
The Gut Microbiota-Immunity Axis in ALS: A Role in Deciphering Disease Heterogeneity?

 ,
,  , , , , , and
, , , , , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Sample Collection
2.3. Molecular Inflammatory Response Evaluation
2.4. SCFA Evaluation
2.5. Microbiota Characterization
2.6. Statistical Analysis
3. Results
3.1. Patients
3.2. Serum Cytokine Profile
3.3. Fecal Cytokine Profile
3.4. Profile of Fecal SCFAs
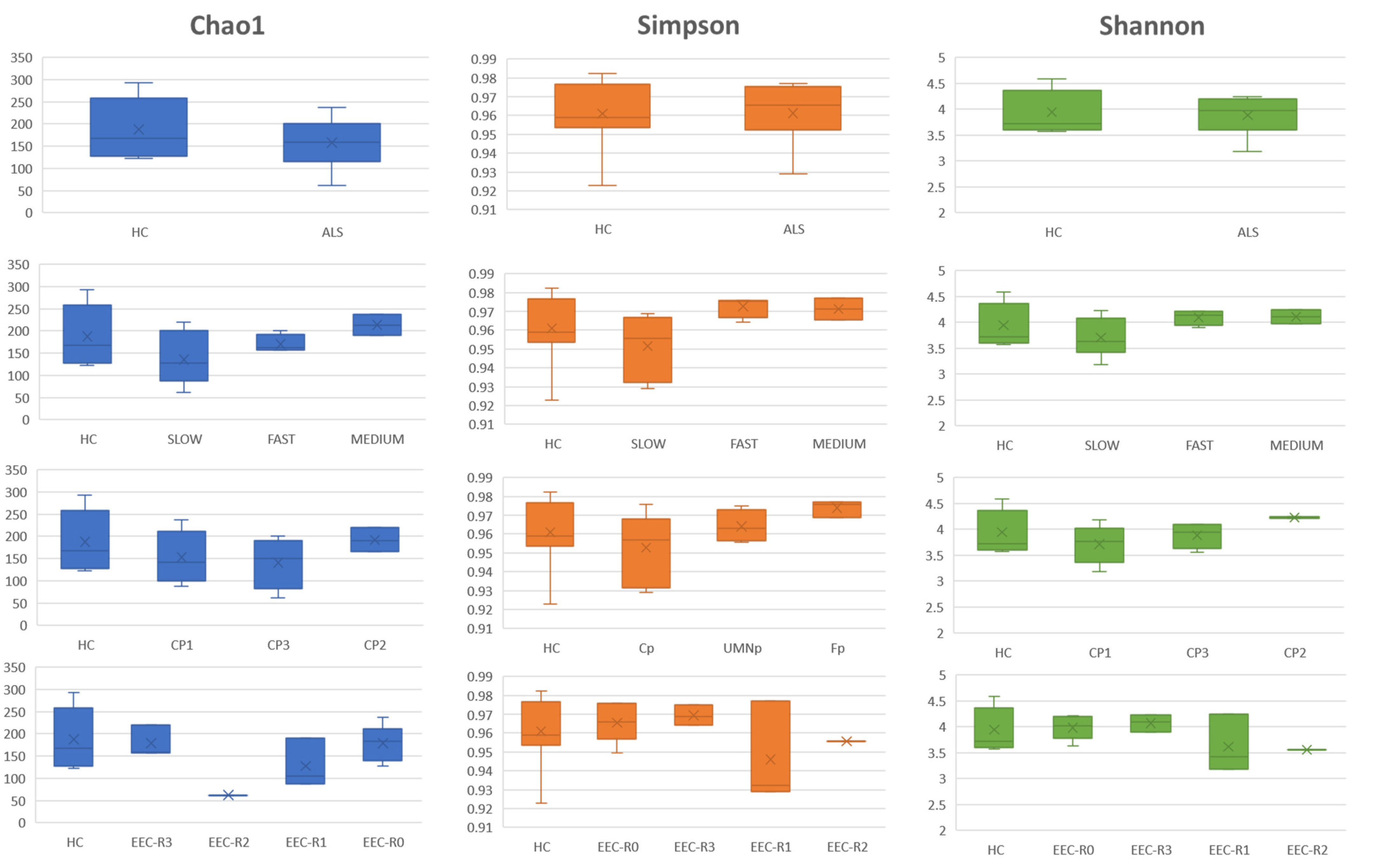
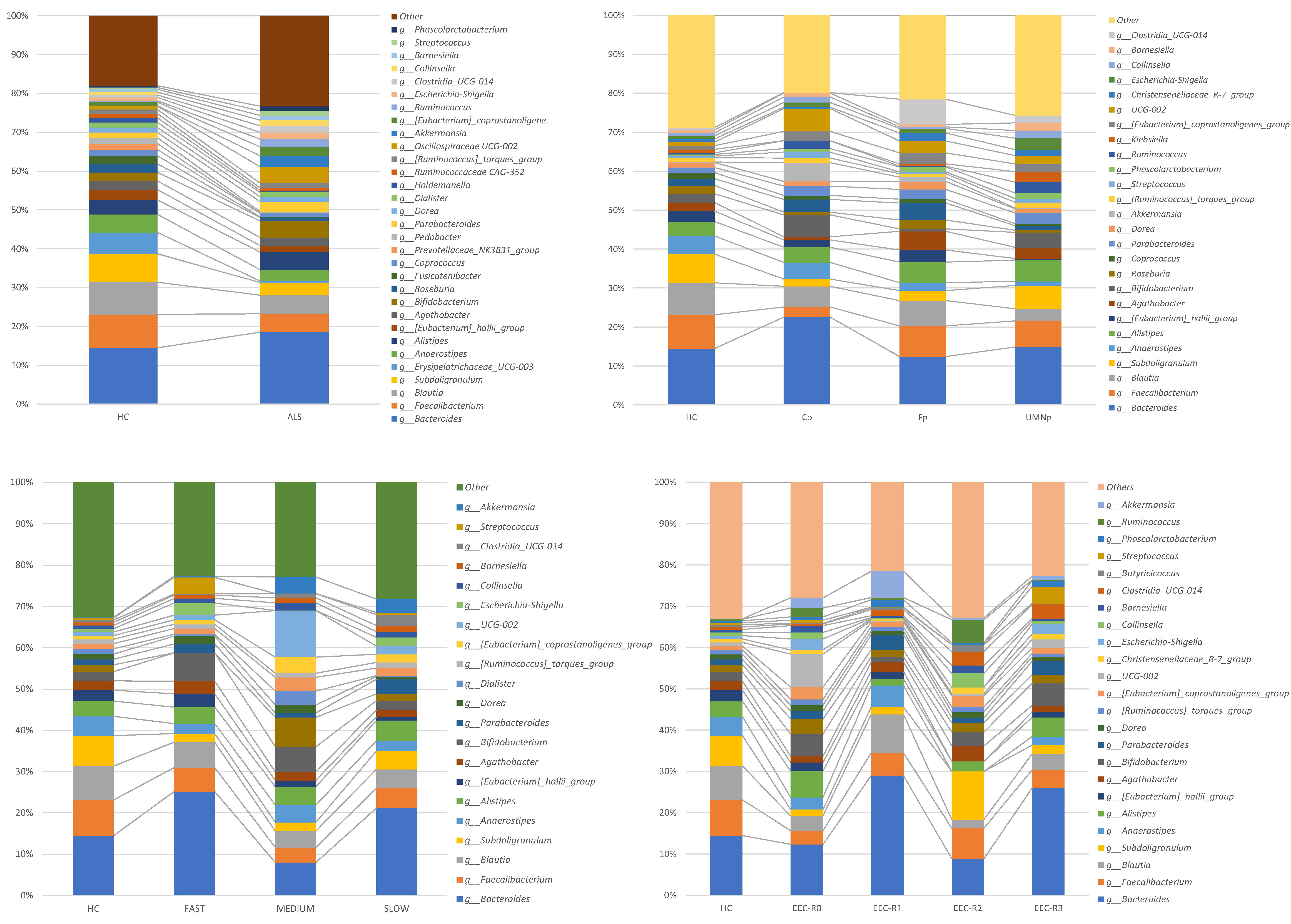
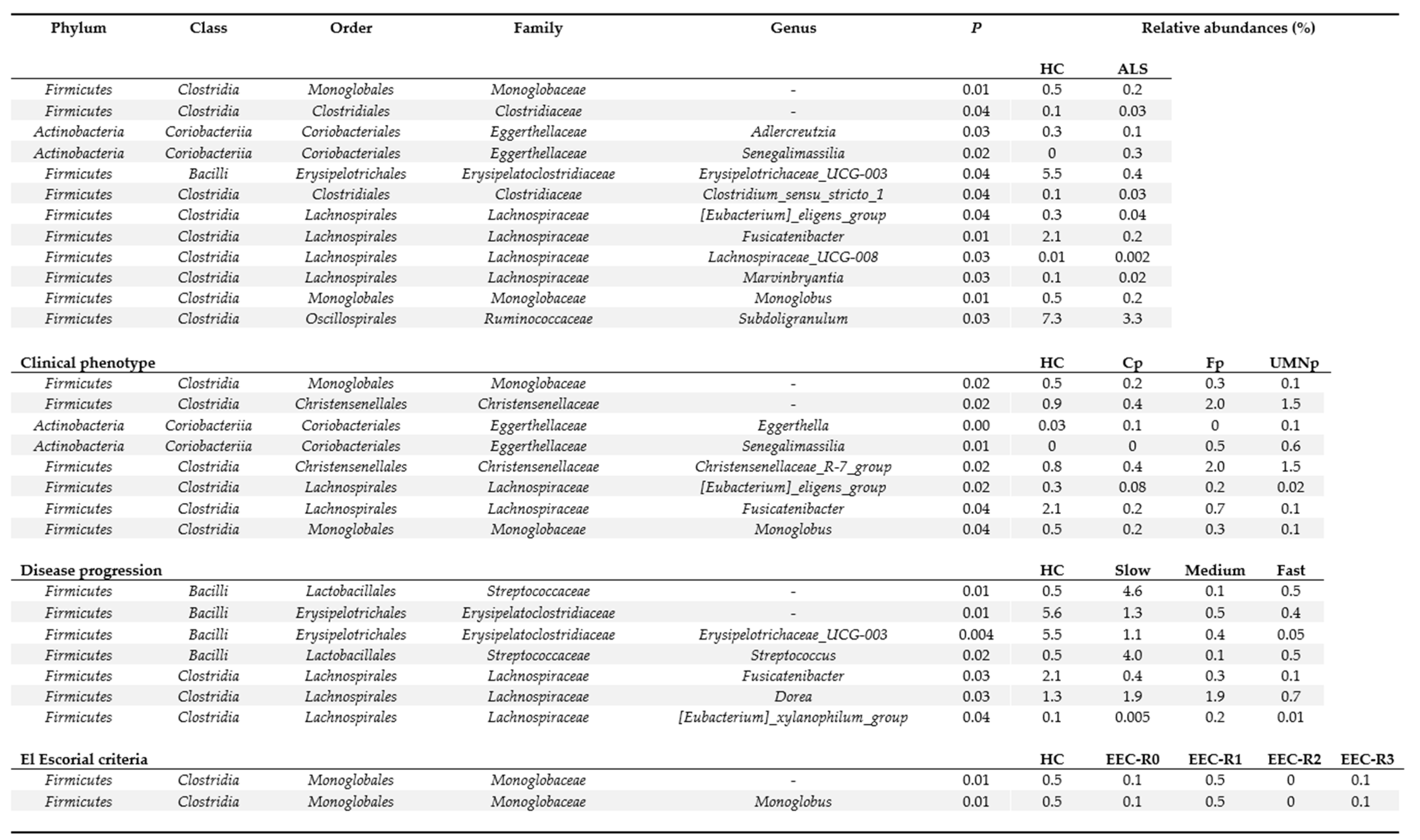
3.5. Definition of the Gut Microbiota Composition
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turner, M.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.S.-Y.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Beghi, E.; Mennini, T.; Bendotti, C.; Bigini, P.; Logroscino, G.; Chiò, A.; Hardiman, O.; Mitchell, U.; Swingler, R.; Traynor, B.J.; et al. The Heterogeneity of Amyotrophic Lateral Sclerosis: A Possible Explanation of Treatment Failure. Curr. Med. Chem. 2007, 14, 3185–3200. [Google Scholar] [CrossRef]
- Lai, S.-L.; Abramzon, Y.; Schymick, J.C.; Stephan, D.A.; Dunckley, T.; Dillman, A.; Cookson, M.; Calvo, A.; Battistini, S.; Giannini, F.; et al. FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2011, 32, 550.e1–550.e4. [Google Scholar] [CrossRef] [Green Version]
- Chio, A.; Borghero, G.; Restagno, G.; Mora, G.; Drepper, C.; Traynor, B.J.; Sendtner, M.; Brunetti, M.; Ossola, I.; Calvo, A.; et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 2012, 135, 784–793. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; Sabatelli, M.; Caponnetto, C.; Traynor, B.J.; Johnson, J.O.; Nalls, M.A.; Calvo, A.; Moglia, C.; Borghero, G.; et al. CHCH10 mutations in an Italian cohort of familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 2015, 36, 1767.e3–1767.e6. [Google Scholar] [CrossRef]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Parish, L.D.; Cau, T.B.; Loi, D.; et al. ATXN2 is a modifier of phenotype in ALS patients of Sardinian ancestry. Neurobiol. Aging 2015, 36, 2906.e1–2906.e5. [Google Scholar] [CrossRef] [Green Version]
- Su, F.-C.; Goutman, S.A.; Chernyak, S.; Mukherjee, B.; Callaghan, B.C.; Batterman, S.; Feldman, E.L. Association of Environmental Toxins With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016, 73, 803–811. [Google Scholar] [CrossRef]
- Béland, L.-C.; Markovinovic, A.; Jakovac, H.; De Marchi, F.; Bilic, E.; Mazzini, L.; Kriz, J.; Munitic, I. Immunity in amyotrophic lateral sclerosis: Blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020, 2, fcaa124. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-Modulated Metabolites at the Interface of Host Immunity. J. Immunol. 2017, 198, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Veiga-Fernandes, H.; Mucida, D. Neuro-Immune Interactions at Barrier Surfaces. Cell 2016, 165, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Erny, D.; De Angelis, A.L.H.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Kehrmann, J.; Effenberg, L.; Wilk, C.; Schoemer, D.; Phuong, N.N.T.; Adamczyk, A.; Pastille, E.; Scholtysik, R.; Klein-Hitpass, L.; Klopfleisch, R.; et al. Depletion of Foxp3 + regulatory T cells is accompanied by an increase in the relative abundance of Firmicutes in the murine gut microbiome. Immunology 2020, 159, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saresella, M.; Piancone, F.; Tortorella, P.; Marventano, I.; Gatti, A.; Caputo, D.; Lunetta, C.; Corbo, M.; Rovaris, M.; Clerici, M. T helper-17 activation dominates the immunologic milieu of both amyotrophic lateral sclerosis and progressive multiple sclerosis. Clin. Immunol. 2013, 148, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2012, 5, 64–79. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef]
- Sun, J.; Huang, T.; Debelius, J.W.; Fang, F. Gut microbiome and amyotrophic lateral sclerosis—A systematic review of current evidence. J. Intern. Med. 2021. [Google Scholar] [CrossRef]
- Zhai, C.-D.; Zheng, J.-J.; An, B.-C.; Huang, H.-F.; Tan, Z.-C. Intestinal microbiota composition in patients with amyotrophic lateral sclerosis. Chin. Med. J. 2019, 132, 1815–1822. [Google Scholar] [CrossRef]
- Fang, X.; Wang, X.; Yang, S.; Meng, F.; Wang, X.; Wei, H.; Chen, T. Evaluation of the Microbial Diversity in Amyotrophic Lateral Sclerosis Using High-Throughput Sequencing. Front. Microbiol. 2016, 7, 1479. [Google Scholar] [CrossRef] [Green Version]
- Rowin, J.; Xia, Y.; Jung, B.; Sun, J. Gut inflammation and dysbiosis in human motor neuron disease. Physiol. Rep. 2017, 5, e13443. [Google Scholar] [CrossRef]
- Brenner, D.; Hiergeist, A.; Adis, C.; Mayer, B.; Gessner, A.; Ludolph, A.C.; Weishaupt, J.H. The fecal microbiome of ALS patients. Neurobiol. Aging 2018, 61, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Restuadi, R.; McCrae, A.F.; Van Eijk, R.P.; Garton, F.; Henderson, R.D.; Wray, N.R.; McCombe, P.A.; Steyn, F.J. Progression and survival of patients with motor neuron disease relative to their fecal microbiota. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 549–562. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Mandrioli, J.; Biguzzi, S.; Guidi, C.; Venturini, E.; Sette, E.; Terlizzi, E.; Ravasio, A.; Casmiro, M.; Salvi, F.; Liguori, R.; et al. Epidemiology of amyotrophic lateral sclerosis in Emilia Romagna Region (Italy): A population based study. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 262–268. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G. PARALS study group Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef]
- Mandrioli, J.; ERRALS Group; Biguzzi, S.; Guidi, C.; Sette, E.; Terlizzi, E.; Ravasio, A.; Casmiro, M.; Salvi, F.; Liguori, R.; et al. Heterogeneity in ALSFRS-R decline and survival: A population-based study in Italy. Neurol. Sci. 2015, 36, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; ERRALS Group; Malerba, S.A.; Beghi, E.; Fini, N.; Fasano, A.; Zucchi, E.; De Pasqua, S.; Guidi, C.; Terlizzi, E.; et al. Riluzole and other prognostic factors in ALS: A population-based registry study in Italy. J. Neurol. 2018, 265, 817–827. [Google Scholar] [CrossRef]
- Fini, N.; Georgoulopoulou, E.; Vinceti, M.; Monelli, M.; Pinelli, G.; Vacondio, P.; Giovannini, M.; Dallari, R.; Marudi, A.; Mandrioli, J. Noninvasive and invasive ventilation and enteral nutrition for ALS in Italy. Muscle Nerve 2014, 50, 508–516. [Google Scholar] [CrossRef]
- Kimura, F.; Fujimura, C.; Ishida, S.; Nakajima, H.; Furutama, D.; Uehara, H.; Shinoda, K.; Sugino, M.; Hanafusa, T. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006, 66, 265–267. [Google Scholar] [CrossRef]
- Niccolai, E.; Baldi, S.; Ricci, F.; Russo, E.; Nannini, G.; Menicatti, M.; Poli, G.; Taddei, A.; Bartolucci, G.; Calabrò, A.S.; et al. Evaluation and comparison of short chain fatty acids composition in gut diseases. World J. Gastroenterol. 2019, 25, 5543–5558. [Google Scholar] [CrossRef] [PubMed]
- Pagliai, G.; Russo, E.; Niccolai, E.; Dinu, M.; Di Pilato, V.; Magrini, A.; Bartolucci, G.; Baldi, S.; Menicatti, M.; Giusti, B.; et al. Influence of a 3-month low-calorie Mediterranean diet compared to the vegetarian diet on human gut microbiota and SCFA: The CARDIVEG Study. Eur. J. Nutr. 2019, 59, 2011–2024. [Google Scholar] [CrossRef]
- Russo, E.; Bacci, G.; Chiellini, C.; Fagorzi, C.; Niccolai, E.; Taddei, A.; Ricci, F.; Ringressi, M.N.; Borrelli, R.; Melli, F.; et al. Preliminary Comparison of Oral and Intestinal Human Microbiota in Patients with Colorectal Cancer: A Pilot Study. Front. Microbiol. 2018, 8, 2699. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Boddy, S.L.; Giovannelli, I.; Sassani, M.; Cooper-Knock, J.; Snyder, M.P.; Segal, E.; Elinav, E.; Barker, L.A.; Shaw, P.J.; McDermott, C.J. The gut microbiome: A key player in the complexity of amyotrophic lateral sclerosis (ALS). BMC Med. 2021, 19, 1–14. [Google Scholar] [CrossRef]
- Guo, J.; Yang, X.; Gao, L.; Zang, D. Evaluating the levels of CSF and serum factors in ALS. Brain Behav. 2017, 7, e00637. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Zitzelsperger, E.; Kuespert, S.; Iberl, S.; Heydn, R.; Johannesen, S.; Petri, S.; Aigner, L.; Thal, D.R.; Hermann, A.; et al. The TGF-β System as a Potential Pathogenic Player in Disease Modulation of Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortelli, R.; Zecca, C.; Piccininni, M.; Benmahamed, S.; Dell’Abate, M.T.; Barulli, M.R.; Capozzo, R.; Battista, P.; Logroscino, G. Plasma Inflammatory Cytokines Are Elevated in ALS. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.-H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflammation 2015, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-H.; Allen, K.; Oei, F.; Leoni, E.; Kuhle, J.; Tree, T.; Fratta, P.; Sharma, N.; Sidle, K.; Howard, R.; et al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflammation 2016, 3, e244. [Google Scholar] [CrossRef] [Green Version]
- Michaelson, N.; Facciponte, D.; Bradley, W.; Stommel, E. Cytokine expression levels in ALS: A potential link between inflammation and BMAA-triggered protein misfolding. Cytokine Growth Factor Rev. 2017, 37, 81–88. [Google Scholar] [CrossRef]
- Baron, P.; Bussini, S.; Cardin, V.; Corbo, M.; Conti, G.; Galimberti, D.; Scarpini, E.; Bresolin, N.; Wharton, S.B.; Shaw, P.; et al. Production of monocyte chemoattractant protein-1 in amyotrophic lateral sclerosis. Muscle Nerve 2005, 32, 541–544. [Google Scholar] [CrossRef]
- Kuhle, J.; Lindberg, R.L.P.; Regeniter, A.; Mehling, M.; Steck, A.J.; Kappos, L.; Czaplinski, A. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 16, 771–774. [Google Scholar] [CrossRef]
- Puma, C.; Danik, M.; Quirion, R.; Ramon, F.; Williams, S. The chemokine interleukin-8 acutely reduces Ca2+ currents in identified cholinergic septal neurons expressing CXCR1 and CXCR2 receptor mRNAs. J. Neurochem. 2001, 78, 960–971. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sironi, M.; Vecchi, A.; Colotta, F.; Mantovani, A.; Locati, M. IL-8 induces a specific transcriptional profile in human neutrophils: Synergism with LPS for IL-1 production. Eur. J. Immunol. 2004, 34, 2286–2292. [Google Scholar] [CrossRef] [PubMed]
- Hesse, R.; Wahler, A.; Gummert, P.; Kirschmer, S.; Otto, M.; Tumani, H.; Lewerenz, J.; Schnack, C.; Von Arnim, C.A.F. Decreased IL-8 levels in CSF and serum of AD patients and negative correlation of MMSE and IL-1β. BMC Neurol. 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-M.; Song, J.; Kim, S.; Han, C.; Park, M.H.; Koh, Y.; Jo, S.A.; Kim, Y.-Y. Identification of peripheral inflammatory markers between normal control and Alzheimer’s disease. BMC Neurol. 2011, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, D.M.; Cotman, C.W. Trophic effects of interleukin-4, -7 and -8 on hippocampal neuronal cultures: Potential involvement of glial-derived factors. Brain Res. 1993, 600, 49–55. [Google Scholar] [CrossRef]
- Gupta, P.K.; Prabhakar, S.; Sharma, S.; Anand, A. Vascular endothelial growth factor-A (VEGF-A) and chemokine ligand-2 (CCL2) in Amyotrophic Lateral Sclerosis (ALS) patients. J. Neuroinflammation 2011, 8, 47–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modgil, S.; Khosla, R.; Tiwari, A.; Sharma, K.; Anand, A. Association of Plasma Biomarkers for Angiogenesis and Proteinopathy in Indian Amyotrophic Lateral Sclerosis Patients. J. Neurosci. Rural. Pract. 2020, 11, 573–580. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, S.; Cai, H.; Gong, Z.; Zang, D. VEGF levels in CSF and serum in mild ALS patients. J. Neurol. Sci. 2014, 346, 216–220. [Google Scholar] [CrossRef]
- Nagata, T.; Nagano, I.; Shiote, M.; Narai, H.; Murakami, T.; Hayashi, T.; Shoji, M.; Abe, K. Elevation of MCP-1 and MCP-1/VEGF ratio in cerebrospinal fluid of amyotrophic lateral sclerosis patients. Neurol. Res. 2007, 29, 772–776. [Google Scholar] [CrossRef]
- Lambrechts, D.; Storkebaum, E.; Morimoto, M.; Del Favero, J.; De Smet, F.; Marklund, S.L.; Wyns, S.; Thijs, V.; Andersson, J.; Van Marion, I.; et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat. Genet. 2003, 34, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Di Gioia, D.; Cionci, N.B.; Baffoni, L.; Amoruso, A.; Pane, M.; Mogna, L.; Gaggìa, F.; Lucenti, M.A.; Bersano, E.; Cantello, R.; et al. A prospective longitudinal study on the microbiota composition in amyotrophic lateral sclerosis. BMC Med. 2020, 18, 153. [Google Scholar] [CrossRef]
- Nicholson, K.; Bjornevik, K.; Abu-Ali, G.; Chan, J.; Cortese, M.; Dedi, B.; Jeon, M.; Xavier, R.; Huttenhower, C.; Ascherio, A.; et al. The human gut microbiota in people with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-G.; Wu, S.; Yi, J.; Xia, Y.; Jin, D.; Zhou, J.; Sun, J. Target Intestinal Microbiota to Alleviate Disease Progression in Amyotrophic Lateral Sclerosis. Clin. Ther. 2017, 39, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphilaand improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2015, 65, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Van Hul, M.; Le Roy, T.; Prifti, E.; Dao, M.C.; Paquot, A.; Zucker, J.-D.; Delzenne, N.M.; Muccioli, G.G.; Clément, K.; Cani, P.D. From correlation to causality: The case of Subdoligranulum. Gut Microbes 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Consolandi, C.; Turroni, S.; Emmi, G.; Severgnini, M.; Fiori, J.; Peano, C.; Biagi, E.; Grassi, A.; Rampelli, S.; Silvestri, E.; et al. Behçet’s syndrome patients exhibit specific microbiome signature. Autoimmun. Rev. 2015, 14, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Segain, J.-P.; De La Blétière, D.R.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottiere, H.; Galmiche, J.-P. Butyrate inhibits inflammatory responses through NFkappa B inhibition: Implications for Crohn’s disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmi, G.; Bettiol, A.; Niccolai, E.; Ramazzotti, M.; Amedei, A.; Pagliai, G.; Taddei, N.; Sofi, F.; Fiorillo, C.; Prisco, D.; et al. Butyrate-Rich Diets Improve Redox Status and Fibrin Lysis in Behçet’s Syndrome. Circ. Res. 2021, 128, 278–280. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, Q.; Dorfman, R.G.; Huang, X.; Fan, T.; Zhang, H.; Zhang, J.; Yu, C. Butyrate inhibits interleukin-17 and generates Tregs to ameliorate colorectal colitis in rats. BMC Gastroenterol. 2016, 16, 84. [Google Scholar] [CrossRef] [Green Version]
- Marrali, G.; Casale, F.; Salamone, P.; Fuda, G.; Caorsi, C.; Amoroso, A.; Brunetti, M.; Restagno, G.; Barberis, M.; Bertuzzo, D.; et al. NADPH oxidase (NOX2) activity is a modifier of survival in ALS. J. Neurol. 2014, 261, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Macfabe, D.F.; Cain, D.P.; Rodriguez-Capote, K.; Franklin, A.E.; Hoffman, J.E.; Boon, F.; Taylor, A.R.; Kavaliers, M.; Ossenkopp, K.-P. Neurobiological effects of intraventricular propionic acid in rats: Possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav. Brain Res. 2007, 176, 149–169. [Google Scholar] [CrossRef] [PubMed]
- El-Ansary, A.; Shaker, G.H.; El-Gezeery, A.R.; Al-Ayadhi, L. The neurotoxic effect of clindamycin-induced gut bacterial imbalance and orally administered propionic acid on DNA damage assessed by the comet assay: Protective potency of carnosine and carnitine. Gut Pathog. 2013, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Al-Orf, N.; El-Ansary, A.; Bjørklund, G.; Moubayed, N.; Bhat, R.S.; Ben Bacha, A. Therapeutic effects of probiotics on neurotoxicity induced by clindamycin and propionic acid in juvenile hamsters. Metab. Brain Dis. 2018, 33, 1811–1820. [Google Scholar] [CrossRef]
- Zeng, Q.; Shen, J.; Chen, K.; Zhou, J.; Liao, Q.; Lu, K.; Yuan, J.; Bi, F.-F. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci. Rep. 2020, 10, 12998. [Google Scholar] [CrossRef]
- Hertzberg, V.S.; Singh, H.; Fournier, C.N.; Moustafa, A.; Polak, M.; Kuelbs, C.A.; Torralba, M.G.; Tansey, M.G.; Nelson, K.E.; Glass, J.D. Gut microbiome differences between amyotrophic lateral sclerosis patients and spouse controls. Amyotroph. Lateral Scler. Front. Degener. 2021, 1–9. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | ALS Patients |
|---|---|
| Sex, %male | 70% |
| Age at enrollment, mean (SD) | 59 (12) |
| Height (cm), mean (SD) | 173 (7.6) |
| Weight (kg), mean (SD) | 80 (13.5) |
| BMI, mean (SD) | 26.8 (4.3) |
| ALSFRS-R at sampling, mean (SD) | 32 (7.4) |
| Clinical Phenotype, n (%) | Bulbar, 1 (5) |
| Classic, 9 (47) | |
| Flail arm/leg, 4 (21) | |
| UMNp, 5 (26) | |
| El Escorial, n (%) | Definite, 7 (37) |
| Clinical probable, 6 (32) | |
| Laboratory-supported, 1 (5) | |
| Fast, 8 (42) | |
| Progression rate, n (%) | Slow, 8 (42) |
| Medium, 3 (16) | |
| Fast, 8 (42) | |
| Survival (days from symptom onset, mean (SD)) | 1929 (1374.7) |
| Site onset, n (%) | Bulbar, 2 (10) |
| Spinal, 17 (90) |
| Condition | Acetic Acid (umol/g) | Propionic Acid (umol/g) | Butyric Acid (umol/g) | Iso-Butyric Acid (umol/g) | Iso-Valeric Acid (umol/g) | Valeric Acid (umol/g) | Total (umol/g) | |
|---|---|---|---|---|---|---|---|---|
| HC | 27.05 ± 10.59 | 8.79 ± 3.48 | 7.28 ± 3.99 | 1.43 ± 0.81 | 1.15 ± 0.89 | 1.28 ± 0.40 | 54.22 ± 19.88 | |
| ALS (all patients) | 22.89 ± 13.9 | 8.05 ± 5.03 | 8.80± 6.92 | 1.49 ± 1.28 | 0.95 ± 1.23 | 1.36 ± 1.21 | 52.93 ± 34.23 | |
| ALS Subgroups | ||||||||
| El Escorial Criteria | EEC-R0 | 17.91 ± 11.06 | 5.72 ± 4.78 | 6.79 ± 6.19 | 1.05 ± 1.48 | 0.95 ± 1.31 | 1.07 ± 1.28 | 37.73 ± 31.80 |
| EEC-R1 | 15.76 ± 12.06 | 7.83 ± 4.22 | 4.84 ± 6.42 | 1.03 ± 1.30 | 0.85 ± 1.49 | 1.72 ± 1.46 | 35.62 ± 30.28 | |
| EEC-R2 | 2.80 | 1.54 | 0.69 | 0.51 | 0.63 | 0.30 | 7.10 | |
| EEC-R3 | 27.22 ± 17.27 | 5.75 ± 6.67 | 7.81 ± 8.67 | 1.76 ± 1.23 | 1.61 ± 1.09 | 1.13 ± 0.88 | 53.18 ± 41.38 | |
| Clinical phenotype | Cp | 16.90 ± 16.54 | 7.83 ± 4.92 | 6.05 ± 7.38 | 0.65 ± 1.19 | 0.54 ± 0.98 | 1.25 ± 1.19 | 37.07 ± 37.51 |
| Bp | 17.68 | 5.72 | 4.54 | 0.35 | 0.28 | 1.07 | 34.08 | |
| Fp | 22.81 ± 5.27 | 5.25 ± 0.71 | 6.38 ± 1.99 | 1.76 ± 0.50 | 1.61 ± 0.49 | 1.13 ± 0.19 | 45.74 ± 10.93 | |
| UMN-p | 20.00 ± 15.20 | 12.17 ± 6.83 | 15.38 ± 8.49 | 3.07 ± 1.53 | 2.70 ± 1.56 | 2.68 ± 1.67 | 75.16 ± 42.11 | |
| Progression rate | Slow | 19.50 ± 12.37 | 6.07 ± 4.62 | 5.85 ± 6.38 | 1.40 ± 1.12 | 1.28 ± 1.04 | 1.26 ± 1.11 | 41.08 ± 31.99 |
| Medium | 17.91 ± 13.28 | 7.48 ± 3.41 | 6.79 ± 5.34 | 1.35 ± 0.46 | 1.12 ± 0.44 | 1.67 ± 0,78 | 37.73 ± 27.31 | |
| Fast | 18.84 ± 17.09 | 8.78 ± 6.28 | 9.09 ± 8.40 | 0.69 ± 1.69 | 0.56 ± 1.64 | 1.05 ± 1.54 | 54.62 ± 41.77 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niccolai, E.; Di Pilato, V.; Nannini, G.; Baldi, S.; Russo, E.; Zucchi, E.; Martinelli, I.; Menicatti, M.; Bartolucci, G.; Mandrioli, J.; et al. The Gut Microbiota-Immunity Axis in ALS: A Role in Deciphering Disease Heterogeneity? Biomedicines 2021, 9, 753. https://doi.org/10.3390/biomedicines9070753
Niccolai E, Di Pilato V, Nannini G, Baldi S, Russo E, Zucchi E, Martinelli I, Menicatti M, Bartolucci G, Mandrioli J, et al. The Gut Microbiota-Immunity Axis in ALS: A Role in Deciphering Disease Heterogeneity? Biomedicines. 2021; 9(7):753. https://doi.org/10.3390/biomedicines9070753
Chicago/Turabian StyleNiccolai, Elena, Vincenzo Di Pilato, Giulia Nannini, Simone Baldi, Edda Russo, Elisabetta Zucchi, Ilaria Martinelli, Marta Menicatti, Gianluca Bartolucci, Jessica Mandrioli, and et al. 2021. "The Gut Microbiota-Immunity Axis in ALS: A Role in Deciphering Disease Heterogeneity?" Biomedicines 9, no. 7: 753. https://doi.org/10.3390/biomedicines9070753