
Epigenetic Regulators of DNA Cytosine Modification: Promising Targets for Cancer Therapy

Abstract
:1. Introduction
2. DNA Methyltransferases and Their Inhibitors
2.1. The Role of DNA Methyltransferases
2.2. Impact of DNMT Aberrations on Cancers
2.3. DNMT Inhibitors
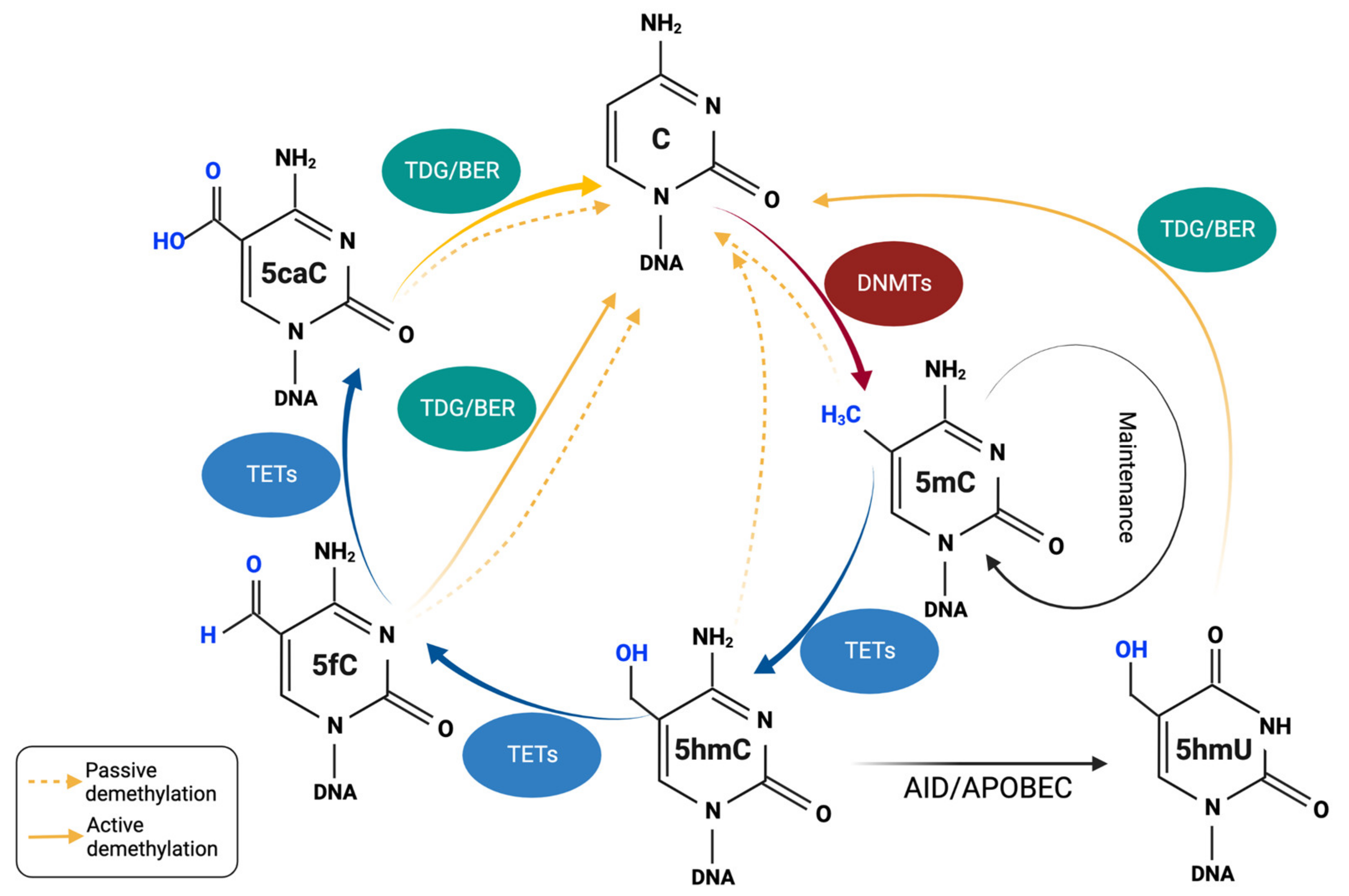
3. DNA Demethylation and TET Proteins
3.1. TETs and the Mechanisms of Passive/Active DNA Demethylation
3.2. LOF of TET and Hematopoietic Cancer
3.3. TET Deficiency and Solid Cancers
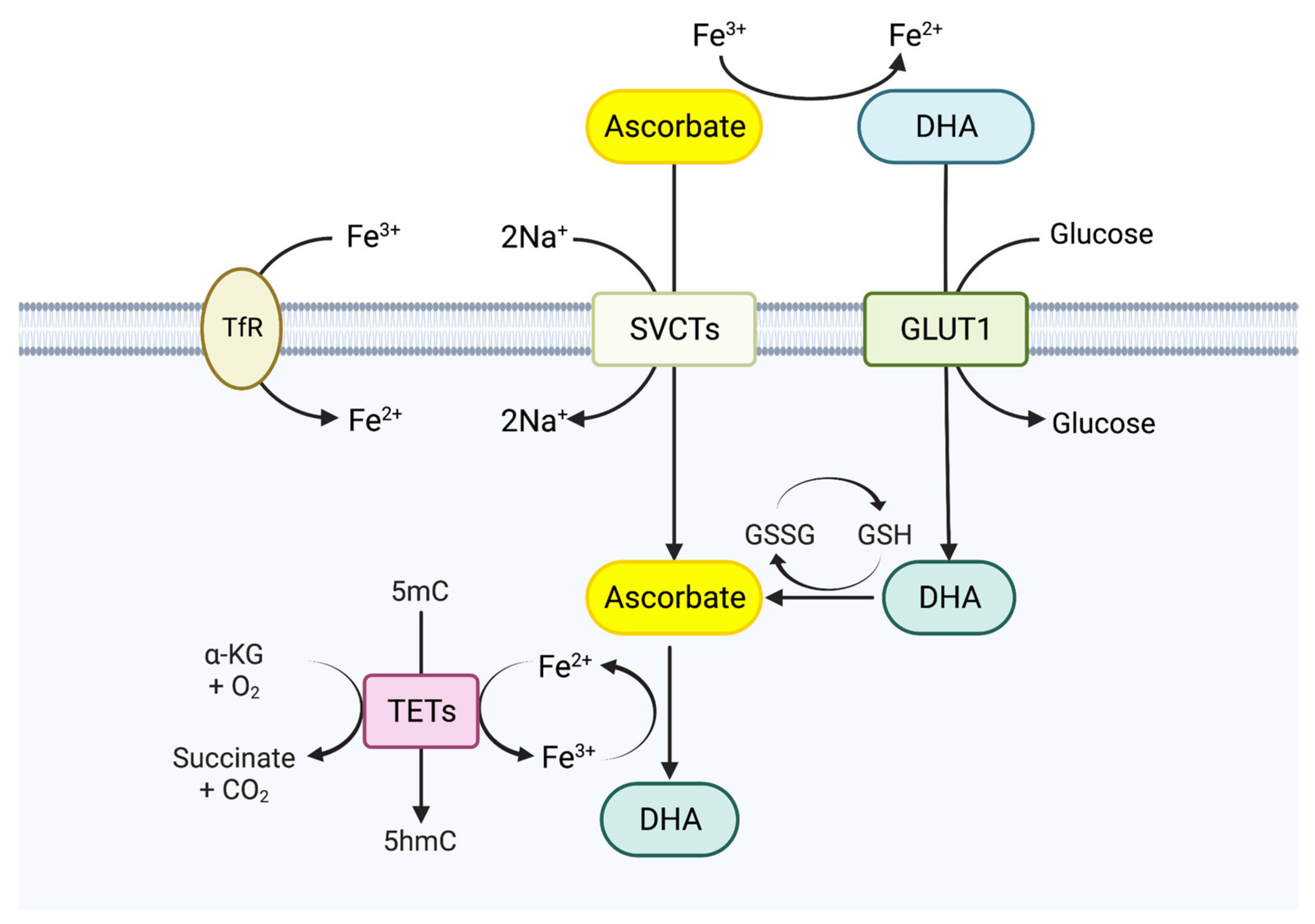
4. TET Modulators and Cancer Therapy: Role of Vitamin C
4.1. Biological Functions of Vitamin C
4.2. TET Enzymes and Vitamin C
4.3. Vitamin C in Cancer Therapy
4.4. Combination Therapy
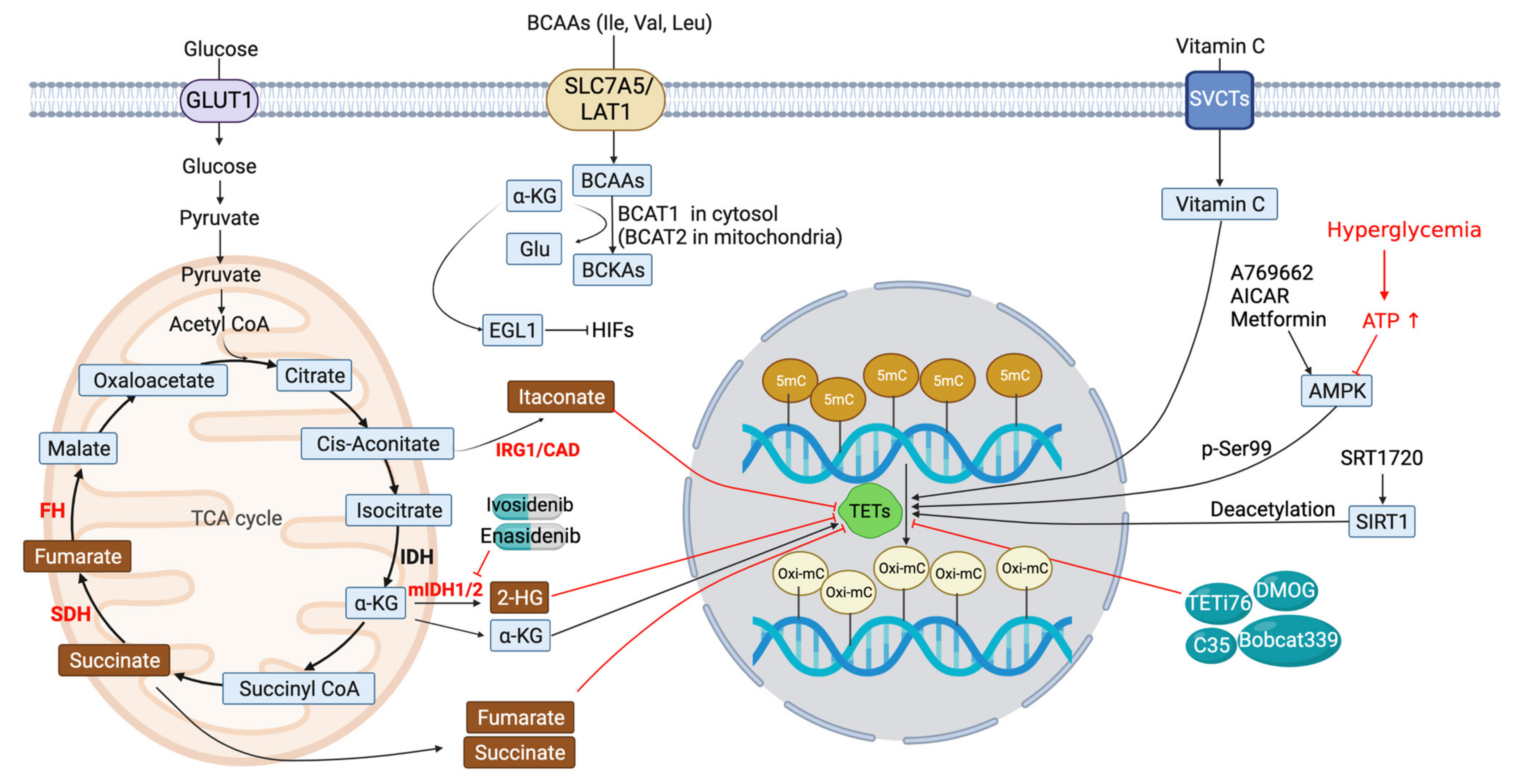
5. Metabolic Modulation of TET Function
5.1. IDH Inhibitors
5.2. Succinate and Fumarate
5.3. Itaconate (ITA)
5.4. BCAA Transaminase 1 (BCAT1)
6. Other TET Modulators
6.1. AMP-Activated Kinase (AMPK)
6.2. Silence Information Regulator 1 (SIRT1)
6.3. Other Potential TET Antagonists (Figure 3 and Table 2)
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brien, G.L.; Gambero, G.; O’connell, D.J.; Jerman, E.; Turner, S.A.; Egan, C.M.; Dunne, E.J.; Jurgens, M.C.; Wynne, K.; Piao, L. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 2012, 19, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fang, J.; Bedford, M.T.; Zhang, Y.; Xu, R.-M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 2006, 312, 748–751. [Google Scholar] [CrossRef]
- Li, H.; Ilin, S.; Wang, W.; Duncan, E.M.; Wysocka, J.; Allis, C.D.; Patel, D.J. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 2006, 442, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Brabson, J.P.; Leesang, T.; Mohammad, S.; Cimmino, L. Epigenetic Regulation of Genomic Stability by Vitamin C. Front. Genet. 2021, 12, 675780. [Google Scholar] [CrossRef]
- Lorsbach, R.; Moore, J.; Mathew, S.; Raimondi, S.; Mukatira, S.; Downing, J. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t (10; 11)(q22; q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Maiques-Diaz, A.; Somervaille, T.C. LSD1: Biologic roles and therapeutic targeting. Epigenomics 2016, 8, 1103–1116. [Google Scholar] [CrossRef] [Green Version]
- Jambhekar, A.; Anastas, J.N.; Shi, Y. Histone Lysine Demethylase Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026484. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Moyado, I.F.; Tsagaratou, A.; Yuita, H.; Seo, H.; Delatte, B.; Heinz, S.; Benner, C.; Rao, A. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc. Natl. Acad. Sci. USA 2019, 116, 16933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouyahya, A.; Mechchate, H.; Oumeslakht, L.; Zeouk, I.; Aboulaghras, S.; Balahbib, A.; Zengin, G.; Kamal, M.A.; Gallo, M.; Montesano, D.; et al. The Role of Epigenetic Modifications in Human Cancers and the Use of Natural Compounds as Epidrugs: Mechanistic Pathways and Pharmacodynamic Actions. Biomolecules 2022, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Kaminskas, E.; Farrell, A.; Abraham, S.; Baird, A.; Hsieh, L.S.; Lee, S.L.; Leighton, J.K.; Patel, H.; Rahman, A.; Sridhara, R.; et al. Approval summary: Azacitidine for treatment of myelodysplastic syndrome subtypes. Clin. Cancer Res. 2005, 11, 3604–3608. [Google Scholar] [CrossRef] [Green Version]
- Inbar-Feigenberg, M.; Choufani, S.; Butcher, D.T.; Roifman, M.; Weksberg, R. Basic concepts of epigenetics. Fertil. Steril. 2013, 99, 607–615. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA Methyltransferases in Cancer: Biology, Paradox, Aberrations, and Targeted Therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.-P.; Guo, F.; Yang, H.; Wu, H.-P.; Xu, G.-F.; Liu, W.; Xie, Z.-G.; Shi, L.; He, X.; Jin, S.-G.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsumura, A.; Hayakawa, T.; Kumaki, Y.; Takebayashi, S.-I.; Sakaue, M.; Matsuoka, C.; Shimotohno, K.; Ishikawa, F.; Li, E.; Ueda, H.R.; et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 2006, 11, 805–814. [Google Scholar] [CrossRef]
- Howard, G.; Eiges, R.; Gaudet, F.; Jaenisch, R.; Eden, A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 2008, 27, 404–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Søes, S.; Daugaard, I.L.; Sørensen, B.S.; Carus, A.; Mattheisen, M.; Alsner, J.; Overgaard, J.; Hager, H.; Hansen, L.L.; Kristensen, L.S. Hypomethylation and increased expression of the putative oncogene ELMO3 are associated with lung cancer development and metastases formation. Oncoscience 2014, 1, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Ushijima, S.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003, 192, 75–82. [Google Scholar] [CrossRef]
- Lue, N.Z.; Garcia, E.M.; Ngan, K.C.; Lee, C.; Doench, J.G.; Liau, B.B. Base editor scanning charts the DNMT3A activity landscape. Nat. Chem. Biol. 2023, 19, 176–186. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Wang, L.; Spitz, M.R.; Hong, W.K.; Mao, L.; Wei, Q. A novel polymorphism in human cytosine DNA-methyltransferase-3B promoter is associated with an increased risk of lung cancer. Cancer Res. 2002, 62, 4992–4995. [Google Scholar] [PubMed]
- Etoh, T.; Kanai, Y.; Ushijima, S.; Nakagawa, T.; Nakanishi, Y.; Sasako, M.; Kitano, S.; Hirohashi, S. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am. J. Pathol. 2004, 164, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.K.; Wu, C.Y.; Chang, J.W.; Juan, L.J.; Hsu, H.S.; Chen, C.Y.; Lu, Y.Y.; Tang, Y.A.; Yang, Y.C.; Yang, P.C.; et al. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res. 2010, 70, 5807–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, J.; Jeltsch, A.; Arimondo, P.B. DNA methyltransferase inhibitors in cancer: A chemical and therapeutic patent overview and selected clinical studies. Expert Opin. Ther. Pat. 2012, 22, 1427–1442. [Google Scholar] [CrossRef]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA Methyltransferases: A Novel Target for Prevention and Therapy. Front. Oncol. 2014, 4, 80. [Google Scholar] [CrossRef]
- Ghoshal, K.; Datta, J.; Majumder, S.; Bai, S.; Kutay, H.; Motiwala, T.; Jacob, S.T. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell. Biol. 2005, 25, 4727–4741. [Google Scholar] [CrossRef] [Green Version]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef]
- Bohl, S.R.; Bullinger, L.; Rücker, F.G. Epigenetic therapy: Azacytidine and decitabine in acute myeloid leukemia. Expert Rev. Hematol. 2018, 11, 361–371. [Google Scholar] [CrossRef]
- Seymour, J.F.; Döhner, H.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. Azacitidine improves clinical outcomes in older patients with acute myeloid leukaemia with myelodysplasia-related changes compared with conventional care regimens. BMC Cancer 2017, 17, 852. [Google Scholar] [CrossRef]
- Kazachenka, A.; Young, G.R.; Attig, J.; Kordella, C.; Lamprianidou, E.; Zoulia, E.; Vrachiolias, G.; Papoutselis, M.; Bernard, E.; Papaemmanuil, E. Epigenetic therapy of myelodysplastic syndromes connects to cellular differentiation independently of endogenous retroelement derepression. Genome Med. 2019, 11, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.; Issa, J.P.J.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; De Castro, C.; Ravandi, F. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2006, 106, 1794–1803. [Google Scholar]
- Kantarjian, H.M.; O’Brien, S.; Huang, X.; Garcia-Manero, G.; Ravandi, F.; Cortes, J.; Shan, J.; Davisson, J.; Bueso-Ramos, C.E.; Issa, J.P. Survival advantage with decitabine versus intensive chemotherapy in patients with higher risk myelodysplastic syndrome: Comparison with historical experience. Cancer 2007, 109, 1133–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, C.B.; Cheng, J.C.; Jones, P.A. Zebularine: A new drug for epigenetic therapy. Biochem. Soc. Trans. 2004, 32, 910–912. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, E.A.; Choy, G.; Redkar, S.; Taverna, P.; Azab, M.; Karpf, A.R. SGI-110: DNA Methyltransferase Inhibitor Oncolytic. Drugs Future 2013, 38, 535–543. [Google Scholar] [PubMed]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer Chemother. Pharmacol. 2014, 74, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Beumer, J.H.; Parise, R.A.; Newman, E.M.; Doroshow, J.H.; Synold, T.W.; Lenz, H.-J.; Egorin, M.J. Concentrations of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine (FdCyd) and its cytotoxic metabolites in plasma of patients treated with FdCyd and tetrahydrouridine (THU). Cancer Chemother. Pharmacol. 2008, 62, 363–368. [Google Scholar] [CrossRef]
- Lee, H.J.; Hore, T.A.; Reik, W. Reprogramming the Methylome: Erasing Memory and Creating Diversity. Cell Stem Cell 2014, 14, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-C.; Wang, Y.; Li, D.-D.; Zhang, Y.; Zhao, T.-C.; Li, C.-F. Procaine is a specific DNA methylation inhibitor with anti-tumor effect for human gastric cancer. J. Cell. Biochem. 2018, 119, 2440–2449. [Google Scholar] [CrossRef]
- Moreira-Silva, F.; Camilo, V.; Gaspar, V.; Mano, J.F.; Henrique, R.; Jerónimo, C. Repurposing Old Drugs into New Epigenetic Inhibitors: Promising Candidates for Cancer Treatment? Pharmaceutics 2020, 12, 410. [Google Scholar] [CrossRef]
- Candelaria, M.; Gallardo-Rincón, D.; Arce, C.; Cetina, L.; Aguilar-Ponce, J.L.; Arrieta, O.; González-Fierro, A.; Chávez-Blanco, A.; de la Cruz-Hernández, E.; Camargo, M.F.; et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann. Oncol. 2007, 18, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Donehower, R.C.; Eisenhauer, E.A.; Wainman, N.; Shah, A.K.; Bonfils, C.; MacLeod, A.R.; Besterman, J.M.; Reid, G.K. A phase I pharmacokinetic and pharmacodynamic study of the DNA methyltransferase 1 inhibitor MG98 administered twice weekly. Ann. Oncol. 2003, 14, 766–774. [Google Scholar] [CrossRef]
- Plummer, R.; Vidal, L.; Griffin, M.; Lesley, M.; de Bono, J.; Coulthard, S.; Sludden, J.; Siu, L.L.; Chen, E.X.; Oza, A.M.; et al. Phase I Study of MG98, an Oligonucleotide Antisense Inhibitor of Human DNA Methyltransferase 1, Given as a 7-Day Infusion in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2009, 15, 3177–3183. [Google Scholar] [CrossRef] [Green Version]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Musch, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic Reactivation of Tumor Suppressor Genes by a Novel Small-Molecule Inhibitor of Human DNA Methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Haffner, M.C.; Zhang, Y.; Lee, B.H.; Brennen, W.N.; Britton, J.; Kachhap, S.K.; Shim, J.S.; Liu, J.O.; Nelson, W.G.; et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate 2011, 71, 333–343. [Google Scholar]
- Sun, J.; He, X.; Zhu, Y.; Ding, Z.; Dong, H.; Feng, Y.; Du, J.; Wang, H.; Wu, X.; Zhang, L.; et al. SIRT1 Activation Disrupts Maintenance of Myelodysplastic Syndrome Stem and Progenitor Cells by Restoring TET2 Function. Cell Stem Cell 2018, 23, 355–369.e9. [Google Scholar] [CrossRef] [Green Version]
- Pappalardi, M.B.; Keenan, K.; Cockerill, M.; Kellner, W.A.; Stowell, A.; Sherk, C.; Wong, K.; Pathuri, S.; Briand, J.; Steidel, M.; et al. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Prébet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Adès, L.; et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef]
- Borthakur, G.; Ahdab, S.E.; Ravandi, F.; Faderl, S.; Ferrajoli, A.; Newman, B.; Issa, J.P.; Kantarjian, H. Activity of decitabine in patients with myelodysplastic syndrome previously treated with azacitidine. Leuk Lymphoma 2008, 49, 690–695. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.K.; Hassan, R.; Yaacob, N.S. Hypomethylating Agents and Immunotherapy: Therapeutic Synergism in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2021, 11, 624742. [Google Scholar] [CrossRef]
- Valente, S.; Trisciuoglio, D.; De Luca, T.; Nebbioso, A.; Labella, D.; Lenoci, A.; Bigogno, C.; Dondio, G.; Miceli, M.; Brosch, G.; et al. 1,3,4-Oxadiazole-Containing Histone Deacetylase Inhibitors: Anticancer Activities in Cancer Cells. J. Med. Chem. 2014, 57, 6259–6265. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hou, J.; Cui, X.; Suo, L.; Lv, Y. RG108 induces the apoptosis of endometrial cancer Ishikawa cell lines by inhibiting the expression of DNMT3B and demethylation of HMLH1. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5056–5064. [Google Scholar]
- Caulfield, T.; Medina-Franco, J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 2011, 176, 185–191. [Google Scholar] [CrossRef]
- Lee, B.H.; Yegnasubramanian, S.; Lin, X.; Nelson, W.G. Procainamide Is a Specific Inhibitor of DNA Methyltransferase 1 *. J. Biol. Chem. 2005, 280, 40749–40756. [Google Scholar] [CrossRef] [Green Version]
- Klisovic, R.B.; Stock, W.; Cataland, S.; Klisovic, M.I.; Liu, S.; Blum, W.; Green, M.; Odenike, O.; Godley, L.; Burgt, J.V.; et al. A Phase I Biological Study of MG98, an Oligodeoxynucleotide Antisense to DNA Methyltransferase 1, in Patients with High-Risk Myelodysplasia and Acute Myeloid Leukemia. Clin. Cancer Res. 2008, 14, 2444–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Loenarz, C.; Schofield, C.J. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 2011, 36, 7–18. [Google Scholar] [CrossRef]
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [Green Version]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [Green Version]
- Oda, M.; Oxley, D.; Dean, W.; Reik, W. Regulation of Lineage Specific DNA Hypomethylation in Mouse Trophectoderm. PLoS ONE 2013, 8, e68846. [Google Scholar] [CrossRef] [Green Version]
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14 (Suppl. 1), R139–R147. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.L.; Fernandez, J.; Koerperich, Z.; Andersen, M.P.; Kotandeniya, D.; Nguyen, M.E.; Sham, Y.Y.; Tretyakova, N.Y. Maintenance DNA Methyltransferase Activity in the Presence of Oxidized Forms of 5-Methylcytosine: Structural Basis for Ten Eleven Translocation-Mediated DNA Demethylation. Biochemistry 2018, 57, 6061–6069. [Google Scholar] [CrossRef] [PubMed]
- Otani, J.; Kimura, H.; Sharif, J.; Endo, T.A.; Mishima, Y.; Kawakami, T.; Koseki, H.; Shirakawa, M.; Suetake, I.; Tajima, S. Cell Cycle-Dependent Turnover of 5-Hydroxymethyl Cytosine in Mouse Embryonic Stem Cells. PLoS ONE 2013, 8, e82961. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Wu, H.; Diep, D.; Yamaguchi, S.; D’Alessio, A.C.; Fung, H.-L.; Zhang, K.; Zhang, Y. Genome-wide analysis reveals TET-and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 2013, 153, 692–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Münzel, M.; Wagner, M.; Müller, M.; Khan, F. Dynamic readers for 5-(hydroxy) methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [Green Version]
- Tovy, A.; Spiro, A.; McCarthy, R.; Shipony, Z.; Aylon, Y.; Allton, K.; Ainbinder, E.; Furth, N.; Tanay, A.; Barton, M. p53 is essential for DNA methylation homeostasis in naive embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017, 31, 959–972. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Gu, T.-P.; Weber, A.R.; Shen, J.-Z.; Li, B.-Z.; Xie, Z.-G.; Yin, R.; Guo, F.; Liu, X.; Tang, F. Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res. 2015, 43, 3986–3997. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Starczak, M.; Gackowski, D. Enigmatic 5-hydroxymethyluracil: Oxidatively modified base, epigenetic mark or both? Mutat. Res./Rev. Mutat. Res. 2016, 767, 59–66. [Google Scholar] [CrossRef]
- Spada, F.; Schiffers, S.; Kirchner, A.; Zhang, Y.; Arista, G.; Kosmatchev, O.; Korytiakova, E.; Rahimoff, R.; Ebert, C.; Carell, T. Active turnover of genomic methylcytosine in pluripotent cells. Nat. Chem. Biol. 2020, 16, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-J.; Zhou, J.-D.; Yang, D.-Q.; Wang, Y.-X.; Wen, X.-M.; Guo, H.; Yang, L.; Lian, X.-Y.; Lin, J.; Qian, J. TET2 expression is a potential prognostic and predictive biomarker in cytogenetically normal acute myeloid leukemia. J. Cell. Physiol. 2018, 233, 5838–5846. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; An, J.; Rao, A. DNA methylation and hydroxymethylation in hematologic differentiation and transformation. Curr. Opin. Cell Biol 2015, 37, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmeister, T.; Meyer, C.; Schwartz, S.; Hofmann, J.; Molkentin, M.; Kowarz, E.; Schneider, B.; Raff, T.; Reinhardt, R.; Gökbuget, N.; et al. The MLL recombinome of adult CD10-negative B-cell precursor acute lymphoblastic leukemia: Results from the GMALL study group. Blood 2009, 113, 4011–4015. [Google Scholar] [CrossRef]
- Antoine, I.; Eric, J.; Catherine, H.; Nathalie, P.; Catherine, H.; Bruno, L.; Isabelle, M.; Caroline, M.-R.; Amandine, L.; Sylvie, T.; et al. First description of the t(10;11)(q22;q23)/MLL-TET1 translocation in a T-cell lymphoblastic lymphoma, with subsequent lineage switch to acute myelomonocytic myeloid leukemia. Haematologica 2013, 98, e166–e168. [Google Scholar]
- Huang, H.; Jiang, X.; Li, Z.; Li, Y.; Song, C.X.; He, C.; Sun, M.; Chen, P.; Gurbuxani, S.; Wang, J.; et al. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 11994–11999. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Chen, M.; Eng, R.; DeJong, J.; Sinha, A.U.; Rahnamay, N.F.; Koche, R.; Al-Shahrour, F.; Minehart, J.C.; Chen, C.-W.; et al. MLL-AF9– and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J. Clin. Investig. 2016, 126, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rao, A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014, 30, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Ko, M. Epigenetic Modification of Cytosines in Hematopoietic Differentiation and Malignant Transformation. Int. J. Mol. Sci. 2023, 24, 1727. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, L.; Jin, S.; Su, S.M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010, 16, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, R.; Koichiro, M.; Sakata-Yanagimoto, M.; Bernard, O.; Matsui, H.; Nakajima-Takagi, Y.; Kato, T.; Koseki, H.; Iwama, A.; Chiba, S. Residual Wild-Type Tet2/Tet3 Allele Is the Savior Preventing Mouse Hematopoietic Progenitor Cells from Leukemia Development. Blood 2018, 132 (Suppl. 1), 1330. [Google Scholar] [CrossRef]
- Neri, F.; Dettori, D.; Incarnato, D.; Krepelova, A.; Rapelli, S.; Maldotti, M.; Parlato, C.; Paliogiannis, P.; Oliviero, S. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 2015, 34, 4168–4176. [Google Scholar] [PubMed] [Green Version]
- Wu, J.; Li, H.; Shi, M.; Zhu, Y.; Ma, Y.; Zhong, Y.; Xiong, C.; Chen, H.; Peng, C. TET1-mediated DNA hydroxymethylation activates inhibitors of the Wnt/β-catenin signaling pathway to suppress EMT in pancreatic tumor cells. J. Exp. Clin. Cancer Res. 2019, 38, 348. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-Y.; Li, P.-L.; Wang, T.-Z.; Zhang, X.-C. Prognostic values of 5-hmC, 5-mC and TET2 in epithelial ovarian cancer. Arch. Gynecol. Obstet. 2015, 292, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Li, J.; Han, X.; Hou, H.; Chen, H.; Zheng, X.; Lu, J.; Wang, L.; Chen, W.; Li, X.; et al. TET3 inhibits TGF-β1-induced epithelial-mesenchymal transition by demethylating miR-30d precursor gene in ovarian cancer cells. J. Exp. Clin. Cancer Res. 2016, 35, 72. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Guo, Y.; Niu, Y.; Jin, J.; Zhang, X.; Shi, X.; Zhang, L.; Li, R.; Chen, L.; Ma, R.Z. Epigenetic silencing of TET2 and TET3 induces an EMT-like process in melanoma. Oncotarget 2016, 8, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Gillberg, L.; Ørskov, A.D.; Liu, M.; Harsløf, L.B.S.; Jones, P.A.; Grønbæk, K. Vitamin C—A new player in regulation of the cancer epigenome. Semin. Cancer Biol. 2018, 51, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C Enhances the Generation of Mouse and Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar]
- Li, X.; Liu, L.; Yang, S.; Song, N.; Zhou, X.; Gao, J.; Yu, N.; Shan, L.; Wang, Q.; Liang, J.; et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. USA 2014, 111, 7096–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawada, H.; Kaneko, M.; Sawanobori, M.; Uno, T.; Matsuzawa, H.; Nakamura, Y.; Matsushita, H.; Ando, K. High Concentrations of L-Ascorbic Acid Specifically Inhibit the Growth of Human Leukemic Cells via Downregulation of HIF-1α Transcription. PLoS ONE 2013, 8, e62717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Quan, Y.; Wang, J.; Wang, F.; Zheng, Y.; Zhou, A. Vitamin C inhibit the proliferation, migration and epithelial-mesenchymal-transition of lens epithelial cells by destabilizing HIF-1α. Int. J. Clin. Exp. Med. 2015, 8, 15155–15163. [Google Scholar] [PubMed]
- Calvo-Asensio, I.; Dillon, E.T.; Lowndes, N.F.; Ceredig, R. The Transcription Factor Hif-1 Enhances the Radio-Resistance of Mouse MSCs. Front. Physiol. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Grosso, S.; Doyen, J.; Parks, S.K.; Bertero, T.; Paye, A.; Cardinaud, B.; Gounon, P.; Lacas-Gervais, S.; Noël, A.; Pouysségur, J.; et al. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013, 4, e544. [Google Scholar] [CrossRef] [Green Version]
- Mingay, M.; Chaturvedi, A.; Bilenky, M.; Cao, Q.; Jackson, L.; Hui, T.; Moksa, M.; Heravi-Moussavi, A.; Humphries, R.K.; Heuser, M.; et al. Vitamin C-induced epigenomic remodelling in IDH1 mutant acute myeloid leukaemia. Leukemia 2018, 32, 11–20. [Google Scholar] [CrossRef]
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic Acid Enhances Tet-Mediated 5-Methylcytosine Oxidation and Promotes DNA Demethylation in Mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef]
- Chen, J.; Guo, L.; Zhang, L.; Wu, H.; Yang, J.; Liu, H.; Wang, X.; Hu, X.; Gu, T.; Zhou, Z.; et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat. Genet. 2013, 45, 1504–1509. [Google Scholar] [CrossRef]
- Koh, K.P.; Yabuuchi, A.; Rao, S.; Huang, Y.; Cunniff, K.; Nardone, J.; Laiho, A.; Tahiliani, M.; Sommer, C.A.; Mostoslavsky, G.; et al. Tet1 and Tet2 Regulate 5-Hydroxymethylcytosine Production and Cell Lineage Specification in Mouse Embryonic Stem Cells. Cell Stem Cell 2011, 8, 200–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; González-Avalos, E.; Chawla, A.; Jeong, M.; López-Moyado, I.F.; Li, W.; Goodell, M.A.; Chavez, L.; Ko, M.; Rao, A. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat. Commun. 2015, 6, 10071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modrzejewska, M.; Gawronski, M.; Skonieczna, M.; Zarakowska, E.; Starczak, M.; Foksinski, M.; Rzeszowska-Wolny, J.; Gackowski, D.; Olinski, R. Vitamin C enhances substantially formation of 5-hydroxymethyluracil in cellular DNA. Free Radic. Biol. Med. 2016, 101, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ohtani, H.; Zhou, W.; Ørskov, A.D.; Charlet, J.; Zhang, Y.W.; Shen, H.; Baylin, S.B.; Liang, G.; Grønbæk, K.; et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA 2016, 113, 10238–10244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [Green Version]
- Chung, T.-L.; Brena, R.M.; Kolle, G.; Grimmond, S.M.; Berman, B.P.; Laird, P.W.; Pera, M.F.; Wolvetang, E.J. Vitamin C Promotes Widespread Yet Specific DNA Demethylation of the Epigenome in Human Embryonic Stem Cells. Stem Cells 2010, 28, 1848–1855. [Google Scholar] [CrossRef]
- Peng, D.; Ge, G.; Gong, Y.; Zhan, Y.; He, S.; Guan, B.; Li, Y.; Xu, Z.; Hao, H.; He, Z.; et al. Vitamin C increases 5-hydroxymethylcytosine level and inhibits the growth of bladder cancer. Clin. Epigenet. 2018, 10, 94. [Google Scholar] [CrossRef]
- Sant, D.W.; Mustafi, S.; Gustafson, C.B.; Chen, J.; Slingerland, J.M.; Wang, G. Vitamin C promotes apoptosis in breast cancer cells by increasing TRAIL expression. Sci. Rep. 2018, 8, 5306. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Langemeijer, S.M.C.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; van Hoogen, P.; van Kessel, A.G.; Raymakers, R.A.P.; et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Huijskens, M.J.; Wodzig, W.K.; Walczak, M.; Germeraad, W.T.; Bos, G.M. Ascorbic acid serum levels are reduced in patients with hematological malignancies. Results Immunol. 2016, 6, 8–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.R.; Qin, H.H.; Wu, W.Y.; He, S.J.; Xu, J.H. Vitamin C Protects Against UV Irradiation-Induced Apoptosis Through Reactivating Silenced Tumor Suppressor Genes p21 and p16 in a Tet-Dependent DNA Demethylation Manner in Human Skin Cancer Cells. Cancer Biother. Radiopharm. 2014, 29, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, C.; Schumacher, F.; Edlich, A.; Wetzel, A.; Yealland, G.; Katharina Neubert, L.; Scholtka, B.; Homann, T.; Kleuser, B. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget 2018, 9, 32822. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.C.; Spencer, E.; Das, A.; Meijer, N.; Lauren, C.; MacPherson, S.; Chambers, S.T. Patients Undergoing Myeloablative Chemotherapy and Hematopoietic Stem Cell Transplantation Exhibit Depleted Vitamin C Status in Association with Febrile Neutropenia. Nutrients 2020, 12, 1879. [Google Scholar] [CrossRef]
- Wulansari, N.; Kim, E.-H.; Sulistio, Y.A.; Rhee, Y.-H.; Song, J.-J.; Lee, S.-H. Vitamin C-Induced Epigenetic Modifications in Donor NSCs Establish Midbrain Marker Expressions Critical for Cell-Based Therapy in Parkinson’s Disease. Stem Cell Rep. 2017, 9, 1192–1206. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, N.; Bhagat, T.D.; Cheville, J.; Lohse, C.; Bhattacharyya, S.; Tischer, A.; Machha, V.; Gordon-Mitchell, S.; Choudhary, G.; Wong, L.F.; et al. Ascorbic acid-induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J. Clin. Investig. 2019, 129, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Ge, G.; Peng, D.; Xu, Z.; Guan, B.; Xin, Z.; He, Q.; Zhou, Y.; Li, X.; Zhou, L.; Ci, W. Restoration of 5-hydroxymethylcytosine by ascorbate blocks kidney tumour growth. EMBO Rep. 2018, 19, e45401. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: A phase 1 study. Blood 2021, 137, 1792–1803. [Google Scholar] [CrossRef]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 MutationsAG-221 Therapy for IDH2-Mutant AML. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.-X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.-P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-L.; Morcelle, C.; Cheng, Z.-L.; Chen, X.; Xu, Y.; Gao, Y.; Song, J.; Li, Z.; Smith, M.D.; Shi, M.; et al. Itaconate inhibits TET DNA dioxygenases to dampen inflammatory responses. Nat. Cell Biol. 2022, 24, 353–363. [Google Scholar] [CrossRef]
- Zhang, D.; An, X.; Li, Z.; Zhang, S. Role of gene promoter methylation regulated by TETs and DNMTs in the overexpression of HLA-G in MCF-7 cells. Exp. Med. 2019, 17, 4709–4714. [Google Scholar] [CrossRef] [PubMed]
- Weirath, N.A.; Hurben, A.K.; Chao, C.; Pujari, S.S.; Cheng, T.; Liu, S.; Tretyakova, N.Y. Small Molecule Inhibitors of TET Dioxygenases: Bobcat339 Activity Is Mediated by Contaminating Copper(II). ACS Med. Chem. Lett. 2022, 13, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Zhao, B.; Liu, X.; Wang, X.; Li, H.; Qin, H.; Wu, X.; Ma, Y.; Horne, D.; Yu, X. Selective targeting of TET catalytic domain promotes somatic cell reprogramming. Proc. Natl. Acad. Sci. USA 2020, 117, 3621–3626. [Google Scholar] [CrossRef]
- Guan, Y.; Tiwari, A.D.; Phillips, J.G.; Hasipek, M.; Grabowski, D.R.; Pagliuca, S.; Gopal, P.; Kerr, C.M.; Adema, V.; Radivoyevitch, T.; et al. A Therapeutic Strategy for Preferential Targeting of TET2-Mutant and TET Dioxygenase–Deficient Cells in Myeloid Neoplasms. Blood Cancer Discov. 2021, 2, 146–161. [Google Scholar] [CrossRef]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-Dose Parenteral Ascorbate Enhanced Chemosensitivity of Ovarian Cancer and Reduced Toxicity of Chemotherapy. Sci. Transl. Med. 2014, 6, ra18–ra222. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2− and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 32, 268. [Google Scholar] [CrossRef] [PubMed]
- Demiray, M. Combinatorial Therapy of High Dose Vitamin C and PARP Inhibitors in DNA Repair Deficiency: A Series of 8 Patients. Integr. Cancer Ther. 2020, 19, 1534735420969812. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef] [PubMed]
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R., Jr.; Levine, M.; Verma, A.; Shenoy, N. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.P.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.C.; Tan, X.M.; Cheng, M.; Li, Z.; Bovino, M.; Aube, J.; et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019, 129, 4316–4331. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Rao, A. TET family dioxygenases and the TET activator vitamin C in immune responses and cancer. Blood 2020, 136, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Pandya, S.S.; Jiang, L.; Liu, G.; Nimkar, T.; Yang, H.; Dai, D. Pharmacokinetics/pharmacodynamics (PK/PD) of ivosidenib in patients with IDH1-mutant advanced solid tumors from a phase 1 study. J. Clin. Oncol. 2018, 36, 2577. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.; Mellinghoff, I.; Maher, E.; Wen, P.; Beeram, M.; Touat, M.; Faris, J.; Azad, N.; Cloughesy, T.; Gore, L. Abstract PL04-05: The first reported results of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in a Phase I study of patients with advanced IDH1-mutant solid tumors, including gliomas. Mol. Cancer Ther. 2015, 14 (Suppl. 2), PL04-05. [Google Scholar] [CrossRef]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406. [Google Scholar]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G., Jr.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes *. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Huynh, J.P.; Lampropoulou, V.; Loginicheva, E.; Esaulova, E.; Gounder, A.P.; Boon, A.C.M.; Schwarzkopf, E.A.; Bradstreet, T.R.; Edelson, B.T.; et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J. Exp. Med. 2018, 215, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tönjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.; Karra, D.; Piro, R.M. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.W.; Wang, Z.; Xie, W.; Cai, Y.; Xia, L.; Easwaran, H.; Luo, J.; Yen, R.C.; Li, Y.; Baylin, S.B. Acetylation Enhances TET2 Function in Protecting against Abnormal DNA Methylation during Oxidative Stress. Mol. Cell 2017, 65, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Deng, C.-X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-Activated AMPK/SIRT1/Autophagy in Cellular Models of Parkinson’s Disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Li, X.; Liu, T.; Wu, T.T.; Feng, Y.; Peng, S.J.; Yin, H.; Wu, Y.C. SIRT1 Deacetylates TET2 and Promotes Its Ubiquitination Degradation to Achieve Neuroprotection Against Parkinson’s Disease. Front. Neurol. 2021, 12, 652882. [Google Scholar] [CrossRef]
- Uh, K.; Ryu, J.; Farrell, K.; Wax, N.; Lee, K. TET family regulates the embryonic pluripotency of porcine preimplantation embryos by maintaining the DNA methylation level of NANOG. Epigenetics 2020, 15, 1228–1242. [Google Scholar] [CrossRef]
- Chua, G.N.L.; Wassarman, K.L.; Sun, H.; Alp, J.A.; Jarczyk, E.I.; Kuzio, N.J.; Bennett, M.J.; Malachowsky, B.G.; Kruse, M.; Kennedy, A.J. Cytosine-Based TET Enzyme Inhibitors. ACS Med. Chem. Lett. 2019, 10, 180–185. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Group | Mechanisms of Action | Drug | Target(s) | Disease(s) and Phase Studies | References |
|---|---|---|---|---|---|
| Nucleoside analogs | Incorporate into DNA instead of cytidine; associate with DNMT1; induce DNMT degradation and DNA demethylation. | 5-azacitidine (5-AZA) | DNMT1 | MDS and AML (FDA-approved); glioma, prostate cancer, pancreatic cancer, ovarian cancer, metastatic melanoma | [38,39,40,41] |
| 5-aza-2′-deoxycytidine (decitabine) | DNMT1 | MDS and AML (FDA-approved); CML, prostate cancer, thyroid cancer | [39,42,43] | ||
| Zebularine | DNMT1 | MDS, solid tumors | [44] | ||
| Guadecitabine (SGI-110) | DNMT1 | AML, MDS, and HCC (phase II); CMML, ovarian cancer, urothelial carcinoma, colorectal cancer, peritoneal cancer | [45] | ||
| 4′-thio-2′-deoxycytidine (TdCyd) | DNMT1 | Refractory solid tumors | [46] | ||
| 5-fluoro-2′-deoxycytidine (FdCyd) | DNMT1 | AML, MDS, head and neck tumors, lung tumors, urinary bladder tumors, breast tumors | [47] | ||
| Non-nucleoside analogs | Block the catalytic site of DNMTs; induce DNA demethylation. | Procainamide | DNMT1 | Prostate cancer, breast cancer, colon cancer, and non-small-cell lung cancer | [48] |
| Procaine | DNMT1, DNMT3A | Breast cancer, gastric cancer, hepatocellular carcinoma, and non-small-cell lung cancer cells | [49] | ||
| Nanaomycin A | DNMT3B | Colon cancer, lung cancer, and bone marrow cells | [50] | ||
| Hydralazine | DNMT1 | Ovarian cancer, cervical cancer, refractory solid tumors, breast cancer (phase II). | [51] | ||
| MG98 | DNMT1 | Solid tumors (phase I) | [52,53] | ||
| N-phthaloyl-L-tryptophan (RG108) | DNMT1 | Solid tumors | [54] | ||
| Disulfiram | DNMT1 | Refractory multiple myeloma, prostate cancer (phase II) | [55] | ||
| SGI-1027 | DNMT1, DNMT3A/B | Hematological cancer, solid tumors | [56] | ||
| GSK3685032 | DNMT1 | AML | [57] |
| Group | Drug | Target(s) | Effect(s) | Disease(s) | References |
|---|---|---|---|---|---|
| TET activators | Vitamin C | TET1/2/3 | Increases 5hmC levels via TET activation; induces DNA hypomethylation | MDS, AML | [125,129] |
| AG-120 (ivosidenib), AG-221 (enasidenib) | mIDH | Reduce the accumulation of 2HG; recover TET-dependent DNA demethylation | AML, hematologic malignancies, glioma, cholangiocarcinoma, chondrosarcoma | [140] | |
| SRT1720 (SIRT1 agonist) | TET2 | SIRT1 agonist; deacetylates TET2 at conserved lysine in its catalytic domain; enhances TET2 activity | MDS | [56] | |
| AMPK activators (AICAR, Metformin, A769662) | TET2 | Phosphorylates TET2; maintains stability of TET2; induces levels of 5hmC | Hyperglycemia-related tumor | [141] | |
| TET inhibitors | 2-Hydroxyglutarate (2HG) | TET2 | Induces DNA hypermethylation, gene silencing, and tumor progression | Hematological malignancies, AML, MDS | [142] |
| Fumarate | TET1/2/3 | Downregulates 5hmC levels; induces DNA hypermethylation | Hematological malignancies, AML, MDS | [143] | |
| Succinate | TET1/2/3 | Downregulates 5hmC levels; induces DNA hypermethylation | Hematological malignancies, AML, MDS | [144] | |
| Itaconate | TET2 | Competes with α-KG to inhibit TET2; dampens LPS-induced inflammatory responses | Hematological malignancies | [145] | |
| Dimethyloxallyl glycine (DMOG) | TET3 | Increases 5mC levels; downregulates pluripotency genes | Solid tumors | [146] | |
| Bobcat339 | TET1/2 | Inhibits TET activity; reduces 5hmC levels | In vitro | [147] | |
| C35 | TET1/2/3 | Inhibits TET activity and somatic cell reprogramming | In vitro | [148] | |
| TETi76 | TET1/2/3 | Reduces 5hmC levels; growth inhibition of TET-deficient leukemic cells | Hematological malignancies, MDS, AML | [149] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, I.; An, J.; Ko, M. Epigenetic Regulators of DNA Cytosine Modification: Promising Targets for Cancer Therapy. Biomedicines 2023, 11, 654. https://doi.org/10.3390/biomedicines11030654
Jung I, An J, Ko M. Epigenetic Regulators of DNA Cytosine Modification: Promising Targets for Cancer Therapy. Biomedicines. 2023; 11(3):654. https://doi.org/10.3390/biomedicines11030654
Chicago/Turabian StyleJung, Inkyung, Jungeun An, and Myunggon Ko. 2023. "Epigenetic Regulators of DNA Cytosine Modification: Promising Targets for Cancer Therapy" Biomedicines 11, no. 3: 654. https://doi.org/10.3390/biomedicines11030654