
Alternative Splicing in Myeloid Malignancies



{kind=link}
{kind=link}
{kind=link}
Abstract
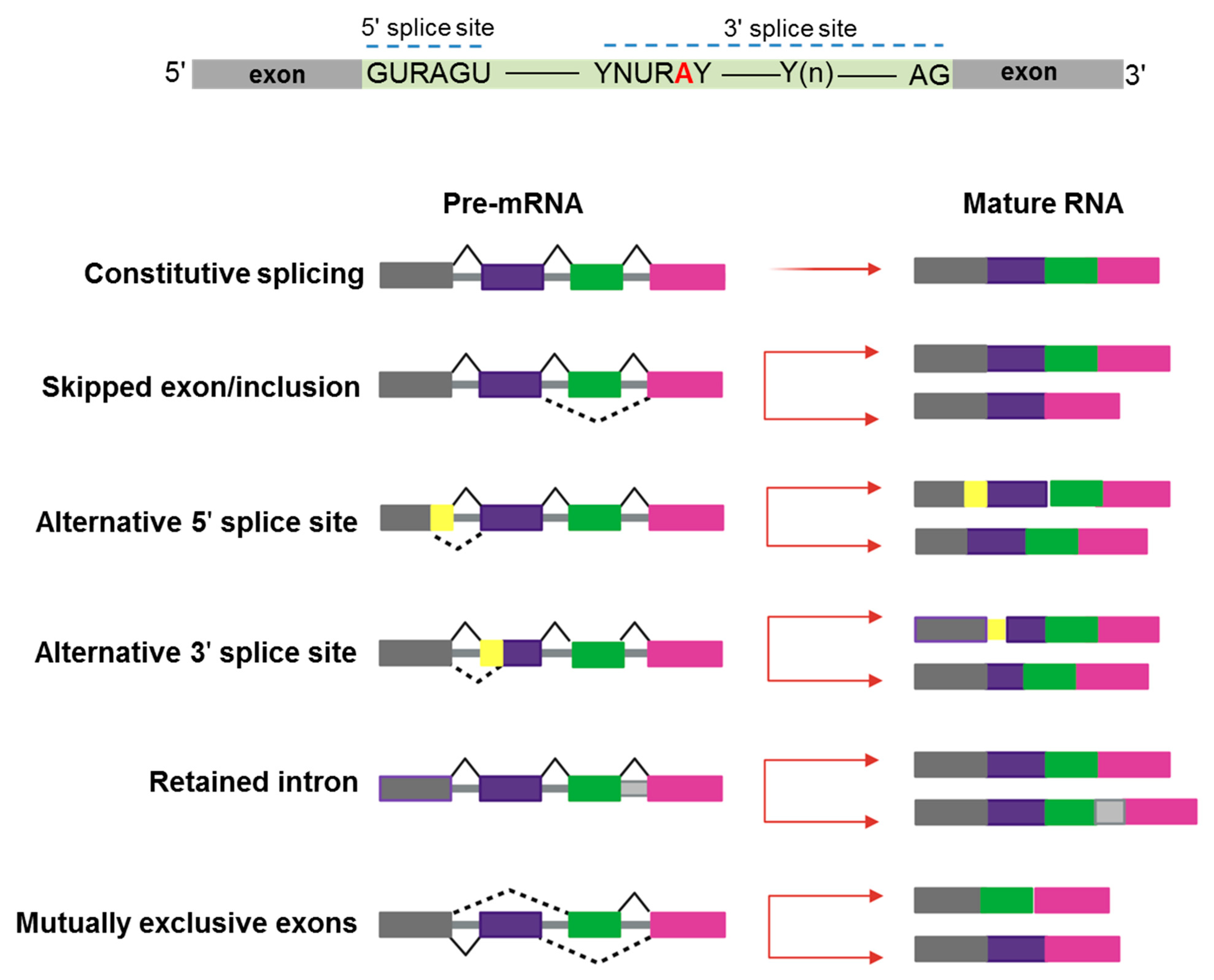
:1. Introduction
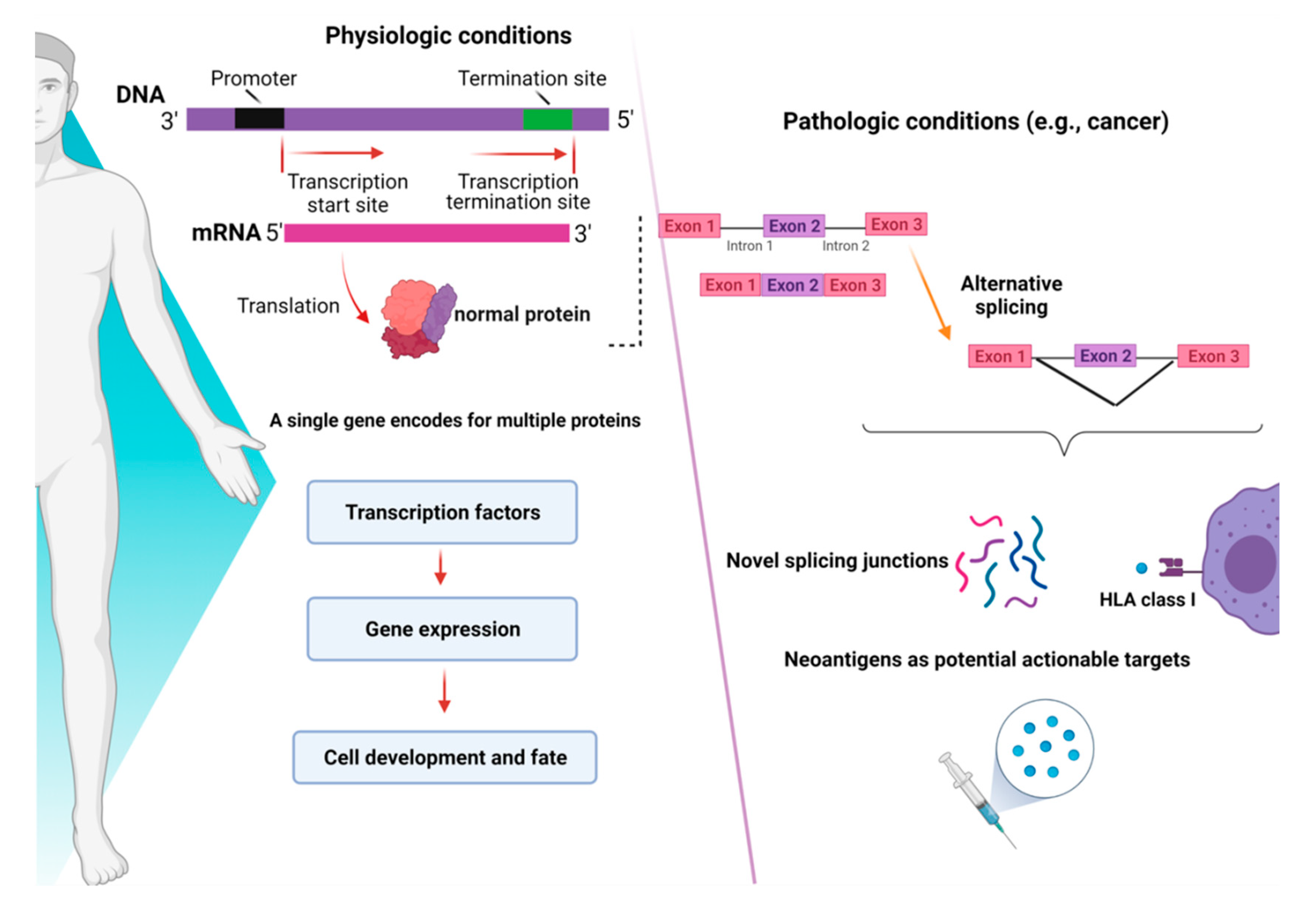
2. De-Regulation of RNA-Splicing in Myeloid Malignancies
3. Alternative Splicing as a Novel Mechanism of Neo-Antigens Production
4. RNA-Splicing-Based Targeted Therapies
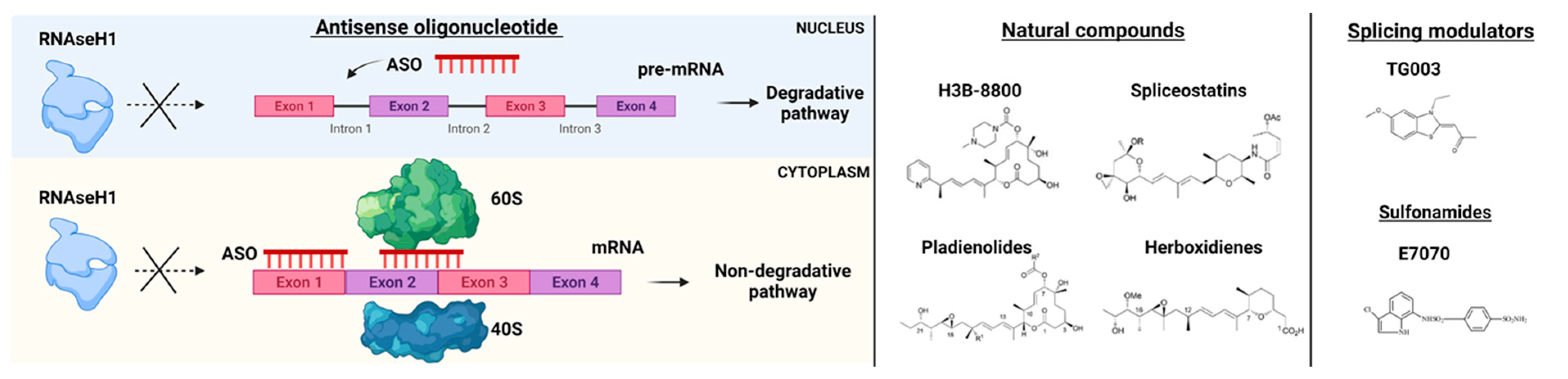
4.1. Antisense Oligonucleotides (ASOs)
4.2. Small Molecules Agents
4.3. Splicing Modulators
5. Future Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, S.; Benbarche, S.; Abdel-Wahab, O. Splicing factor mutations in hematologic malignancies. Blood 2021, 138, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.C.; Lopez-Oreja, I.; Valcarcel, J. Roles and mechanisms of alternative splicing in cancer-implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Trinh, B.Q.; Ummarino, S.; Zhang, Y.; Ebralidze, A.K.; Bassal, M.A.; Nguyen, T.M.; Heller, G.; Coffey, R.; Tenen, D.E.; van der Kouwe, E.; et al. Myeloid lncRNA LOUP mediates opposing regulatory effects of RUNX1 and RUNX1-ETO in t(8;21) AML. Blood 2021, 138, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Belluti, S.; Rigillo, G.; Imbriano, C. Transcription factors in cancer: When alternative splicing determines opposite cell fates. Cells 2020, 9, 760. [Google Scholar] [CrossRef] [Green Version]
- Anufrieva, K.S.; Shender, V.O.; Arapidi, G.P.; Pavlyukov, M.S.; Shakhparonov, M.I.; Shnaider, P.V.; Butenko, I.O.; Lagarkova, M.A.; Govorun, V.M. Therapy-induced stress response is associated with downregulation of pre-mRNA splicing in cancer cells. Genome Med. 2018, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Anczukow, O.; Krainer, A.R. Splicing-factor alterations in cancers. RNA 2016, 22, 1285–1301. [Google Scholar] [CrossRef] [Green Version]
- Chlon, T.M.; Stepanchick, E.; Hershberger, C.E.; Daniels, N.J.; Hueneman, K.M.; Kuenzi Davis, A.; Choi, K.; Zheng, Y.; Gurnari, C.; Haferlach, T.; et al. Germline DDX41 mutations cause ineffective hematopoiesis and myelodysplasia. Cell Stem Cell 2021, 28, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Komeno, Y.; Yan, M.; Matsuura, S.; Lam, K.; Lo, M.C.; Huang, Y.J.; Tenen, D.G.; Downing, J.R.; Zhang, D.E. Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood 2014, 123, 3760–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Vasic, R.; Halene, S. Role of alternative splicing in hematopoietic stem cells during development. Stem Cell Investig. 2018, 5, 26. [Google Scholar] [CrossRef]
- Goldstein, O.; Meyer, K.; Greenshpan, Y.; Bujanover, N.; Feigin, M.; Ner-Gaon, H.; Shay, T.; Gazit, R. Mapping whole-transcriptome splicing in mouse hematopoietic stem cells. Stem Cell Rep. 2017, 8, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.J.; Ritchie, W.; Ebner, O.A.; Selbach, M.; Wong, J.W.; Huang, Y.; Gao, D.; Pinello, N.; Gonzalez, M.; Baidya, K.; et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013, 154, 583–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesana, M.; Guo, M.H.; Cacchiarelli, D.; Wahlster, L.; Barragan, J.; Doulatov, S.; Vo, L.T.; Salvatori, B.; Trapnell, C.; Clement, K.; et al. A CLK3-HMGA2 alternative splicing axis impacts human hematopoietic stem cell molecular identity throughout development. Cell Stem Cell 2018, 22, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Lovci, M.T.; Bengtson, M.H.; Massirer, K.B. Post-translational modifications and RNA-binding proteins. Adv. Exp. Med. Biol. 2016, 907, 297–317. [Google Scholar]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Hodson, D.J.; Screen, M.; Turner, M. RNA-binding proteins in hematopoiesis and hematological malignancy. Blood 2019, 133, 2365–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Jiang, T.; Jia, D.; Han, Y.; Liu, F.; Huang, Y.; Qu, Z.; Zhao, Y.; Tu, J.; Lv, Y.; et al. BCAS2 is essential for hematopoietic stem and progenitor cell maintenance during zebrafish embryogenesis. Blood 2019, 133, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative splicing of hTERT Pre-mRNA: A potential strategy for the regulation of telomerase activity. Int. J. Mol. Sci. 2017, 18, 567. [Google Scholar] [CrossRef] [Green Version]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco Targets Ther. 2013, 7, 57–68. [Google Scholar] [PubMed] [Green Version]
- Adamia, S.; Haibe-Kains, B.; Pilarski, P.M.; Bar-Natan, M.; Pevzner, S.; Avet-Loiseau, H.; Lode, L.; Verselis, S.; Fox, E.A.; Burke, J.; et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin. Cancer Res. 2014, 20, 1135–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.D.; Kelley, J.M.; Gocayne, J.D.; Dubnick, M.; Polymeropoulos, M.H.; Xiao, H.; Merril, C.R.; Wu, A.; Olde, B.; Moreno, R.F.; et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 1991, 252, 1651–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.T.; Chiu, Y.C.; Kao, C.J.; Hou, H.A.; Lin, C.C.; Tsai, C.H.; Tseng, M.H.; Chou, W.C.; Tien, H.F. The prognostic significance of global aberrant alternative splicing in patients with myelodysplastic syndrome. Blood Cancer J. 2018, 8, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: Dysregulated genes/pathways and clinical associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Malcovati, L.; Galli, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 3649. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Avishai, N.; Mahfouz, R.; Tabarroki, A.; Cowen, J.; Sharghi-Moshtaghin, R.; Hitomi, M.; Rogers, H.J.; Hasrouni, E.; Phillips, J.; et al. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia 2015, 29, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Dolatshad, H.; Pellagatti, A.; Fernandez-Mercado, M.; Yip, B.H.; Malcovati, L.; Attwood, M.; Przychodzen, B.; Sahgal, N.; Kanapin, A.A.; Lockstone, H.; et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 2015, 29, 1092–1103. [Google Scholar] [CrossRef]
- Conte, S.; Katayama, S.; Vesterlund, L.; Karimi, M.; Dimitriou, M.; Jansson, M.; Mortera-Blanco, T.; Unneberg, P.; Papaemmanuil, E.; Sander, B.; et al. Aberrant splicing of genes involved in haemoglobin synthesis and impaired terminal erythroid maturation in SF3B1 mutated refractory anaemia with ring sideroblasts. Br. J. Haematol. 2015, 171, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Lin, K.T.; Bradley, R.K.; Abdel-Wahab, O.; Krainer, A.R. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev. 2020, 34, 413–427. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H.; Steeples, V.; Repapi, E.; Armstrong, R.N.; Llorian, M.; Roy, S.; Shaw, J.; Dolatshad, H.; Taylor, S.; Verma, A.; et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J. Clin. Investig. 2017, 127, 2206–2221. [Google Scholar] [CrossRef] [Green Version]
- Rivera, O.D.; Mallory, M.J.; Quesnel-Vallieres, M.; Chatrikhi, R.; Schultz, D.C.; Carroll, M.; Barash, Y.; Cherry, S.; Lynch, K.W. Alternative splicing redefines landscape of commonly mutated genes in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2021, 118, e2014967118. [Google Scholar] [CrossRef] [PubMed]
- David, J.K.; Maden, S.K.; Weeder, B.R.; Thompson, R.F.; Nellore, A. Putatively cancer-specific exon-exon junctions are shared across patients and present in developmental and other non-cancer cells. NAR Cancer 2020, 2, zcaa001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahles, A.; Ong, C.S.; Zhong, Y.; Ratsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive analysis of alternative splicing across tumors from 8705 patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) Groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; et al. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Saha, A.; Kim, Y.; Gewirtz, A.D.H.; Jo, B.; Gao, C.; McDowell, I.C.; Consortium, G.T.; Engelhardt, B.E.; Battle, A. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 2017, 27, 1843–1858. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Zhang, J.; Lee, H.; Batista, M.T.; Johnston, S.A. RNA transcription and splicing errors as a source of cancer frameshift neoantigens for vaccines. Sci. Rep. 2019, 9, 14184. [Google Scholar] [CrossRef]
- Hershberger, C.E.; Moyer, D.C.; Adema, V.; Kerr, C.M.; Walter, W.; Hutter, S.; Meggendorfer, M.; Baer, C.; Kern, W.; Nadarajah, N.; et al. Complex landscape of alternative splicing in myeloid neoplasms. Leukemia 2020, 35, 1108–1120. [Google Scholar] [CrossRef]
- Biernacki, M.; Foster, K.A.; Clough, C.; Busch, S.; Cummings, C.; Oehler, V.G.; Stirewalt, D.; Doulatov, S.; Bleakley, M. A shared SF3B1 neoantigen is presented on primary malignant cells and induced pluripotent stem cell-derived hematopoietic cells. Blood 2020, 136, 13–14. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Cote, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-Gonzalez, H.; Chai, S.; Wang, F.; et al. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep. 2018, 23, 270–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef]
- Schischlik, F.; Jager, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Milosevic Feenstra, J.D.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell 2017, 171, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef]
- Inoue, D.; Chew, G.L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-dependent antisense oligonucleotides are robustly active in directing RNA cleavage in both the cytoplasm and the nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [Green Version]
- Kierlin-Duncan, M.N.; Sullenger, B.A. Using 5’-PTMs to repair mutant beta-globin transcripts. RNA 2007, 13, 1317–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Kitano, A.; Jiang, Y.; Luu, V.; Hoegenauer, K.A.; Nakada, D. Clonal expansion and myeloid leukemia progression modeled by multiplex gene editing of murine hematopoietic progenitor cells. Exp. Hematol. 2018, 64, 33–44. [Google Scholar] [CrossRef]
- Buonamici, S.; Yoshimi, A.; Thomas, M.; Seiler, M.; Chan, B.; Caleb, B.; Darmanet, R.; Fekkes, P.; Karr, C.; Keaney, G.F.; et al. H3B-8800, an orally bioavailable modulator of the SF3b complex, shows efficacy in spliceosome-mutant myeloid malignancies. Blood 2016, 128, 966. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I first-in-human dose escalation study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021, 35, 3542–3550. [Google Scholar] [CrossRef]
- Bonnal, S.; Vigevani, L.; Valcarcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012, 11, 847–859. [Google Scholar] [CrossRef]
- Folco, E.G.; Coil, K.E.; Reed, R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011, 25, 440–444. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef]
- Hsiehchen, D.; Goralski, M.; Kim, J.; Xie, Y.; Nijhawan, D. Biomarkers for RBM39 degradation in acute myeloid leukemia. Leukemia 2020, 34, 1924–1928. [Google Scholar] [CrossRef]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell 2019, 35, 369–384. [Google Scholar] [CrossRef] [Green Version]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikas, I.P.; Themistocleous, S.C.; Paschou, S.A.; Tsamis, K.I.; Ryu, H.S. Serine-arginine protein Kinase 1 (SRPK1) as a prognostic factor and potential therapeutic target in cancer: Current evidence and future perspectives. Cells 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliuca, S.; Gurnari, C.; Visconte, V. Molecular targeted therapy in Myelodysplastic syndromes: New options for tailored treatments. Cancers 2021, 13, 784. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurnari, C.; Pagliuca, S.; Visconte, V. Alternative Splicing in Myeloid Malignancies. Biomedicines 2021, 9, 1844. https://doi.org/10.3390/biomedicines9121844
Gurnari C, Pagliuca S, Visconte V. Alternative Splicing in Myeloid Malignancies. Biomedicines. 2021; 9(12):1844. https://doi.org/10.3390/biomedicines9121844
Chicago/Turabian StyleGurnari, Carmelo, Simona Pagliuca, and Valeria Visconte. 2021. "Alternative Splicing in Myeloid Malignancies" Biomedicines 9, no. 12: 1844. https://doi.org/10.3390/biomedicines9121844