VPAgs-Dataset4ML: A Dataset to Predict Viral Protective Antigens for Machine Learning-Based Reverse Vaccinology



Abstract
:1. Introduction
2. Methods
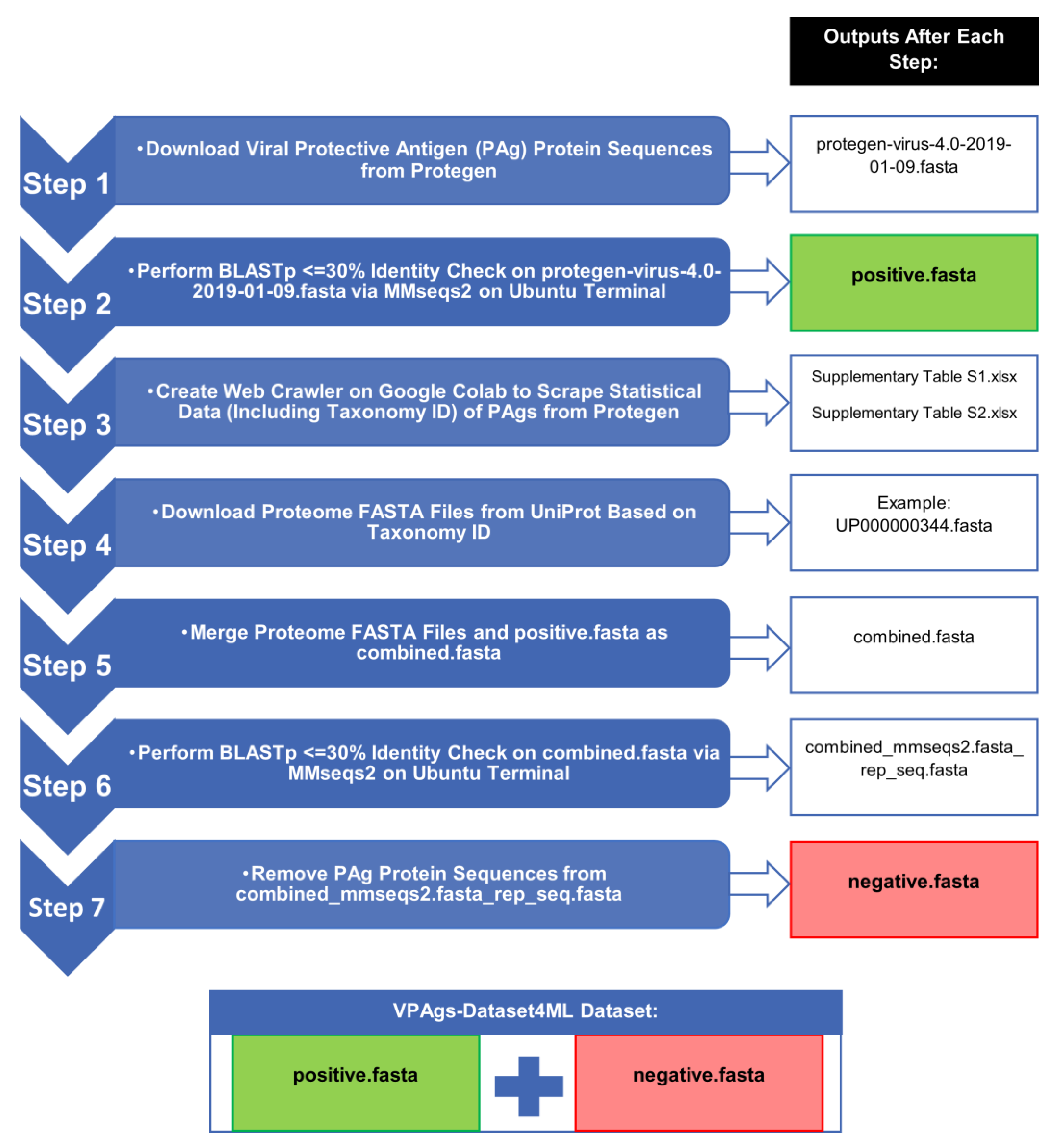
2.1. Step 1: Download Viral Protective Antigen (PAg) Protein Sequences from Protegen
2.2. Step 2: Perform BLASTp ≤ 30% Identity Check on protegen-virus-4.0-2019-01-09.fasta via Many-Against-Many Sequence (MMseqs2) on the Ubuntu Terminal
2.2.1. A Brief Overview of BLAST
2.2.2. A Brief Overview of MMseqs2
2.3. Step 3: Create Web Crawler on Google Colab to Scrape Statistical Data (Including Taxonomy ID) of PAgs from Protegen
2.4. Step 4: Download Proteome FASTA Files from UniProt Based on Taxonomy ID
2.5. Step 5: Merge Proteome FASTA Files and positive.fasta as combined.fasta
2.6. Step 6: Perform BLASTp ≤30% Identity Check on combined.fasta via MMseqs2 on Ubuntu Terminal
2.7. Step 7: Remove PAg Protein Sequences from combined_mmseqs2.fasta_rep_seq.fasta
2.8. Ethics and Permission
3. Data Description
3.1. Quantitative Overview of VPAgs-Dataset4ML
3.2. A Snapshot of VPAgs-Dataset4ML
4. User Notes
4.1. Usage of the VPAgs-Dataset4ML Dataset
4.2. Imbalanced Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Our World in Data. Death Rate from Infectious Diseases, 1990 to 2019. Available online: https://ourworldindata.org/grapher/infectious-disease-death-rates (accessed on 10 January 2023).
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.E.; Gowtage-Sequeria, S. Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 2005, 11, 1842. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The mother of all pandemics. Rev. Biomed. 2006, 17, 69–79. [Google Scholar] [CrossRef]
- Frost, W.H. Statistics of Influenza Morbidity: With Special Reference to Certain Factors in Case Incidence and Case Fatality. Public Heal. Rep. 1896–1970 1920, 35, 584. [Google Scholar] [CrossRef]
- Johnson, N.P.A.S.; Mueller, J. Updating the Accounts: Global Mortality of the 1918–1920 “Spanish” Influenza Pandemic. Bull. Hist. Med. 2002, 76, 105–115. [Google Scholar] [CrossRef]
- World Health Organization. Ebola Virus Disease. Available online: https://www.who.int/news-room/fact-sheets/detail/ebola-virus-disease (accessed on 22 October 2022).
- Merson, M.H. The HIV–AIDS pandemic at 25—The global response. N. Engl. J. Med. 2006, 354, 2414–2417. [Google Scholar] [CrossRef]
- World Health Organization. HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 10 November 2022).
- Hon, E.; Li, A.; Nelson, E.; Leung, C. Severe Acute Respiratory Syndrome (SARS) In: Textbook of Paediatric Infectious Diseases, Feigin, R.D.; Cherry, J.D., Demmler, G.J., Kaplan, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- World Health Organization. Summary of Probable SARS Cases with Onset of Illness from 1 November 2002 to 31 July 2003. Available online: https://www.who.int/publications/m/item/summary-of-probable-sars-cases-with-onset-of-illness-from-1-november-2002-to-31-july-2003 (accessed on 10 November 2022).
- World Health Organization. Consensus Document on the Epidemiology of Severe Acute Respiratory syndrome (SARS); World Health Organization: Geneva, Switzerland, 2003. [Google Scholar]
- Worldometers. COVID-19 Coronavirus Pandemic. Available online: https://www.worldometers.info/coronavirus/ (accessed on 10 November 2022).
- Ehreth, J. The global value of vaccination. Vaccine 2003, 21, 596–600. [Google Scholar] [CrossRef]
- Carter, A.; Msemburi, W.; Sim, S.Y.; AM Gaythorpe, K.; Lindstrand, A.; Hutubessy, R.C. Modeling the impact of vaccination for the immunization agenda 2030: Deaths averted due to vaccination against 14 pathogens in 194 countries from 2021–2030. Ann Hutubessy Raymond CW Model. Impact Vaccin. Immun. Agenda 2021, 2030, 1–41. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Fast Facts on Global Immunization. Available online: https://www.cdc.gov/globalhealth/immunization/data/fast-facts.html#:~:text=Immunization%20Prevents%20Death%20Worldwide,save%20nearly%2019%20million%20lives (accessed on 5 November 2022).
- Koff, W.C.; Burton, D.R.; Johnson, P.R.; Walker, B.D.; King, C.R.; Nabel, G.J.; Ahmed, R.; Bhan, M.K.; Plotkin, S.A. Accelerating Next-Generation Vaccine Development for Global Disease Prevention. Science 2013, 340, 1232910. [Google Scholar] [CrossRef]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Dalsass, M.; Brozzi, A.; Medini, D.; Rappuoli, R. Comparison of open-source reverse vaccinology programs for bacterial vaccine antigen discovery. Front. Immunol. 2019, 10, 113. [Google Scholar] [CrossRef]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Aricò, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B.; et al. Identification of Vaccine Candidates Against Serogroup B Meningococcus by Whole-Genome Sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Folaranmi, T.; Rubin, L.; Martin, S.W.; Patel, M.; MacNeil, J.R. Use of serogroup B meningococcal vaccines in persons aged ≥10 years at increased risk for serogroup B meningococcal disease: Recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR. Morb. Mortal. Wkly. Rep. 2015, 64, 608. [Google Scholar] [PubMed]
- Vernikos, G.; Medini, D. Bexsero® chronicle. Pathog. Glob. Health 2014, 108, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Ong, E.; Wang, H.; Wong, M.U.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign-ML: Supervised machine learning reverse vaccinology model for improved prediction of bacterial protective antigens. Bioinformatics 2020, 36, 3185–3191. [Google Scholar] [CrossRef] [PubMed]
- Kotsiantis, S.B.; Zaharakis, I.; Pintelas, P. Supervised machine learning: A review of classification techniques. Emerg. Artif. Intell. Appl. Comput. Eng. 2007, 160, 3–24. [Google Scholar]
- Vapnik, V.N. The Nature of Statistical Learning Theory; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1995. [Google Scholar]
- Burges, C.J.C. A Tutorial on Support Vector Machines for Pattern Recognition. Data Min. Knowl. Discov. 1998, 2, 121–167. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef]
- Bowman, B.N.; McAdam, P.R.; Vivona, S.; Zhang, J.X.; Luong, T.; Belew, R.K.; Sahota, H.; Guiney, D.; Valafar, F.; Fierer, J.; et al. Improving reverse vaccinology with a machine learning approach. Vaccine 2011, 29, 8156–8164. [Google Scholar] [CrossRef] [PubMed]
- Heinson, A.I.; Gunawardana, Y.; Moesker, B.; Hume, C.C.D.; Vataga, E.; Hall, Y.; Stylianou, E.; McShane, H.; Williams, A.; Niranjan, M.; et al. Enhancing the Biological Relevance of Machine Learning Classifiers for Reverse Vaccinology. Int. J. Mol. Sci. 2017, 18, 312. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Rahman, K.; Saha, S.; Kaykobad, M.; Rahman, M.S. Antigenic: An improved prediction model of protective antigens. Artif. Intell. Med. 2019, 94, 28–41. [Google Scholar] [CrossRef]
- Kohavi, R. A Study of Cross-Validation and Bootstrap for Accuracy Estimation and Model Selection. In Proceedings of the The 1995 International Joint Conference, Montreal, QC, Canada, 20–25 August 1995; pp. 1137–1145. [Google Scholar]
- Vivona, S.; Bernante, F.; Filippini, F. NERVE: New enhanced reverse vaccinology environment. BMC Biotechnol. 2006, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xiang, Z.; Mobley, H.L.T. Vaxign: The First Web-Based Vaccine Design Program for Reverse Vaccinology and Applications for Vaccine Development. J. Biomed. Biotechnol. 2010, 2010, 297505. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Chanumolu, S.K.; Gupta, A.; Chauhan, R.S.; Rout, C. Jenner-predict server: Prediction of protein vaccine candidates (PVCs) in bacteria based on host-pathogen interactions. BMC Bioinform. 2013, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef]
- Ong, E.; Cooke, M.F.; Huffman, A.; Xiang, Z.; Wong, M.U.; Wang, H.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign2: The second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. 2021, 49, W671–W678. [Google Scholar] [CrossRef]
- Yang, B.; Sayers, S.; Xiang, Z.; He, Y. Protegen: A web-based protective antigen database and analysis system. Nucleic Acids Res. 2011, 39, D1073–D1078. [Google Scholar] [CrossRef]
- UniProt Consortium. The universal protein resource (UniProt). Nucleic Acids Res. 2007, 36, D190–D195. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef]
- Anaconda Software Distribution. Conda. Available online: https://www.anaconda.com/ (accessed on 30 October 2022).
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.; Steinegger, M.; Söding, J. MMseqs software suite for fast and deep clustering and searching of large protein sequence sets. Bioinformatics 2016, 32, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Reback, J.; McKinney, W.; Van Den Bossche, J.; Augspurger, T.; Cloud, P.; Klein, A.; Hawkins, S.; Roeschke, M.; Tratner, J.; She, C. pandas-dev/pandas: Pandas 1.0. 5. Zenodo 2020. [Google Scholar]
- McKinney, W. Data structures for Statistical Computing in Python. In Proceedings of the Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar]
- McKinney, W. Pandas: A foundational Python library for data analysis and statistics. Python High Perform. Sci. Comput. 2011, 14, 1–9. [Google Scholar]
- Richardson, L. Beautiful Soup Documentation. Available online: https://www.crummy.com/software/BeautifulSoup/bs4/doc/ (accessed on 30 October 2022).
- Bisong, E. Google Colaboratory. In Building Machine Learning and Deep Learning Models on Google Cloud Platform: A Comprehensive Guide for Beginners; Apress: Berkeley, CA, USA, 2019; pp. 59–64. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. UniProt. Available online: https://www.uniprot.org/ (accessed on 11 March 2022).
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. arXiv 2015, arXiv:1603.04467. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Paszke, A.; Gross, S.; Chintala, S.; Chanan, G.; Yang, E.; DeVito, Z.; Lin, Z.; Desmaison, A.; Antiga, L.; Lerer, A. Automatic differentiation in pytorch. NIPS 2017 Workshop Autodiff 2017. [Google Scholar]
- Frank, E.; Hall, M.A.; Witten, I.H. Online Appendix for “Data Mining: Practical Machine Learning Tools and Techniques, 4th ed.; Morgan Kaufmann: Burlington, MA, USA, 2016. [Google Scholar]
- Berthold, M.; Cebron, N.; Dill, F.; Gabriel, T.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner in Data Analysis, Machine Learning and Applications SE-38; Preisach, C., Burkhardt, H., Schmidt-Thieme, L., Decker, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. AcM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A. Orange: Data mining toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A grammar of data manipulation. R Package Version 0.4 2015, 3, 156. [Google Scholar]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 6 November 2022).
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- He, H.; Bai, Y.; Garcia, E.A.; Li, S. ADASYN: Adaptive Synthetic Sampling Approach for Imbalanced Learning. In Proceedings of the 2008 IEEE International Joint Conference on Neural Networks (IEEE World Congress on Computational Intelligence), Hong Kong, China, 1–8 June 2008; pp. 1322–1328. [Google Scholar]
- Lin, W.-C.; Tsai, C.-F.; Hu, Y.-H.; Jhang, J.-S. Clustering-based undersampling in class-imbalanced data. Inf. Sci. 2017, 409-410, 17–26. [Google Scholar] [CrossRef]
- Tomek, I. An Experiment with The Edited Nearest-Nieghbor Rule. IEEE Trans. Syst. Man Cybern. 1976, 6, 448–452. [Google Scholar]
- Chen, C.; Liaw, A.; Breiman, L. Using random forest to learn imbalanced data. Univ. Calif. Berkeley 2004, 110, 24. [Google Scholar]
- Maclin, R.; Opitz, D. An empirical evaluation of bagging and boosting. AAAI/IAAI 1997, 1997, 546–551. [Google Scholar]


{kind=link}
| Year | Dataset | Dataset Size | Protective Antigens (PAgs) (Positives) | Non-Protective Protein Sequences (Negatives) | Viral Protein Sequence Data File(s)Available for Positives and Negatives? |
|---|---|---|---|---|---|
| 2007 | In: VaxiJen [28] | 200 | 100 | 100 | No |
| 2010 | In: ANTIGENpro [29] | 1324 * | 576 * | 748 * | No |
| 2019 | In: Antigenic [32] | 1324 * | 576 * | 748 * | No |
| 2023 | VPAgs-Dataset4ML (Our dataset) | 2145 | 210 | 1935 | Yes |
| >Protegen: 579|VO: VO_0011168|NP_056918.1 nucleocapsid protein [Measles morbillivirus] MATLLRSLALFKRNKDKPPITSGSGGAIRGIKHIIIVPIPGDSSITTRSRLLDRLVRLIGNPDVSGPKLTGALIGILSLFVESPGQLIQRITDDPDVSIRLLEVVQSDQSQSGLTFASRGTNMEDEADQYFSHDDPSSSDQSRSGWFENKEISDIEVQDPEGFNMILGTILAQIWVLLAKAVTAPDTAADSELRRWIKYTQQRRVVGEFRLERKWLDVVRNRIAEDLSLRRFMVALILDIKRTPGNKPRIAEMICDIDTYIVEAGLASFILTIKFGIETMYPALGLHEFAGELSTLESLMNLYQQMGETAPYMVILENSIQNKFSAGSYPLLWSYAMGVGVELENSMGGLNFGRSYFDPAYFRLGQEMVRRSAGKVSSTLASELGITAEDARLVSEIAMHTTEDRISRAVGPRQAQVSFLHGDQSENELPGLGGKEDRRVKQGRGEARESYRETGSSRASDARAAHPPTSMPLDIDTASESGQDPQDSRRSADALLRLQAMAGILEEQGSDTDTPRVYNDRDLLD >Protegen: 580|VO: VO_0011169|NP_056919.1 phosphoprotein [Measles morbillivirus] MAEEQARHVKNGLECIRALKAEPIGSLAVEEAMAAWSEISDNPGQDRATCKEEEAGSSGLSKPCLSAIGSTEGGAPRIRGQGSGESDDDAETLGIPSRNLQASSTGLQCYHVYDHSGEAVKGIQDADSIMVQSGLDGDSTLSGGDDESENSDVDIGEPDTEGYAITDRGSAPISMGFRASDVETAEGGEIHELLKLQSRGNNFPKLGKTLNVPPPPNPSRASTSETPIKKGTDARLASFGTEIASLLTGGATQCARKSPSEPSGPGAPAGNVPECVSNAALIQEWTPESGTTISPRSQNNEEGGDYYDDELFSDVQDIKTALAKIHEDNQKIISKLESLLLLKGEVESIKKQINRQNISISTLEGHLSSIMIAIPGLGKDPNDPTADVELNPDLKPIIGRDSGRALAEVLKKPVASRQLQGMTNGRTSSRGQLLKEFQLKPIGKKVSSAVGFVPDTGPASRSVIRSIIKSSRLEEDRKRYLMTLLDDIKGANDLAKFHQMLMKIIMK >Protegen: 581|VO: VO_0011170|NP_056921.1 matrix protein [Measles morbillivirus] MTEIYDFDKSAWDIKGSIAPIQPTTYSDGRLVPQVRVIDPGLGDRKDECFMYMFLLGVVEDSDPLGPPIGRAFGSLPLGVGRSTAKPEELLKEATELDIVVRRTAGLNEKLVFYNNTPLTLLTPWRKVLTTGSVFNANQVCNAVNLIPLDTPQRFRVVYMSITRLSDNGYYTVPRRMLEFRSVNAVAFNLLVTLRIDKAIGPGKIIDNAEQLPEATFMVHIGNFRRKKSEVYSADYCKMKIEKMGLVFALGGIGGTSLHIRSTGKMSKTLHAQLGFKKTLCYPLMDINEDLNRLLWRSRCKIVRIQAVLQPSVPQEFRIYDDVIINDDQGLFKVL >Protegen: 648|VO: VO_0011232|NP_056793.1 nucleoprotein N [Rabies lyssavirus] MDADKIVFKVNNQVVSLKPEIIVDQYEYKYPAIKDLKKPCITLGKAPDLNKAYKSVLSCMSAAKLDPDDVCSYLAAAMQFFEGTCPEDWTSYGIVIARKGDKITPGSLVEIKRTDVEGNWALTGGMELTRDPTVPEHASLVGLLLSLYRLSKISGQSTGNYKTNIADRIEQIFETAPFVKIVEHHTLMTTHKMCANWSTIPNFRFLAGTYDMFFSRIEHLYSAIRVGTVVTAYEDCSGLVSFTGFIKQINLTAREAILYFFHKNFEEEIRRMFEPGQETAVPHSYFIHFRSLGLSGKSPYSSNAVGHVFNLIHFVGCYMGQVRSLNATVIAACAPHEMSVLGGYLGEEFFGKGTFERRFFRDEKELQEYEAAELTKTDVALADDGTVNSDDEDYFSGETRSPEAVYTRIIMNGGRLKRSHIRRYVSVSSNHQARPNSFAEFLNKTYSSDS >Protegen: 654|VO: VO_0011238|CAA09075.1 gB, partial [Suid alphaherpesvirus 1] ESEDPDAL |
| >tr|Q3I803|Q3I803_MONPV Uncharacterized protein OS=Monkeypox virus OX=10244 GN=MPXV_LIB1970_184_168 PE=4 SV=1 UPId=UP000127566 PPId=UP000000344 MMFIHCVVFPDLSNPSKTINAPRDILVNLTYFLQFMRIIKLKIIYGHPRRLGLSTFIDHG >tr|A0A650BTW0|A0A650BTW0_MONPV Uncharacterized protein OS=Monkeypox virus OX=10244 GN=PDLMKLCO_00006 PE=4 SV=1 UPId=UP000424348 PPId=UP000000344 MILSINLLTVPFSHTINYDDQTYDNDIKSLLEVTTRFHRHYHN >tr|A0A650BUA7|A0A650BUA7_MONPV A-type inclusion protein A25 OS=Monkeypox virus OX=10244 GN=PDLMKLCO_00143 PE=4 SV=1 UPId=UP000424348 PPId=UP000000344 MLQRLQSRISDLEIQLNDCERNNEINADMEKR >tr|A0A650BUV3|A0A650BUV3_MONPV A-type inclusion protein OS=Monkeypox virus OX=10244 GN=PDLMKLCO_00142 PE=4 SV=1 UPId=UP000424348 PPId=UP000000344 MDLDRHLNDCKNGNGASSEEVNRLKTRIRDLERSLEIFSKDESELYSAYKTELGNAREQI SNLQESLRRERESDKTDSYYRRELTRERNKIVELKKRT >tr|E2FL62|E2FL62_MONPV Uncharacterized protein OS=Monkeypox virus OX=10244 PE=4 SV=1 UPId=UP000168391 PPId=UP000000344 MYIIYRHLSFLTMNSLIENSVLHVRKLLYMIHFNDIDHAPTTATSRNCEDQYLKK |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salod, Z.; Mahomed, O. VPAgs-Dataset4ML: A Dataset to Predict Viral Protective Antigens for Machine Learning-Based Reverse Vaccinology. Data 2023, 8, 41. https://doi.org/10.3390/data8020041
Salod Z, Mahomed O. VPAgs-Dataset4ML: A Dataset to Predict Viral Protective Antigens for Machine Learning-Based Reverse Vaccinology. Data. 2023; 8(2):41. https://doi.org/10.3390/data8020041
Chicago/Turabian StyleSalod, Zakia, and Ozayr Mahomed. 2023. "VPAgs-Dataset4ML: A Dataset to Predict Viral Protective Antigens for Machine Learning-Based Reverse Vaccinology" Data 8, no. 2: 41. https://doi.org/10.3390/data8020041