
Selective Inhibition of HDAC Class I Sensitizes Leukemia and Neuroblastoma Cells to Anticancer Drugs

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cellular Assays
2.2. Drugs and Compounds
2.3. GEO Datasets
2.4. Elastic NET Regularization
2.5. qRT-PCR
2.6. Immunocytochemistry and Confocal Imaging
2.7. Quantification of ERK Activity with ERK-KTR Reporter
2.8. Statistical Analysis
2.9. Code Availability
3. Results
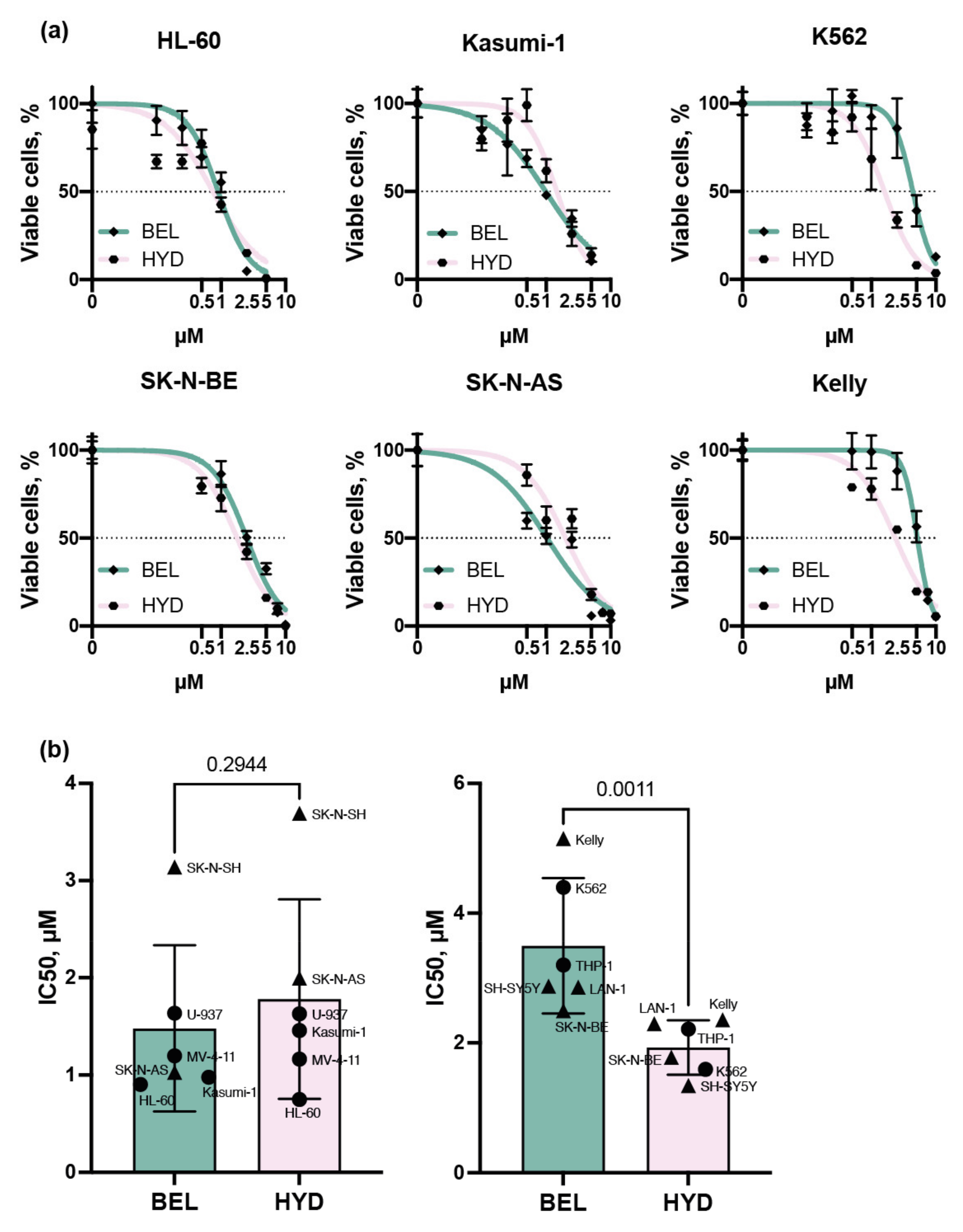
3.1. Leukemia and Neuroblastoma Cells Have Different Sensitivity to Belinostat and Hydrazostat
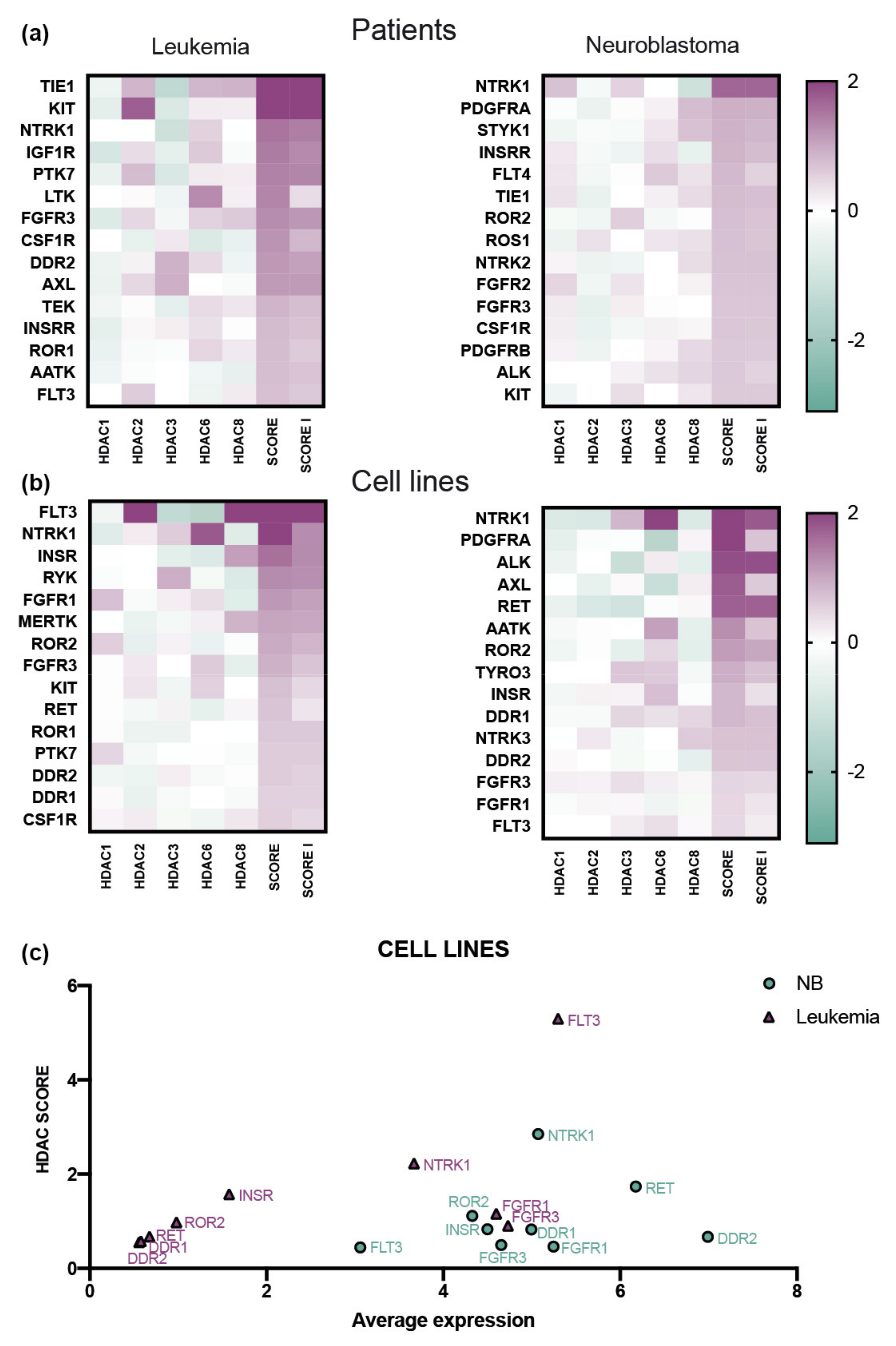
3.2. The Expression of HDAC Class I Correlates with the Expression of Kinases NTRK1 and FGFR3 in Leukemia and NB Patients and Cell Lines
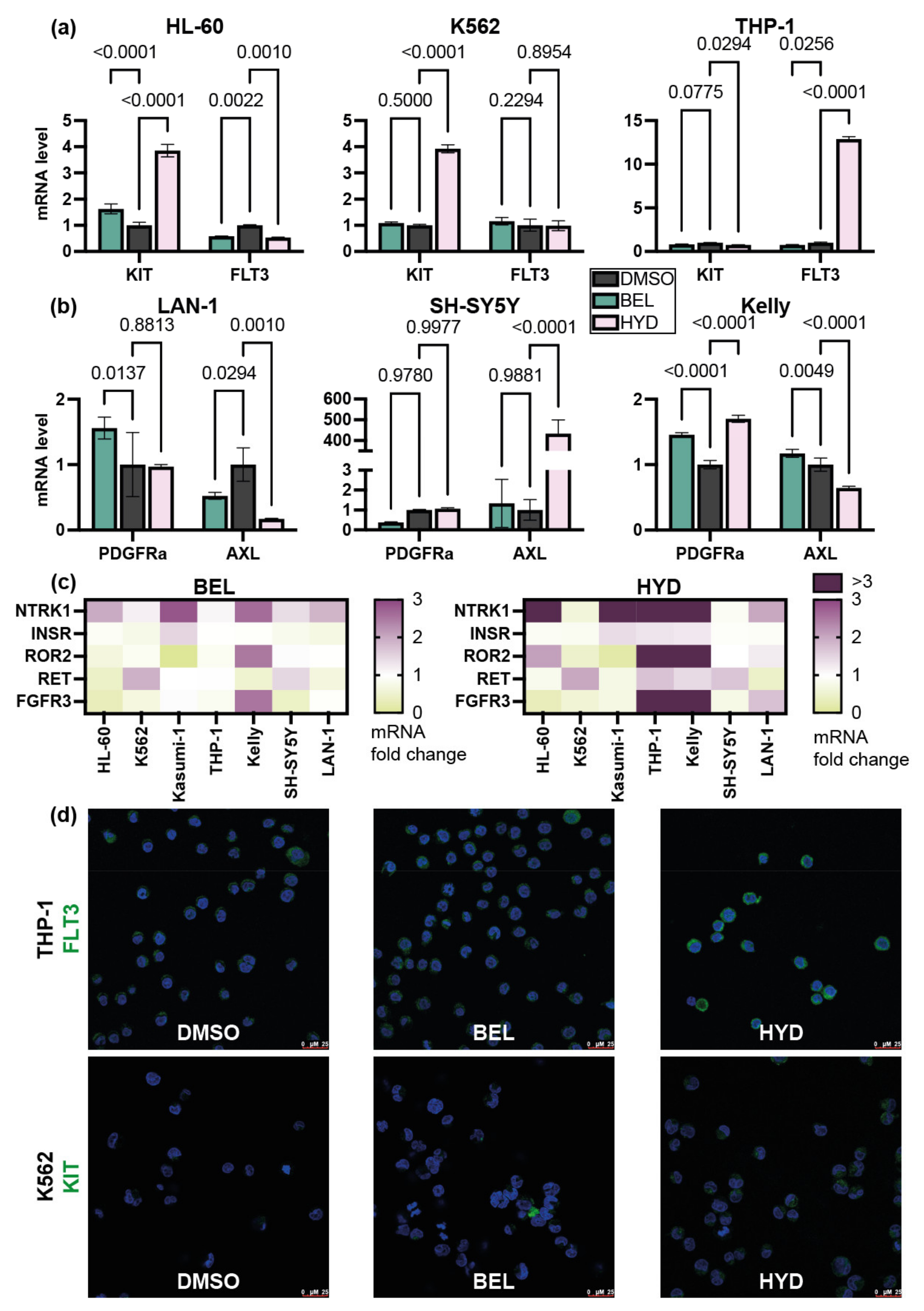
3.3. Hydrazostat Induce the Expression of RTK-Coding Genes in NB and Leukemia Cells
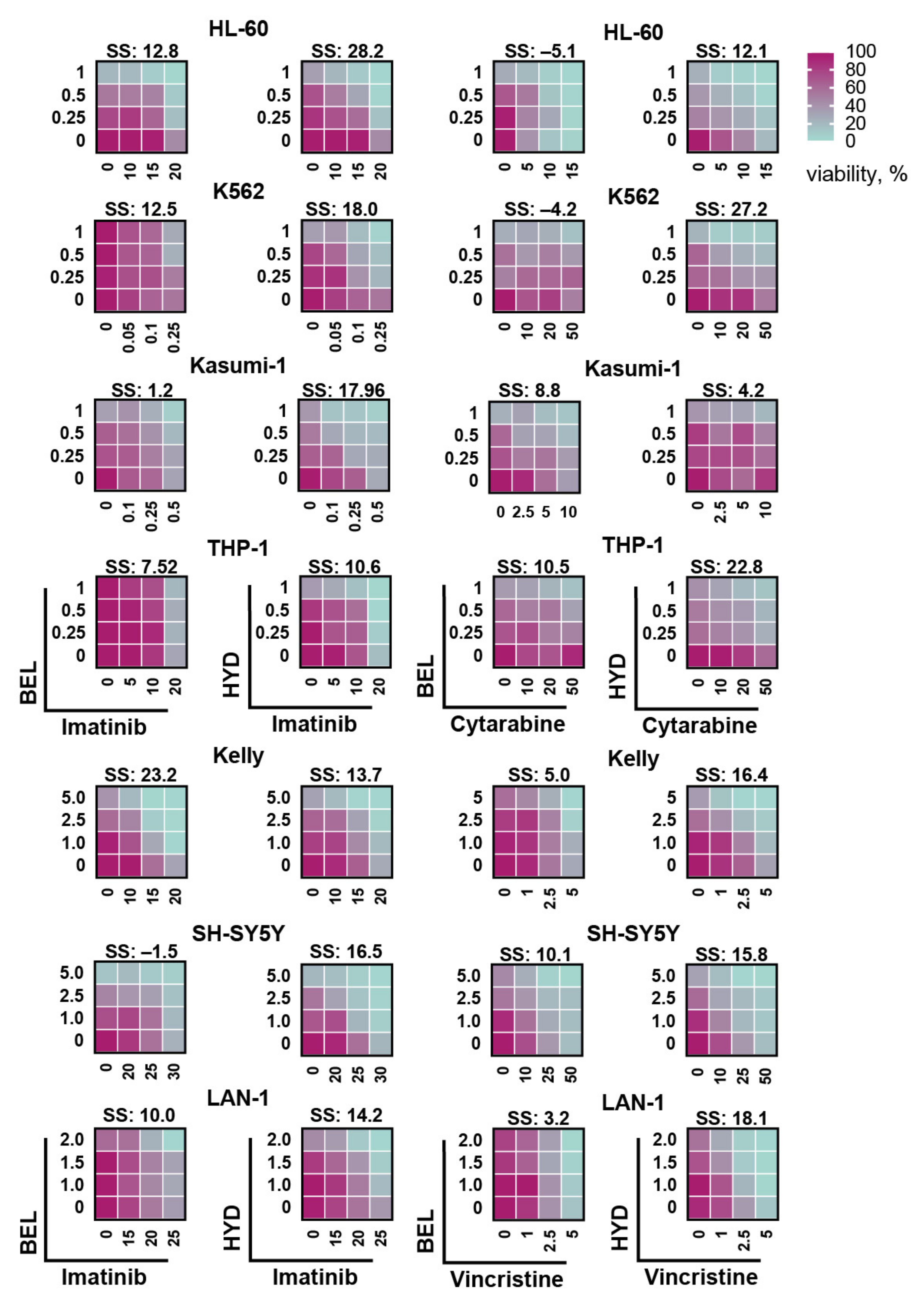
3.4. Hydrazostat Makes Cells More Sensitive to RTK Inhibitor Imatinib and Cytotoxic Drugs
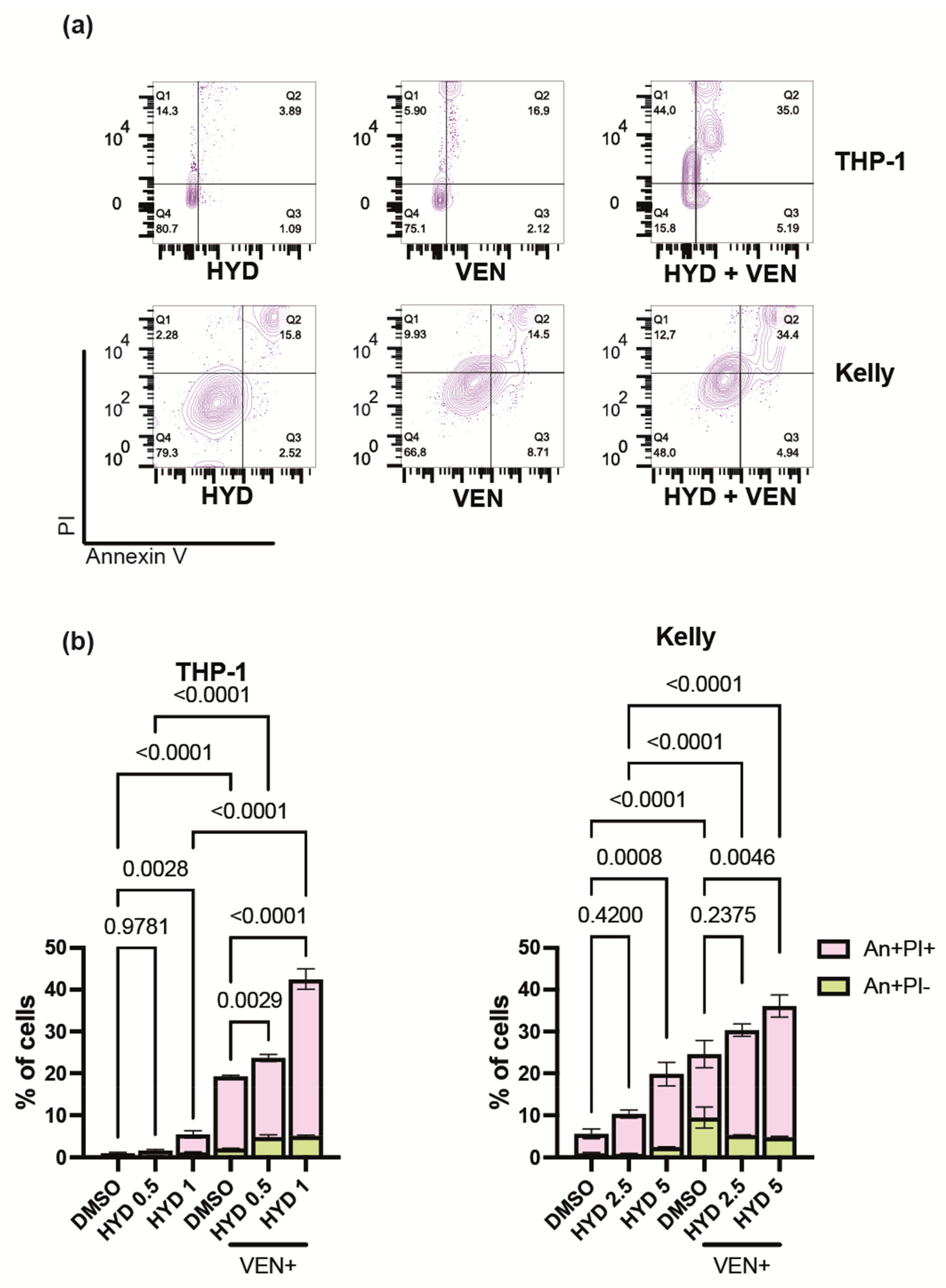
3.5. Hydrazostat Enhances Venetoclax-Induced Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lebedev, T.D.; Vagapova, E.R.; Popenko, V.I.; Leonova, O.G.; Spirin, P.V.; Prassolov, V.S. Two Receptors, Two Isoforms, Two Cancers: Comprehensive Analysis of KIT and TrkA Expression in Neuroblastoma and Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1046. [Google Scholar] [CrossRef]
- Cortes, J.; Perl, A.E.; Döhner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 Inhibitor, as Monotherapy in Patients with Relapsed or Refractory Acute Myeloid Leukaemia: An Open-Label, Multicentre, Single-Arm, Phase 2 Trial. Lancet. Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Levis, M.; Allebach, J.; Tse, K.-F.; Zheng, R.; Baldwin, B.R.; Smith, B.D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-Targeted Tyrosine Kinase Inhibitor Is Cytotoxic to Leukemia Cells in Vitro and in Vivo. Blood 2002, 99, 3885–3891. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Varela, C.R.; Light, J.E.; Kolla, V.; Evans, A.E. Trk Receptor Expression and Inhibition in Neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [Google Scholar] [CrossRef] [Green Version]
- Beppu, K.; Jaboine, J.; Merchant, M.S.; Mackall, C.L.; Thiele, C.J. Effect of Imatinib Mesylate on Neuroblastoma Tumorigenesis and Vascular Endothelial Growth Factor Expression. J. Natl. Cancer Inst. 2004, 96, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Q.; Halilovic, E.; Li, X.; Liang, J.; Cao, Y.; Rakiec, D.P.; Ruddy, D.A.; Jeay, S.; Wuerthner, J.U.; Timple, N.; et al. Combined ALK and MDM2 Inhibition Increases Antitumor Activity and Overcomes Resistance in Human ALK Mutant Neuroblastoma Cell Lines and Xenograft Models. Elife 2017, 6, e17137. [Google Scholar] [CrossRef] [Green Version]
- Calafiore, L.; Amoroso, L.; Della Casa Alberighi, O.; Luksch, R.; Zanazzo, G.; Castellano, A.; Podda, M.; Dominici, C.; Haupt, R.; Corrias, M.V.; et al. Two-Stage Phase II Study of Imatinib Mesylate in Subjects with Refractory or Relapsing Neuroblastoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 1406–1413. [Google Scholar] [CrossRef]
- Morandi, F.; Amoroso, L.; Dondero, A.; Castriconi, R.; Parodi, S.; Luksch, R.; Casale, F.; Castellano, A.; Garaventa, A.; Moretta, A.; et al. Updated Clinical and Biological Information from the Two-Stage Phase II Study of Imatinib Mesylate in Subjects with Relapsed/Refractory Neuroblastoma. Oncoimmunology 2018, 7, e1468953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, J.H.; Voss, S.D.; Hall, D.C.; Minard, C.G.; Balis, F.M.; Wilner, K.; Berg, S.L.; Fox, E.; Adamson, P.C.; Blaney, S.; et al. Activity of Crizotinib in Patients with ALK-Aberrant Relapsed/Refractory Neuroblastoma: A Children’s Oncology Group Study (ADVL0912). Clin. Cancer Res. 2021, 27, 3543–3548. [Google Scholar] [CrossRef]
- Vitali, R.; Mancini, C.; Cesi, V.; Tanno, B.; Piscitelli, M.; Mancuso, M.; Sesti, F.; Pasquali, E.; Calabretta, B.; Dominici, C.; et al. Activity of Tyrosine Kinase Inhibitor Dasatinib in Neuroblastoma Cells in Vitro and in Orthotopic Mouse Model. Int. J. Cancer 2009, 125, 2547–2555. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary Mutations as Mediators of Resistance to Targeted Therapy in Leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.R.; Breccia, M.; Abruzzese, E.; Castagnetti, F.; Luciano, L.; Gozzini, A.; Annunziata, M.; Martino, B.; Stagno, F.; Cavazzini, F.; et al. Outcome of 82 Chronic Myeloid Leukemia Patients Treated with Nilotinib or Dasatinib after Failure of Two Prior Tyrosine Kinase Inhibitors. Haematologica 2013, 98, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; La Rosée, P.; Stoffregen, E.P.; Druker, B.J.; Deininger, M.W. Several Bcr-Abl Kinase Domain Mutants Associated with Imatinib Mesylate Resistance Remain Sensitive to Imatinib. Blood 2003, 101, 4611–4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in the Blast Crisis of Chronic Myeloid Leukemia and Acute Lymphoblastic Leukemia with the Philadelphia Chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyers, C.L.; Hochhaus, A.; Feldman, E.; Goldman, J.M.; Miller, C.B.; Ottmann, O.G.; Schiffer, C.A.; Talpaz, M.; Guilhot, F.; Deininger, M.W.N.; et al. Imatinib Induces Hematologic and Cytogenetic Responses in Patients with Chronic Myelogenous Leukemia in Myeloid Blast Crisis: Results of a Phase II Study. Blood 2002, 99, 3530–3539. [Google Scholar] [CrossRef]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, Mechanism, and Inhibition of Histone Deacetylases and Related Metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Napper, A.D.; Hixon, J.; McDonagh, T.; Keavey, K.; Pons, J.-F.; Barker, J.; Yau, W.T.; Amouzegh, P.; Flegg, A.; Hamelin, E.; et al. Discovery of Indoles as Potent and Selective Inhibitors of the Deacetylase SIRT1. J. Med. Chem. 2005, 48, 8045–8054. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC Family: What Are the Cancer Relevant Targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Greer, C.B.; Tanaka, Y.; Kim, Y.J.; Xie, P.; Zhang, M.Q.; Park, I.-H.; Kim, T.H. Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery. Cell Rep. 2015, 13, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- San José-Enériz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef] [Green Version]
- McDermott, J.; Jimeno, A. Belinostat for the Treatment of Peripheral T-Cell Lymphomas. Drugs Today 2014, 50, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Su, Y.; Madlambayan, G.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Ma, J.; Knight, T.; et al. Antileukemic Activity and Mechanism of Action of the Novel PI3K and Histone Deacetylase Dual Inhibitor CUDC-907 in Acute Myeloid Leukemia. Haematologica 2019, 104, 2225–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Zha, J.; Shi, Y.; Li, Y.; Yuan, D.; Chen, Q.; Lin, F.; Fang, Z.; Yu, Y.; Dai, Y.; et al. Co-Inhibition of HDAC and MLL-Menin Interaction Targets MLL-Rearranged Acute Myeloid Leukemia Cells via Disruption of DNA Damage Checkpoint and DNA Repair. Clin. Epigenet. 2019, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Dedoni, S.; Marras, L.; Olianas, M.C.; Ingianni, A.; Onali, P. Downregulation of TrkB Expression and Signaling by Valproic Acid and Other Histone Deacetylase Inhibitors. J. Pharmacol. Exp. Ther. 2019, 370, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Fang, W.Y.; Chang, L.; Gao, W.H.; Shen, Y.; Jia, M.Y.; Zhang, Y.X.; Wang, Y.; Dou, H.B.; Zhang, W.J.; et al. Targeting HDAC3, a New Partner Protein of AKT in the Reversal of Chemoresistance in Acute Myeloid Leukemia via DNA Damage Response. Leukemia 2017, 31, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.-L.; Bonine-Summers, A.R.; et al. Hdac3 Is Essential for the Maintenance of Chromatin Structure and Genome Stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W.R. Structure of HDAC3 Bound to Co-Repressor and Inositol Tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in Normal and Malignant Hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelmetti, V.; Zhang, J.; Fanelli, M.; Minucci, S.; Pelicci, P.G.; Lazar, M.A. Aberrant Recruitment of the Nuclear Receptor Corepressor-Histone Deacetylase Complex by the Acute Myeloid Leukemia Fusion Partner ETO. Mol. Cell. Biol. 1998, 18, 7185–7191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, H.; Scaglioni, P.P.; Bhaumik, M.; Rego, E.M.; Cai, L.F.; Majid, S.M.; Miyachi, H.; Kakizuka, A.; Miller, W.H.; Pandolfi, P.P.; et al. In Vivo Analysis of the Role of Aberrant Histone Deacetylase Recruitment and RAR Alpha Blockade in the Pathogenesis of Acute Promyelocytic Leukemia. J. Exp. Med. 2006, 203, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlov, M.V.; Konduktorov, K.A.; Shcherbakova, A.S.; Kochetkov, S.N. Synthesis of N′-Propylhydrazide Analogs of Hydroxamic Inhibitors of Histone Deacetylases (HDACs) and Evaluation of Their Impact on Activities of HDACs and Replication of Hepatitis C Virus (HCV). Bioorg. Med. Chem. Lett. 2019, 29, 2369–2374. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; He, L.; Aittokallio, T.; Tang, J. SynergyFinder: A Web Application for Analyzing Drug Combination Dose-Response Matrix Data. Bioinformatics 2017, 33, 2413–2415. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Sanders, M.A.; Gilissen, C.; Tönnissen, E.; van der Heijden, A.; Döhner, K.; Bullinger, L.; Jansen, J.H.; Valk, P.J.M.; van der Reijden, B.A. High BRE Expression Predicts Favorable Outcome in Adult Acute Myeloid Leukemia, in Particular among MLL-AF9-Positive Patients. Blood 2011, 118, 5613–5621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of Neuroblastoma Identifies Chromothripsis and Defects in Neuritogenesis Genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef] [PubMed]
- R2: Genomics Analysis and Visualization Platform. Available online: http://r2.amc.nl (accessed on 26 September 2021).
- CancerCellBiology at GitHub. Available online: https://github.com/CancerCellBiology/Vagapova-et-al.-2021-Hydrazostat (accessed on 28 November 2021).
- Lebedev, T.; Vagapova, E.; Spirin, P.; Rubtsov, P.; Astashkova, O.; Mikheeva, A.; Sorokin, M.; Vladimirova, U.; Suntsova, M.; Konovalov, D.; et al. Growth Factor Signaling Predicts Therapy Resistance Mechanisms and Defines Neuroblastoma Subtypes. Oncogene 2021, 40, 6258–6272. [Google Scholar] [CrossRef] [PubMed]
- Regot, S.; Hughey, J.J.; Bajar, B.T.; Carrasco, S.; Covert, M.W. High-Sensitivity Measurements of Multiple Kinase Activities in Live Single Cells. Cell 2014, 157, 1724–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentiviral Gene Ontology Vectors. Available online: http://www.lentigo-vectors.de (accessed on 1 September 2021).
- Spangle, J.M.; Roberts, T.M. Epigenetic Regulation of RTK Signaling. J. Mol. Med. 2017, 95, 791–798. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.T.; Mojumdar, K.; Ng, P.K.-S.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018, 25, 1304–1317.e5. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Tefferi, A. Imatinib Targets Other than Bcr/Abl and Their Clinical Relevance in Myeloid Disorders. Blood 2004, 104, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- CancerRxGene. Available online: https://www.cancerrxgene.org (accessed on 23 November 2021).
- Long, J.; Jia, M.-Y.; Fang, W.-Y.; Chen, X.-J.; Mu, L.-L.; Wang, Z.-Y.; Shen, Y.; Xiang, R.-F.; Wang, L.-N.; Wang, L.; et al. FLT3 Inhibition Upregulates HDAC8 via FOXO to Inactivate P53 and Promote Maintenance of FLT3-ITD+ Acute Myeloid Leukemia. Blood 2020, 135, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-Generation of Selective Histone Deacetylase Inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Stowe, R.; Pinello, C.E.; Tian, G.; Madoux, F.; Li, D.; Zhao, L.Y.; Li, J.-L.; Wang, Y.; Wang, Y.; et al. Identification of Histone Deacetylase Inhibitors with Benzoylhydrazide Scaffold That Selectively Inhibit Class I Histone Deacetylases. Chem. Biol. 2015, 22, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Chen, Q.; Shimada, K.; Tang, M.; Li, H.; Gurumurthy, A.; Khoury, J.D.; Xu, B.; Huang, S.; Qiu, Y. Histone Deacetylase Inhibitor Targets CD123/CD47-Positive Cells and Reverse Chemoresistance Phenotype in Acute Myeloid Leukemia. Leukemia 2018, 1, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective Inhibition of HDAC8 Decreases Neuroblastoma Growth in Vitro and in Vivo and Enhances Retinoic Acid-Mediated Differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef] [PubMed]
- Iraci, N.; Diolaiti, D.; Papa, A.; Porro, A.; Valli, E.; Gherardi, S.; Herold, S.; Eilers, M.; Bernardoni, R.; Della Valle, G.; et al. A SP1/MIZ1/MYCN Repression Complex Recruits HDAC1 at the TRKA and P75NTR Promoters and Affects Neuroblastoma Malignancy by Inhibiting the Cell Response to NGF. Cancer Res. 2011, 71, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Frerich, J.M.; Turtzo, L.C.; Li, S.; Chiang, J.; Yang, C.; Wang, X.; Zhang, C.; Wu, C.; Sun, Z.; et al. Histone Deacetylase Inhibitors Are Neuroprotective and Preserve NGF-Mediated Cell Survival Following Traumatic Brain Injury. Proc. Natl. Acad. Sci. USA 2013, 110, 10747–10752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meel, M.H.; de Gooijer, M.C.; Metselaar, D.S.; Sewing, A.C.P.; Zwaan, K.; Waranecki, P.; Breur, M.; Buil, L.C.M.; Lagerweij, T.; Wedekind, L.E.; et al. Combined Therapy of AXL and HDAC Inhibition Reverses Mesenchymal Transition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2020, 26, 3319–3332. [Google Scholar] [CrossRef] [Green Version]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.-K.; Gray, N.S.; George, R.E. ALK Inhibitor Resistance in ALKF1174L-Driven Neuroblastoma Is Associated with AXL Activation and Induction of EMT. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lernoux, M.; Schnekenburger, M.; Losson, H.; Vermeulen, K.; Hahn, H.; Gérard, D.; Lee, J.-Y.; Mazumder, A.; Ahamed, M.; Christov, C.; et al. Novel HDAC Inhibitor MAKV-8 and Imatinib Synergistically Kill Chronic Myeloid Leukemia Cells via Inhibition of BCR-ABL/MYC-Signaling: Effect on Imatinib Resistance and Stem Cells. Clin. Epigenet. 2020, 12, 69. [Google Scholar] [CrossRef]
- Pietschmann, K.; Bolck, H.A.; Buchwald, M.; Spielberg, S.; Polzer, H.; Spiekermann, K.; Bug, G.; Heinzel, T.; Böhmer, F.-D.; Krämer, O.H. Breakdown of the FLT3-ITD/STAT5 Axis and Synergistic Apoptosis Induction by the Histone Deacetylase Inhibitor Panobinostat and FLT3-Specific Inhibitors. Mol. Cancer Ther. 2012, 11, 2373–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bali, P.; George, P.; Cohen, P.; Tao, J.; Guo, F.; Sigua, C.; Vishvanath, A.; Scuto, A.; Annavarapu, S.; Fiskus, W.; et al. Superior Activity of the Combination of Histone Deacetylase Inhibitor LAQ824 and the FLT-3 Kinase Inhibitor PKC412 against Human Acute Myelogenous Leukemia Cells with Mutant FLT-3. Clin. Cancer Res. 2004, 10, 4991–4997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyrenne, B.M.; Lewis, J.M.; Weed, J.G.; Carlson, K.R.; Mirza, F.N.; Foss, F.M.; Girardi, M. Synergy of BCL2 and Histone Deacetylase Inhibition against Leukemic Cells from Cutaneous T-Cell Lymphoma Patients. Blood 2017, 130, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Berghauser Pont, L.M.E.; Spoor, J.K.H.; Venkatesan, S.; Swagemakers, S.; Kloezeman, J.J.; Dirven, C.M.F.; van der Spek, P.J.; Lamfers, M.L.M.; Leenstra, S. The Bcl-2 Inhibitor Obatoclax Overcomes Resistance to Histone Deacetylase Inhibitors SAHA and LBH589 as Radiosensitizers in Patient-Derived Glioblastoma Stem-like Cells. Genes Cancer 2014, 5, 445–459. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vagapova, E.; Kozlov, M.; Lebedev, T.; Ivanenko, K.; Leonova, O.; Popenko, V.; Spirin, P.; Kochetkov, S.; Prassolov, V. Selective Inhibition of HDAC Class I Sensitizes Leukemia and Neuroblastoma Cells to Anticancer Drugs. Biomedicines 2021, 9, 1846. https://doi.org/10.3390/biomedicines9121846
Vagapova E, Kozlov M, Lebedev T, Ivanenko K, Leonova O, Popenko V, Spirin P, Kochetkov S, Prassolov V. Selective Inhibition of HDAC Class I Sensitizes Leukemia and Neuroblastoma Cells to Anticancer Drugs. Biomedicines. 2021; 9(12):1846. https://doi.org/10.3390/biomedicines9121846
Chicago/Turabian StyleVagapova, Elmira, Maxim Kozlov, Timofey Lebedev, Karina Ivanenko, Olga Leonova, Vladimir Popenko, Pavel Spirin, Sergey Kochetkov, and Vladimir Prassolov. 2021. "Selective Inhibition of HDAC Class I Sensitizes Leukemia and Neuroblastoma Cells to Anticancer Drugs" Biomedicines 9, no. 12: 1846. https://doi.org/10.3390/biomedicines9121846