling. J. Fungi 2023, 9, 485. https://doi.org/10.3390/jof9040485
Zhao C, Feng X-l, Wang Z-x, Qi J. The First Whole Genome Sequencing of Agaricus bitorquis and Its Metabolite Profiling. Journal of Fungi. 2023; 9(4):485. https://doi.org/10.3390/jof9040485
Chicago/Turabian StyleZhao, Chunhua, Xi-long Feng, Zhen-xin Wang, and Jianzhao Qi. 2023. "The First Whole Genome Sequencing of Agaricus bitorquis and Its Metabolite Profiling" Journal of Fungi 9, no. 4: 485. https://doi.org/10.3390/jof9040485