Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme

 ,
,  ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
1.1. The Discovery of IDE: An Historical Perspective
1.2. The Function of IDE as a Protease of Insulin
1.3. Other Proteolytic Functions of IDE
1.4. Non-Proteolytic Functions of IDE
1.5. Molecular and Biochemical Characteristics of IDE
1.6. Subcellular Localization of IDE
1.7. Transcriptional and Posttranscriptional Regulation of IDE
1.8. Pharmacological Modulation of IDE
2. Studies of Global Manipulation of Ide on Insulin and Glucose Tolerance In Vivo
2.1. Effects of Pancellular Genetic Deletion of Ide on Insulin and Glucose Tolerance In Vivo
2.2. Effects of Pharmacological Inhibition of IDE on Insulin and Glucose Tolerance In Vivo
3. Role of IDE in Pancreatic β-Cells
3.1. IDE Protein Expression in Pancreatic β-Cells
3.2. Effects of Genetic Deletion of Ide on Insulin Secretion In Vitro
3.3. Impaired Insulin Secretion and β-Cell Immaturity in the B-IDE-KO Mouse
4. Role of IDE in Liver
4.1. Metabolic Phenotype of the L-IDE-KO Mouse
4.2. Metabolic Phenotype of Hepatic IDE Gain of Function in Mice
4.3. Novel Insights into the Etiology and Pathophysiology of Hepatic Insulin Resistance: Lessons from Knockout Mouse Models
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Najjar, S.M.; Perdomo, G. Hepatic insulin clearance: Mechanism and physiology. Physiology 2019, 34, 198–215. [Google Scholar] [CrossRef]
- Kuo, W.L.; Montag, A.G.; Rosner, M.R. Insulin-degrading enzyme is differentially expressed and developmentally regulated in various rat tissues. Endocrinology 1993, 132, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Hooper, N.M. Families of zinc metalloproteases. FEBS Lett. 1994, 354, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Morton, F.R.; Kok, C.Y.; Kong, J.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2008, 36, D320–D325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Gamba, A.; Leal, M.C.; Morelli, L.; Castaño, E.M. Insulin-degrading enzyme: Structure-function relationship and its possible roles in health and disease. Curr. Pharm. Des. 2009, 15, 3644–3655. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J. Homologues of insulinase, a new superfamily of metalloendopeptidases. Biochem. J. 1991, 275 Pt 2, 389–391. [Google Scholar] [CrossRef] [Green Version]
- Kole, H.K.; Muthukumar, G.; Lenard, J. Purification and properties of a membrane-bound insulin binding protein, a putative receptor, from Neurospora crassa. Biochemistry 1991, 30, 682–688. [Google Scholar] [CrossRef]
- Kole, H.K.; Smith, D.R.; Lenard, J. Characterization and partial purification of an insulinase from Neurospora crassa. Arch. Biochem. Biophys. 1992, 297, 199–204. [Google Scholar] [CrossRef]
- Fricke, B.; Betz, R.; Friebe, S. A periplasmic insulin-cleaving proteinase (ICP) from Acinetobacter calcoaceticus sharing properties with protease III from Escherichia coli and IDE from eucaryotes. J. Basic Microbiol. 1995, 35, 21–31. [Google Scholar] [CrossRef]
- Cheng, Y.S.; Zipser, D. Purification and characterization of protease III from Escherichia coli. J. Biol. Chem. 1979, 254, 4698–4706. [Google Scholar]
- Dykstra, C.C.; Kushner, S.R. Physical characterization of the cloned protease III gene from Escherichia coli K-12. J. Bacteriol. 1985, 163, 1055–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lapham, A.N.; Freedman, C.G.; Reed, T.L.; Schmidt, W.K. Yeast as a tractable genetic system for functional studies of the insulin-degrading enzyme. J. Biol. Chem. 2005, 280, 27481–27490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adames, N.; Blundell, K.; Ashby, M.N.; Boone, C. Role of yeast insulin-degrading enzyme homologs in propheromone processing and bud site selection. Science 1995, 270, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Pierotti, A.R.; Chesneau, V.; Foulon, T.; Prat, A. N-arginine dibasic convertase. Methods Enzymol. 1995, 248, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Sato, Y.; Ohno, M.; Hiraoka, Y.; Saijo, S.; Sakamoto, J.; Chen, P.M.; Morita, Y.; Matsuda, S.; Iwasaki, K.; et al. Nardilysin Is Required for Maintaining Pancreatic β-Cell Function. Diabetes 2016, 65, 3015–3027. [Google Scholar] [CrossRef] [Green Version]
- Ishizu-Higashi, S.; Seno, H.; Nishi, E.; Matsumoto, Y.; Ikuta, K.; Tsuda, M.; Kimura, Y.; Takada, Y.; Kimura, Y.; Nakanishi, Y.; et al. Deletion of nardilysin prevents the development of steatohepatitis and liver fibrotic changes. PLoS ONE 2014, 9, e98017. [Google Scholar] [CrossRef]
- Hiraoka, Y.; Matsuoka, T.; Ohno, M.; Nakamura, K.; Saijo, S.; Matsumura, S.; Nishi, K.; Sakamoto, J.; Chen, P.M.; Inoue, K.; et al. Critical roles of nardilysin in the maintenance of body temperature homoeostasis. Nat. Commun. 2014, 5, 3224. [Google Scholar] [CrossRef] [Green Version]
- Nishi, E.; Prat, A.; Hospital, V.; Elenius, K.; Klagsbrun, M. N-arginine dibasic convertase is a specific receptor for heparin-binding EGF-like growth factor that mediates cell migration. EMBO J. 2001, 20, 3342–3350. [Google Scholar] [CrossRef] [Green Version]
- Mirsky, I.A.; Broh-Kahn, R.H. The inactivation of insulin by tissue extracts; the distribution and properties of insulin inactivating extracts. Arch. Biochem. 1949, 20, 1–9. [Google Scholar]
- Affholter, J.A.; Fried, V.A.; Roth, R.A. Human insulin-degrading enzyme shares structural and functional homologies with E. coli protease III. Science 1988, 242, 1415–1418. [Google Scholar] [CrossRef] [Green Version]
- Duckworth, W.C.; Hamel, F.G.; Bennett, R.; Ryan, M.P.; Roth, R.A. Human red blood cell insulin-degrading enzyme and rat skeletal muscle insulin protease share antigenic sites and generate identical products from insulin. J. Biol. Chem. 1990, 265, 2984–2987. [Google Scholar] [CrossRef]
- Broh-Kahn, R.H.; Mirsky, I.A. The inactivation of insulin by tissue extracts; the effect of fasting on the insulinase content of rat liver. Arch. Biochem. 1949, 20, 10–14. [Google Scholar] [PubMed]
- Broh-Kahn, R.H.; Simkin, B.; Mirsky, A. The inactivation of insulin by tissue extracts. V. The effect of the composition of the diet on the restoration of the liver insulinase activity of the fasted rat. Arch. Biochem. 1950, 27, 174–184. [Google Scholar] [PubMed]
- Mirsky, I.A.; Simkin, B.; Broh-Kahn, R.H. The inactivation of insulin by tissue extracts. VI. The existence, distribution and properties of an insulinase inhibitor. Arch. Biochem. 1950, 28, 415–423. [Google Scholar]
- Mirsky, I.A.; Perisutti, G.; Diengott, D. Effect of insulinase-inhibitor on destruction of insulin by intact mouse. Proc. Soc. Exp. Biol. Med. 1955, 88, 76–78. [Google Scholar] [CrossRef]
- Mirsky, I.A.; Perisutti, G.; Jinks, R. The destruction of insulin by intact mice. Endocrinology 1955, 56, 484–488. [Google Scholar] [CrossRef]
- Brush, J.S.; Shah, R.J. Purification and characterization of inhibitors of insulin specific protease in human serum. Biochem. Biophys. Res. Commun. 1973, 53, 894–903. [Google Scholar] [CrossRef]
- Ogawa, W.; Shii, K.; Yonezawa, K.; Baba, S.; Yokono, K. Affinity purification of insulin-degrading enzyme and its endogenous inhibitor from rat liver. J. Biol. Chem. 1992, 267, 1310–1316. [Google Scholar] [CrossRef]
- McKenzie, R.A.; Burghen, G.A. Partial purification and characterization of insulin protease and its intracellular inhibitor from rat liver. Arch. Biochem. Biophys. 1984, 229, 604–611. [Google Scholar] [CrossRef]
- Ryan, M.P.; Duckworth, W.C. Partial characterization of an endogenous inhibitor of a calcium-dependent form of insulin protease. Biochem. Biophys. Res. Commun. 1983, 116, 195–203. [Google Scholar] [CrossRef]
- Saric, T.; Müller, D.; Seitz, H.J.; Pavelic, K. Non-covalent interaction of ubiquitin with insulin-degrading enzyme. Mol. Cell. Endocrinol. 2003, 204, 11–20. [Google Scholar] [CrossRef]
- Mirsky, I.A. The hypoglycemic action of insulinase-inhibitors by mouth in patients with diabetes mellitus. Trans. Assoc. Am. Physicians 1956, 69, 262–275. [Google Scholar] [PubMed]
- News of Science. Science 1956, 123, 258–262. [CrossRef] [PubMed]
- Mering, J.; Minkowski, O. Diabetes mellitus nach Pankreasexstirpation. Arch. Exp. Pathol. Pharmakol. 1890, 26, 371–387. [Google Scholar] [CrossRef] [Green Version]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A. Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [Google Scholar]
- Mirsky, I.A. Insulinase. Diabetes 1957, 6, 448–449. [Google Scholar] [CrossRef] [Green Version]
- Duckworth, W.C. Insulin degradation: Mechanisms, products, and significance. Endocr. Rev. 1988, 9, 319–345. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin degradation: Progress and potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar] [CrossRef] [Green Version]
- Manolopoulou, M.; Guo, Q.; Malito, E.; Schilling, A.B.; Tang, W.J. Molecular basis of catalytic chamber-assisted unfolding and cleavage of human insulin by human insulin-degrading enzyme. J. Biol. Chem. 2009, 284, 14177–14188. [Google Scholar] [CrossRef] [Green Version]
- Grasso, G.; Rizzarelli, E.; Spoto, G. AP/MALDI-MS complete characterization of the proteolytic fragments produced by the interaction of insulin degrading enzyme with bovine insulin. J. Mass Spectrom. 2007, 42, 1590–1598. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Heinemann, M.A.; Kitabchi, A.E. Purification of insulin-specific protease by affinity chromatography. Proc. Natl. Acad. Sci. USA 1972, 69, 3698–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misbin, R.I.; Almira, E.C.; Duckworth, W.C.; Mehl, T.D. Inhibition of insulin degradation by insulin-like growth factors. Endocrinology 1983, 113, 1525–1527. [Google Scholar] [CrossRef] [PubMed]
- Authier, F.; Bergeron, J.J.; Ou, W.J.; Rachubinski, R.A.; Posner, B.I.; Walton, P.A. Degradation of the cleaved leader peptide of thiolase by a peroxisomal proteinase. Proc. Natl. Acad. Sci. USA 1995, 92, 3859–3863. [Google Scholar] [CrossRef] [Green Version]
- Frank, B.H.; Peavy, D.E.; Hooker, C.S.; Duckworth, W.C. Receptor binding properties of monoiodotyrosyl insulin isomers purified by high performance liquid chromatography. Diabetes 1983, 32, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.P.; Duckworth, W.C. Insulin degradation: Assays and enzymes. In The Insulin Receptor; Kahn, C., Harrison, L., Eds.; Alan R Liss Inc.: New York, NY, USA, 1988; Volume 1, pp. 29–57. [Google Scholar]
- Song, E.S.; Mukherjee, A.; Juliano, M.A.; Pyrek, J.S.; Goodman, J.P., Jr.; Juliano, L.; Hersh, L.B. Analysis of the subsite specificity of rat insulysin using fluorogenic peptide substrates. J. Biol. Chem. 2001, 276, 1152–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fosam, A.; Sikder, S.; Abel, B.S.; Tella, S.H.; Walter, M.F.; Mari, A.; Muniyappa, R. Reduced Insulin Clearance and Insulin-Degrading Enzyme Activity Contribute to Hyperinsulinemia in African Americans. J. Clin. Endocrinol. Metab. 2020, 105, e1835–e1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.S.; Hersh, L.B. Insulysin: An allosteric enzyme as a target for Alzheimer’s disease. J. Mol. Neurosci. 2005, 25, 201–206. [Google Scholar] [CrossRef]
- Song, E.S.; Juliano, M.A.; Juliano, L.; Fried, M.G.; Wagner, S.L.; Hersh, L.B. ATP effects on insulin-degrading enzyme are mediated primarily through its triphosphate moiety. J. Biol. Chem. 2004, 279, 54216–54220. [Google Scholar] [CrossRef] [Green Version]
- Im, H.; Manolopoulou, M.; Malito, E.; Shen, Y.; Zhao, J.; Neant-Fery, M.; Sun, C.Y.; Meredith, S.C.; Sisodia, S.S.; Leissring, M.A.; et al. Structure of substrate-free human insulin-degrading enzyme (IDE) and biophysical analysis of ATP-induced conformational switch of IDE. J. Biol. Chem. 2007, 282, 25453–25463. [Google Scholar] [CrossRef] [Green Version]
- Cabrol, C.; Huzarska, M.A.; Dinolfo, C.; Rodriguez, M.C.; Reinstatler, L.; Ni, J.; Yeh, L.A.; Cuny, G.D.; Stein, R.L.; Selkoe, D.J.; et al. Small-molecule activators of insulin-degrading enzyme discovered through high-throughput compound screening. PLoS ONE 2009, 4, e5274. [Google Scholar] [CrossRef]
- Song, E.S.; Juliano, M.A.; Juliano, L.; Hersh, L.B. Substrate activation of insulin-degrading enzyme (insulysin). A potential target for drug development. J. Biol. Chem. 2003, 278, 49789–49794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leissring, M.A.; Lu, A.; Condron, M.M.; Teplow, D.B.; Stein, R.L.; Farris, W.; Selkoe, D.J. Kinetics of amyloid beta-protein degradation determined by novel fluorescence- and fluorescence polarization-based assays. J. Biol. Chem. 2003, 278, 37314–37320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suire, C.N.; Lane, S.; Leissring, M.A. Development and Characterization of Quantitative, High-Throughput-Compatible Assays for Proteolytic Degradation of Glucagon. SLAS Discov. 2018, 23, 1060–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suire, C.N.; Brizuela, M.K.; Leissring, M.A. Quantitative, High-Throughput Assays for Proteolytic Degradation of Amylin. Methods Protoc. 2020, 3, 81. [Google Scholar] [CrossRef]
- Clot, J.P.; Janicot, M.; Fouque, F.; Desbuquois, B.; Haumont, P.Y.; Lederer, F. Characterization of insulin degradation products generated in liver endosomes: In vivo and in vitro studies. Mol. Cell. Endocrinol. 1990, 72, 175–185. [Google Scholar] [CrossRef]
- Hamel, F.G.; Posner, B.I.; Bergeron, J.J.; Frank, B.H.; Duckworth, W.C. Isolation of insulin degradation products from endosomes derived from intact rat liver. J. Biol. Chem. 1988, 263, 6703–6708. [Google Scholar] [CrossRef]
- Seabright, P.J.; Smith, G.D. The characterization of endosomal insulin degradation intermediates and their sequence of production. Biochem. J. 1996, 320 Pt 3, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Authier, F.; Metioui, M.; Fabrega, S.; Kouach, M.; Briand, G. Endosomal proteolysis of internalized insulin at the C-terminal region of the B chain by cathepsin D. J. Biol. Chem. 2002, 277, 9437–9446. [Google Scholar] [CrossRef] [Green Version]
- Kouach, M.; Desbuquois, B.; Authier, F. Endosomal proteolysis of internalised [ArgA0]-human insulin at neutral pH generates the mature insulin peptide in rat liver in vivo. Diabetologia 2009, 52, 2621–2632. [Google Scholar] [CrossRef] [Green Version]
- Authier, F.; Posner, B.I.; Bergeron, J.J. Endosomal proteolysis of internalized proteins. FEBS Lett. 1996, 389, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Authier, F.; Posner, B.I.; Bergeron, J.J. Insulin-degrading enzyme. Clin. Investig. Med. 1996, 19, 149–160. [Google Scholar]
- Shen, Y.; Joachimiak, A.; Rosner, M.R.; Tang, W.J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanidis, L.; Fusco, N.D.; Cooper, S.E.; Smith-Carpenter, J.E.; Alper, B.J. Molecular Determinants of Substrate Specificity in Human Insulin-Degrading Enzyme. Biochemistry 2018, 57, 4903–4914. [Google Scholar] [CrossRef] [PubMed]
- Affholter, J.A.; Cascieri, M.A.; Bayne, M.L.; Brange, J.; Casaretto, M.; Roth, R.A. Identification of residues in the insulin molecule important for binding to insulin-degrading enzyme. Biochemistry 1990, 29, 7727–7733. [Google Scholar] [CrossRef] [PubMed]
- Schäffer, L. A model for insulin binding to the insulin receptor. Eur. J. Biochem. 1994, 221, 1127–1132. [Google Scholar] [CrossRef]
- Kristensen, C.; Kjeldsen, T.; Wiberg, F.C.; Schäffer, L.; Hach, M.; Havelund, S.; Bass, J.; Steiner, D.F.; Andersen, A.S. Alanine scanning mutagenesis of insulin. J. Biol. Chem. 1997, 272, 12978–12983. [Google Scholar] [CrossRef] [Green Version]
- De Meyts, P. Insulin/receptor binding: The last piece of the puzzle? What recent progress on the structure of the insulin/receptor complex tells us (or not) about negative cooperativity and activation. BioEssays 2015, 37, 389–397. [Google Scholar] [CrossRef]
- Macháčková, K.; Mlčochová, K.; Potalitsyn, P.; Hanková, K.; Socha, O.; Buděšínský, M.; Muždalo, A.; Lepšík, M.; Černeková, M.; Radosavljević, J.; et al. Mutations at hypothetical binding site 2 in insulin and insulin-like growth factors 1 and 2 result in receptor- and hormone-specific responses. J. Biol. Chem. 2019, 294, 17371–17382. [Google Scholar] [CrossRef]
- Affholter, J.A.; Hsieh, C.L.; Francke, U.; Roth, R.A. Insulin-degrading enzyme: Stable expression of the human complementary DNA, characterization of its protein product, and chromosomal mapping of the human and mouse genes. Mol. Endocrinol. 1990, 4, 1125–1135. [Google Scholar] [CrossRef] [Green Version]
- Kuo, W.L.; Gehm, B.D.; Rosner, M.R. Regulation of insulin degradation: Expression of an evolutionarily conserved insulin-degrading enzyme increases degradation via an intracellular pathway. Mol. Endocrinol. 1991, 5, 1467–1476. [Google Scholar] [CrossRef] [Green Version]
- Kuo, W.L.; Gehm, B.D.; Rosner, M.R. Cloning and expression of the cDNA for a Drosophila insulin-degrading enzyme. Mol. Endocrinol. 1990, 4, 1580–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, W.L.; Gehm, B.D.; Rosner, M.R.; Li, W.; Keller, G. Inducible expression and cellular localization of insulin-degrading enzyme in a stably transfected cell line. J. Biol. Chem. 1994, 269, 22599–22606. [Google Scholar] [PubMed]
- Gehm, B.D.; Rosner, M.R. Regulation of insulin, epidermal growth factor, and transforming growth factor-alpha levels by growth factor-degrading enzymes. Endocrinology 1991, 128, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Kayalar, C.; Wong, W.T. Metalloendoprotease inhibitors which block the differentiation of L6 myoblasts inhibit insulin degradation by the endogenous insulin-degrading enzyme. J. Biol. Chem. 1989, 264, 8928–8934. [Google Scholar] [CrossRef]
- Kayalar, C.; Wong, W.T.; Hendrickson, L. Differentiation of BC3H1 and primary skeletal muscle cells and the activity of their endogenous insulin-degrading enzyme are inhibited by the same metalloendoprotease inhibitors. J. Cell. Biochem. 1990, 44, 137–151. [Google Scholar] [CrossRef]
- Shii, K.; Roth, R.A. Inhibition of insulin degradation by hepatoma cells after microinjection of monoclonal antibodies to a specific cytosolic protease. Proc. Natl. Acad. Sci. USA 1986, 83, 4147–4151. [Google Scholar] [CrossRef] [Green Version]
- Fawcett, J.; Permana, P.A.; Levy, J.L.; Duckworth, W.C. Regulation of protein degradation by insulin-degrading enzyme: Analysis by small interfering RNA-mediated gene silencing. Arch. Biochem. Biophys. 2007, 468, 128–133. [Google Scholar] [CrossRef]
- Louie, S.; Lakkyreddy, J.; Castellano, B.M.; Haley, B.; Dang, A.N.; Lam, C.; Tang, D.; Lang, S.; Snedecor, B.; Misaghi, S. Insulin Degrading Enzyme (IDE) Expressed by Chinese Hamster Ovary (CHO) Cells Is Responsible for Degradation of Insulin in Culture Media. J. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Li, L.; Leissring, M.A. Insulin-degrading enzyme is exported via an unconventional protein secretion pathway. Mol. Neurodegener. 2009, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. Insulin-degrading enzyme is not secreted from cultured cells. Sci. Rep. 2018, 8, 2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Hay, S.O.; Kang, D.; McBride, M.; Li, L.; Zhao, J.; Leissring, M.A. Deletion of insulin-degrading enzyme elicits antipodal, age-dependent effects on glucose and insulin tolerance. PLoS ONE 2011, 6, e20818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.C.; Eckman, E.A.; Sambamurti, K.; Dobbs, N.; Chow, K.M.; Eckman, C.B.; Hersh, L.B.; Thiele, D.L. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 6221–6226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steneberg, P.; Bernardo, L.; Edfalk, S.; Lundberg, L.; Backlund, F.; Ostenson, C.G.; Edlund, H. The type 2 diabetes-associated gene ide is required for insulin secretion and suppression of alpha-synuclein levels in beta-cells. Diabetes 2013, 62, 2004–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa-Pérez, P.; Merino, B.; Fernandez-Diaz, C.M.; Cidad, P.; Lobaton, C.D.; Moreno, A.; Muturi, H.T.; Ghadieh, H.E.; Najjar, S.M.; Leissring, M.A.; et al. Liver-specific ablation of insulin-degrading enzyme causes hepatic insulin resistance and glucose intolerance, without affecting insulin clearance in mice. Metab. Clin. Exp. 2018, 88, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Merino, B.; Fernández-Díaz, C.M.; Parrado-Fernández, C.; González-Casimiro, C.M.; Postigo-Casado, T.; Lobaton, C.D.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Hepatic insulin-degrading enzyme regulates glucose and insulin homeostasis in diet-induced obese mice. Metab. Clin. Exp. 2020, 113, 154352. [Google Scholar] [CrossRef]
- Duckworth, W.C. Insulin and glucagon degradation by the kidney. I. Subcellular distribution under different assay condition. Biochim. Biophys. Acta 1976, 437, 518–530. [Google Scholar] [CrossRef]
- Ciaccio, C.; Tundo, G.R.; Grasso, G.; Spoto, G.; Marasco, D.; Ruvo, M.; Gioia, M.; Rizzarelli, E.; Coletta, M. Somatostatin: A novel substrate and a modulator of insulin-degrading enzyme activity. J. Mol. Biol. 2009, 385, 1556–1567. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.G.; Duckworth, W.C.; Hamel, F.G. Degradation of amylin by insulin-degrading enzyme. J. Biol. Chem. 2000, 275, 36621–36625. [Google Scholar] [CrossRef] [Green Version]
- Kurochkin, I.V.; Goto, S. Alzheimer’s beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994, 345, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Edbauer, D.; Willem, M.; Lammich, S.; Steiner, H.; Haass, C. Insulin-degrading enzyme rapidly removes the beta-amyloid precursor protein intracellular domain (AICD). J. Biol. Chem. 2002, 277, 13389–13393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, L.; Llovera, R.E.; Alonso, L.G.; Frangione, B.; de Prat-Gay, G.; Ghiso, J.; Castano, E.M. Insulin-degrading enzyme degrades amyloid peptides associated with British and Danish familial dementia. Biochem. Biophys. Res. Commun. 2005, 332, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.; Schulze, C.; Baumeister, H.; Buck, F.; Richter, D. Rat insulin-degrading enzyme: Cleavage pattern of the natriuretic peptide hormones ANP, BNP, and CNP revealed by HPLC and mass spectrometry. Biochemistry 1992, 31, 11138–11143. [Google Scholar] [CrossRef] [PubMed]
- Tundo, G.R.; Di Muzio, E.; Ciaccio, C.; Sbardella, D.; Di Pierro, D.; Polticelli, F.; Coletta, M.; Marini, S. Multiple allosteric sites are involved in the modulation of insulin-degrading-enzyme activity by somatostatin. FEBS J. 2016, 283, 3755–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malito, E.; Ralat, L.A.; Manolopoulou, M.; Tsay, J.L.; Wadlington, N.L.; Tang, W.J. Molecular bases for the recognition of short peptide substrates and cysteine-directed modifications of human insulin-degrading enzyme. Biochemistry 2008, 47, 12822–12834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safavi, A.; Miller, B.C.; Cottam, L.; Hersh, L.B. Identification of gamma-endorphin-generating enzyme as insulin-degrading enzyme. Biochemistry 1996, 35, 14318–14325. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.V.; Gehm, B.D.; Rosner, M.R. An evolutionarily conserved enzyme degrades transforming growth factor-alpha as well as insulin. J. Cell Biol. 1989, 109, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Fagan, J.M.; Waxman, L. Purification of a protease in red blood cells that degrades oxidatively damaged haemoglobin. Biochem. J. 1991, 277 Pt 3, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Werlen, R.C.; Offord, R.E.; Rose, K. Preparation and characterization of novel substrates of insulin proteinase (EC 3.4.99.45). Biochem. J. 1994, 302 Pt 3, 907–911. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.G.; Ren, M.; Zhao, F.; Tang, W.J. Structures of human CCL18, CCL3, and CCL4 reveal molecular determinants for quaternary structures and sensitivity to insulin-degrading enzyme. J. Mol. Biol. 2015, 427, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Hahn, F.; Schmalen, A.; Setz, C.; Friedrich, M.; Schlosser, S.; Kolle, J.; Spranger, R.; Rauch, P.; Fraedrich, K.; Reif, T.; et al. Proteolysis of mature HIV-1 p6 Gag protein by the insulin-degrading enzyme (IDE) regulates virus replication in an Env-dependent manner. PLoS ONE 2017, 12, e0174254. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Lang, Y.; Speck, E.R.; Delovitch, T.L. Processing and presentation of insulin. III. Insulin degrading enzyme: A neutral metalloendoproteinase that is non-homologous to classical endoproteinases mediates the processing of insulin epitopes for helper T cells. Int. Immunol. 1992, 4, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Ellis, J.; Delovitch, T.L. Processing and presentation of insulin. II. Evidence for intracellular, plasma membrane-associated and extracellular degradation of human insulin by antigen-presenting B cells. J. Immunol. 1989, 142, 4184–4193. [Google Scholar] [PubMed]
- Parmentier, N.; Stroobant, V.; Colau, D.; de Diesbach, P.; Morel, S.; Chapiro, J.; van Endert, P.; Van den Eynde, B.J. Production of an antigenic peptide by insulin-degrading enzyme. Nat. Immunol. 2010, 11, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Culina, S.; Mauvais, F.X.; Hsu, H.T.; Burgevin, A.; Guenette, S.; Moser, A.; van Endert, P. No major role for insulin-degrading enzyme in antigen presentation by MHC molecules. PLoS ONE 2014, 9, e88365. [Google Scholar] [CrossRef]
- Leissring, M.A.; Farris, W.; Wu, X.; Christodoulou, D.C.; Haigis, M.C.; Guarente, L.; Selkoe, D.J. Alternative translation initiation generates a novel isoform of insulin-degrading enzyme targeted to mitochondria. Biochem. J. 2004, 383, 439–446. [Google Scholar] [CrossRef]
- Grasso, G.; Mielczarek, P.; Niedziolka, M.; Silberring, J. Metabolism of cryptic peptides derived from neuropeptide FF precursors: The involvement of insulin-degrading enzyme. Int. J. Mol. Sci. 2014, 15, 16787–16799. [Google Scholar] [CrossRef] [Green Version]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimer’s Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Zingale, G.A.; Bellia, F.; Ahmed, I.M.M.; Mielczarek, P.; Silberring, J.; Grasso, G. IDE Degrades Nociceptin/Orphanin FQ through an Insulin Regulated Mechanism. Int. J. Mol. Sci. 2019, 20, 4447. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ali, M.A.; Cohen, J.I. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 2006, 127, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ali, M.A.; Wang, K.; Sayre, D.; Hamel, F.G.; Fischer, E.R.; Bennett, R.G.; Cohen, J.I. Insulin degrading enzyme induces a conformational change in varicella-zoster virus gE, and enhances virus infectivity and stability. PLoS ONE 2010, 5, e11327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berarducci, B.; Rajamani, J.; Zerboni, L.; Che, X.; Sommer, M.; Arvin, A.M. Functions of the unique N-terminal region of glycoprotein E in the pathogenesis of varicella-zoster virus infection. Proc. Natl. Acad. Sci. USA 2010, 107, 282–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmalen, A.; Karius-Fischer, J.; Rauch, P.; Setz, C.; Korn, K.; Henklein, P.; Fossen, T.; Schubert, U. The N-Terminus of the HIV-1 p6 Gag Protein Regulates Susceptibility to Degradation by IDE. Viruses 2018, 10, 710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maianti, J.P.; McFedries, A.; Foda, Z.H.; Kleiner, R.E.; Du, X.Q.; Leissring, M.A.; Tang, W.J.; Charron, M.J.; Seeliger, M.A.; Saghatelian, A.; et al. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature 2014, 511, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupfer, S.R.; Wilson, E.M.; French, F.S. Androgen and glucocorticoid receptors interact with insulin degrading enzyme. J. Biol. Chem. 1994, 269, 20622–20628. [Google Scholar]
- Harada, S.; Smith, R.M.; Hu, D.Q.; Jarett, L. Dexamethasone inhibits insulin binding to insulin-degrading enzyme and cytosolic insulin-binding protein p82. Biochem. Biophys. Res. Commun. 1996, 218, 154–158. [Google Scholar] [CrossRef]
- Protzek, A.O.; Rezende, L.F.; Costa-Junior, J.M.; Ferreira, S.M.; Cappelli, A.P.; de Paula, F.M.; de Souza, J.C.; Kurauti, M.A.; Carneiro, E.M.; Rafacho, A.; et al. Hyperinsulinemia caused by dexamethasone treatment is associated with reduced insulin clearance and lower hepatic activity of insulin-degrading enzyme. J. Steroid Biochem. Mol. Biol. 2016, 155, 1–8. [Google Scholar] [CrossRef]
- Caravaggio, J.W.; Hasu, M.; MacLaren, R.; Thabet, M.; Raizman, J.E.; Veinot, J.P.; Marcel, Y.L.; Milne, R.W.; Whitman, S.C. Insulin-degrading enzyme deficiency in bone marrow cells increases atherosclerosis in LDL receptor-deficient mice. Cardiovasc. Pathol. 2013, 22, 458–464. [Google Scholar] [CrossRef]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007, 282, 33583–33592. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Díaz, C.M.; Merino, B.; López-Acosta, J.F.; Cidad, P.; de la Fuente, M.A.; Lobaton, C.D.; Moreno, A.; Leissring, M.A.; Perdomo, G.; Cózar-Castellano, I. Pancreatic beta-cell-specific deletion of insulin-degrading enzyme leads to dysregulated insulin secretion and beta-cell functional immaturity. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E805–E819. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.H.; Kuo, W.L.; Rosner, M.R.; Tang, W.J.; Goldman, R.D. Structural changes in intermediate filament networks alter the activity of insulin-degrading enzyme. FASEB J. 2009, 23, 3734–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 2017, 45, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.M.; Jeong, K.; Lim, S.S. FAK Family Kinases in Vascular Diseases. Int. J. Mol. Sci. 2020, 21, 3630. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Loijens, J.C.; Martin, K.H.; Karginov, A.V.; Parsons, J.T. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol. Biol. Cell 2002, 13, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Chorell, E.; Steneberg, P.; Vernersson-Lindahl, E.; Edlund, H.; Wittung-Stafshede, P. Insulin-degrading enzyme prevents alpha-synuclein fibril formation in a nonproteolytical manner. Sci. Rep. 2015, 5, 12531. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Chorell, E.; Wittung-Stafshede, P. Insulin-degrading enzyme is activated by the C-terminus of alpha-synuclein. Biochem. Biophys. Res. Commun. 2015, 466, 192–195. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Cullen, P.J.; Korswagen, H.C. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat. Cell Biol. 2011, 14, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Wunder, C. The SNXy flavours of endosomal sorting. Nat. Cell Biol. 2011, 13, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yang, J.; Villar, V.A.M.; Asico, L.D.; Ma, X.; Armando, I.; Sanada, H.; Yoneda, M.; Felder, R.A.; Jose, P.A.; et al. Loss of renal SNX5 results in impaired IDE activity and insulin resistance in mice. Diabetologia 2017. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.F.; Chuang, L.M.; Hsiao, C.F.; Hung, Y.J.; Lin, M.W.; Chen, Y.T.; Grove, J.; Jorgenson, E.; Quertermous, T.; Risch, N.; et al. An autosomal genome-wide scan for loci linked to pre-diabetic phenotypes in nondiabetic Chinese subjects from the Stanford Asia-Pacific Program of Hypertension and Insulin Resistance Family Study. Diabetes 2005, 54, 1200–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Yang, J.; Jones, J.E.; Villar, V.A.; Yu, P.; Armando, I.; Felder, R.A.; Jose, P.A. Sorting nexin 5 and dopamine d1 receptor regulate the expression of the insulin receptor in human renal proximal tubule cells. Endocrinology 2015, 156, 2211–2221. [Google Scholar] [CrossRef] [Green Version]
- Rubin, S.M.; Sage, J.; Skotheim, J.M. Integrating Old and New Paradigms of G1/S Control. Mol. Cell 2020, 80, 183–192. [Google Scholar] [CrossRef]
- Radulescu, R.T.; Duckworth, W.C.; Levy, J.L.; Fawcett, J. Retinoblastoma protein co-purifies with proteasomal insulin-degrading enzyme: Implications for cell proliferation control. Biochem. Biophys. Res. Commun. 2010, 395, 196–199. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Z.; Ren, M.; Yang, X.; Liu, B.; Qi, H.; Yu, M.; Song, S.; Chen, S.; Liu, L.; et al. SIRT4 regulates PTEN stability through IDE in response to cellular stresses. FASEB J. 2019, 33, 5535–5547. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Bianculli, A.; Orlandi, A.; Desimio, M.G.; Arcuri, G.; Coletta, M.; Marini, S. Insulin-degrading enzyme (IDE): A novel heat shock-like protein. J. Biol. Chem. 2013, 288, 2281–2289. [Google Scholar] [CrossRef] [Green Version]
- Fakhrai-Rad, H.; Nikoshkov, A.; Kamel, A.; Fernström, M.; Zierath, J.R.; Norgren, S.; Luthman, H.; Galli, J. Insulin-degrading enzyme identified as a candidate diabetes susceptibility gene in GK rats. Hum. Mol. Genet. 2000, 9, 2149–2158. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Leissring, M.A.; Eckman, E.A.; Bertram, L.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J. Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid beta-protein. Am. J. Pathol. 2004, 164, 1425–1434. [Google Scholar] [CrossRef]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. A monomeric variant of insulin degrading enzyme (IDE) loses its regulatory properties. PLoS ONE 2010, 5, e9719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noinaj, N.; Bhasin, S.K.; Song, E.S.; Scoggin, K.E.; Juliano, M.A.; Juliano, L.; Hersh, L.B.; Rodgers, D.W. Identification of the allosteric regulatory site of insulysin. PLoS ONE 2011, 6, e20864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, R.K.; Gehm, B.D.; Kuo, W.L.; Rosner, M.R. Functional analysis of conserved residues in the active site of insulin-degrading enzyme. J. Biol. Chem. 1993, 268, 21538–21544. [Google Scholar] [CrossRef]
- Li, P.; Kuo, W.L.; Yousef, M.; Rosner, M.R.; Tang, W.J. The C-terminal domain of human insulin degrading enzyme is required for dimerization and substrate recognition. Biochem. Biophys. Res. Commun. 2006, 343, 1032–1037. [Google Scholar] [CrossRef]
- Gehm, B.D.; Kuo, W.L.; Perlman, R.K.; Rosner, M.R. Mutations in a zinc-binding domain of human insulin-degrading enzyme eliminate catalytic activity but not insulin binding. J. Biol. Chem. 1993, 268, 7943–7948. [Google Scholar] [CrossRef]
- Neant-Fery, M.; Garcia-Ordoñez, R.D.; Logan, T.P.; Selkoe, D.J.; Li, L.; Reinstatler, L.; Leissring, M.A. Molecular basis for the thiol sensitivity of insulin-degrading enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 9582–9587. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.J. Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2016, 27, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Leissring, M.A.; Selkoe, D.J. Structural biology: Enzyme target to latch on to. Nature 2006, 443, 761–762. [Google Scholar] [CrossRef]
- McCord, L.A.; Liang, W.G.; Dowdell, E.; Kalas, V.; Hoey, R.J.; Koide, A.; Koide, S.; Tang, W.J. Conformational states and recognition of amyloidogenic peptides of human insulin-degrading enzyme. Proc. Natl. Acad. Sci. USA 2013, 110, 13827–13832. [Google Scholar] [CrossRef] [Green Version]
- Huising, M.O. Paracrine regulation of insulin secretion. Diabetologia 2020, 63, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Leissring, M.A.; Hemming, M.L.; Chang, A.Y.; Selkoe, D.J. Alternative splicing of human insulin-degrading enzyme yields a novel isoform with a decreased ability to degrade insulin and amyloid beta-protein. Biochemistry 2005, 44, 6513–6525. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, H.; Müller, D.; Rehbein, M.; Richter, D. The rat insulin-degrading enzyme. Molecular cloning and characterization of tissue-specific transcripts. FEBS Lett. 1993, 317, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Runyan, K.; Duckworth, W.C.; Kitabchi, A.E.; Huff, G. The effect of age on insulin-degrading activity in rat tissue. Diabetes 1979, 28, 324–325. [Google Scholar] [CrossRef] [PubMed]
- Sudoh, S.; Frosch, M.P.; Wolf, B.A. Differential effects of proteases involved in intracellular degradation of amyloid beta-protein between detergent-soluble and -insoluble pools in CHO-695 cells. Biochemistry 2002, 41, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Díaz, C.M.; Escobar-Curbelo, L.; López-Acosta, J.F.; Lobaton, C.D.; Moreno, A.; Sanz-Ortega, J.; Perdomo, G.; Cózar-Castellano, I. Insulin degrading enzyme is up-regulated in pancreatic beta cells by insulin treatment. Histol. Histopathol. 2018, 33, 1167–1180. [Google Scholar] [CrossRef]
- Akiyama, H.; Shii, K.; Yokono, K.; Yonezawa, K.; Sato, S.; Watanabe, K.; Baba, S. Cellular localization of insulin-degrading enzyme in rat liver using monoclonal antibodies specific for this enzyme. Biochem. Biophys. Res. Commun. 1988, 155, 914–922. [Google Scholar] [CrossRef]
- Song, E.S.; Jang, H.; Guo, H.F.; Juliano, M.A.; Juliano, L.; Morris, A.J.; Galperin, E.; Rodgers, D.W.; Hersh, L.B. Inositol phosphates and phosphoinositides activate insulin-degrading enzyme, while phosphoinositides also mediate binding to endosomes. Proc. Natl. Acad. Sci. USA 2017, 114, E2826–E2835. [Google Scholar] [CrossRef] [Green Version]
- Hamel, F.G.; Mahoney, M.J.; Duckworth, W.C. Degradation of intraendosomal insulin by insulin-degrading enzyme without acidification. Diabetes 1991, 40, 436–443. [Google Scholar] [CrossRef]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Duckworth, W.C. Insulin degradation by liver cell membranes. Endocrinology 1979, 104, 1758–1764. [Google Scholar] [CrossRef] [PubMed]
- Yokono, K.; Imamura, Y.; Sakai, H.; Baba, S. Insulin-degrading activity of plasma membranes from rat skeletal muscle: Its isolation, characterization, and biologic significance. Diabetes 1979, 28, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Yokono, K.; Roth, R.A.; Baba, S. Identification of insulin-degrading enzyme on the surface of cultured human lymphocytes, rat hepatoma cells, and primary cultures of rat hepatocytes. Endocrinology 1982, 111, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, I.D.; Williams, J.A.; Bailey, A.C.; Wong, K.Y.; Iwamoto, Y.; Yokono, K.; Baba, S.; Roth, R.A. Degradation of insulin by isolated mouse pancreatic acini. Evidence for cell surface protease activity. Diabetes 1984, 33, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, R.D.; Bandari, S.K.; Vlodavsky, I. Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix Biol. 2019, 75–76, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Folstein, M.F. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Harada, S.; Loten, E.G.; Smith, R.M.; Jarett, L. Nonreceptor mediated nuclear accumulation of insulin in H35 rat hepatoma cells. J. Cell. Physiol. 1992, 153, 607–613. [Google Scholar] [CrossRef]
- Harada, S.; Smith, R.M.; Jarett, L. Mechanisms of nuclear translocation of insulin. Cell Biochem. Biophys. 1999, 31, 307–319. [Google Scholar] [CrossRef]
- Harada, S.; Smith, R.M.; Smith, J.A.; Jarett, L. Inhibition of insulin-degrading enzyme increases translocation of insulin to the nucleus in H35 rat hepatoma cells: Evidence of a cytosolic pathway. Endocrinology 1993, 132, 2293–2298. [Google Scholar] [CrossRef]
- Shah, N.; Zhang, S.; Harada, S.; Smith, R.M.; Jarett, L. Electron microscopic visualization of insulin translocation into the cytoplasm and nuclei of intact H35 hepatoma cells using covalently linked Nanogold-insulin. Endocrinology 1995, 136, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Smith, R.M.; Jarett, L. 1,10-Phenanthroline increases nuclear accumulation of insulin in response to inhibiting insulin degradation but has a biphasic effect on insulin’s ability to increase mRNA levels. DNA Cell Biol. 1994, 13, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Smith, R.M.; Smith, J.A.; Shah, N.; Jarett, L. Demonstration of specific insulin binding to cytosolic proteins in H35 hepatoma cells, rat liver and skeletal muscle. Biochem. J. 1995, 306 Pt 1, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Pivovarova, O.; Gogebakan, O.; Pfeiffer, A.F.; Rudovich, N. Glucose inhibits the insulin-induced activation of the insulin-degrading enzyme in HepG2 cells. Diabetologia 2009, 52, 1656–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Ke, B.; Zhao, Z.; Ye, X.; Gao, Z.; Ye, J. Regulation of insulin degrading enzyme activity by obesity-associated factors and pioglitazone in liver of diet-induced obese mice. PLoS ONE 2014, 9, e95399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Liu, J.; Chen, J.; Yao, C.; Yang, Y.; Wang, J.; Zhuang, H.; Hua, Z.C. FADD Phosphorylation Modulates Blood Glucose Levels by Decreasing the Expression of Insulin-Degrading Enzyme. Mol. Cells 2020, 43, 373–383. [Google Scholar] [CrossRef]
- de Kloet, A.D.; Woods, S.C. Minireview: Endocannabinoids and their receptors as targets for obesity therapy. Endocrinology 2009, 150, 2531–2536. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, L.; Xiong, K.; Godlewski, G.; Mukhopadhyay, B.; Tam, J.; Yin, S.; Gao, P.; Shan, X.; Pickel, J.; et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology 2012, 142, 1218–1228. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Febbraio, M.A.; Whitham, M. From cytokine to myokine: The emerging role of interleukin-6 in metabolic regulation. Immunol. Cell Biol. 2014, 92, 331–339. [Google Scholar] [CrossRef]
- Kurauti, M.A.; Costa-Junior, J.M.; Ferreira, S.M.; Santos, G.J.; Sponton, C.H.G.; Carneiro, E.M.; Telles, G.D.; Chacon-Mikahil, M.P.T.; Cavaglieri, C.R.; Rezende, L.F.; et al. Interleukin-6 increases the expression and activity of insulin-degrading enzyme. Sci. Rep. 2017, 7, 46750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camberos, M.C.; Pérez, A.A.; Udrisar, D.P.; Wanderley, M.I.; Cresto, J.C. ATP inhibits insulin-degrading enzyme activity. Exp. Biol. Med. 2001, 226, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Ivancic, V.A.; Krasinski, C.A.; Zheng, Q.; Meservier, R.J.; Spratt, D.E.; Lazo, N.D. Enzyme kinetics from circular dichroism of insulin reveals mechanistic insights into the regulation of insulin-degrading enzyme. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camberos, M.C.; Cresto, J.C. Insulin-degrading enzyme hydrolyzes ATP. Exp. Biol. Med. 2007, 232, 281–292. [Google Scholar]
- Grasso, G.; Satriano, C.; Milardi, D. A neglected modulator of insulin-degrading enzyme activity and conformation: The pH. Biophys. Chem. 2015, 203–204, 33–40. [Google Scholar] [CrossRef]
- Beffy, P.; Lajoix, A.D.; Masiello, P.; Dietz, S.; Péraldi-Roux, S.; Chardès, T.; Ribes, G.; Gross, R. A constitutive nitric oxide synthase modulates insulin secretion in the INS-1 cell line. Mol. Cell. Endocrinol. 2001, 183, 41–48. [Google Scholar] [CrossRef]
- Fujimoto, M.; Shimizu, N.; Kunii, K.; Martyn, J.A.; Ueki, K.; Kaneki, M. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes 2005, 54, 1340–1348. [Google Scholar] [CrossRef] [Green Version]
- Natali, A.; Santini, E.; Delbarba, A.; Baldi, S.; Venturi, E.; Tulipani, A.; Nisoli, E.; Ferrannini, E. Effects of short and prolonged mild intracellular nitric oxide manipulations on various aspects of insulin secretion in INS-1E β-cells. Exp. Clin. Endocrinol. Diabetes 2012, 120, 210–216. [Google Scholar] [CrossRef]
- Cordes, C.M.; Bennett, R.G.; Siford, G.L.; Hamel, F.G. Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation. Biochem. Pharmacol. 2009, 77, 1064–1073. [Google Scholar] [CrossRef] [Green Version]
- Cordes, C.M.; Bennett, R.G.; Siford, G.L.; Hamel, F.G. Redox regulation of insulin degradation by insulin-degrading enzyme. PLoS ONE 2011, 6, e18138. [Google Scholar] [CrossRef] [Green Version]
- Natali, A.; Ribeiro, R.; Baldi, S.; Tulipani, A.; Rossi, M.; Venturi, E.; Mari, A.; Macedo, M.P.; Ferrannini, E. Systemic inhibition of nitric oxide synthesis in non-diabetic individuals produces a significant deterioration in glucose tolerance by increasing insulin clearance and inhibiting insulin secretion. Diabetologia 2013, 56, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Oddo, S.; Sugarman, M.C.; Akbari, Y.; LaFerla, F.M. Age- and region-dependent alterations in Abeta-degrading enzymes: Implications for Abeta-induced disorders. Neurobiol. Aging 2005, 26, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Shinall, H.; Song, E.S.; Hersh, L.B. Susceptibility of amyloid beta peptide degrading enzymes to oxidative damage: A potential Alzheimer’s disease spiral. Biochemistry 2005, 44, 15345–15350. [Google Scholar] [CrossRef]
- Minamiyama, Y.; Takemura, S.; Bito, Y.; Shinkawa, H.; Tsukioka, T.; Nakahira, A.; Suehiro, S.; Okada, S. Supplementation of alpha-tocopherol improves cardiovascular risk factors via the insulin signalling pathway and reduction of mitochondrial reactive oxygen species in type II diabetic rats. Free Radic. Res. 2008, 42, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Venturini, P.R.; Thomazini, B.F.; Oliveira, C.A.; Alves, A.A.; Camargo, T.F.; Domingues, C.E.C.; Barbosa-Sampaio, H.C.L.; do Amaral, M.E.C. Vitamin E supplementation and caloric restriction promotes regulation of insulin secretion and glycemic homeostasis by different mechanisms in rats. Biochem. Cell Biol. 2018, 96, 777–785. [Google Scholar] [CrossRef]
- Rezende, L.F.; Camargo, R.L.; Branco, R.C.; Cappelli, A.P.; Boschero, A.C.; Carneiro, E.M. Reduced insulin clearance and lower insulin-degrading enzyme expression in the liver might contribute to the thrifty phenotype of protein-restricted mice. Br. J. Nutr. 2014, 112, 900–907. [Google Scholar] [CrossRef] [Green Version]
- Kurauti, M.A.; Freitas-Dias, R.; Ferreira, S.M.; Vettorazzi, J.F.; Nardelli, T.R.; Araujo, H.N.; Santos, G.J.; Carneiro, E.M.; Boschero, A.C.; Rezende, L.F.; et al. Acute Exercise Improves Insulin Clearance and Increases the Expression of Insulin-Degrading Enzyme in the Liver and Skeletal Muscle of Swiss Mice. PLoS ONE 2016, 11, e0160239. [Google Scholar] [CrossRef] [Green Version]
- Item, F.; Konrad, D. Visceral fat and metabolic inflammation: The portal theory revisited. Obes. Rev. 2012, 13 (Suppl. 2), 30–39. [Google Scholar] [CrossRef] [Green Version]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Sonne, O. Increased inhibitory potency of free fatty acid-poor albumin on the released and activity of insulin-degrading enzymes from isolated rat adipocytes and hepatocytes. Anal. Biochem. 1985, 151, 109–117. [Google Scholar] [CrossRef]
- Juul, S.M.; Jones, R.H. Evidence for a direct effect of bacitracin on cell-mediated insulin degradation in isolated hepatocytes. Biochem. J. 1982, 206, 295–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svedberg, J.; Björntorp, P.; Smith, U.; Lönnroth, P. Free-fatty acid inhibition of insulin binding, degradation, and action in isolated rat hepatocytes. Diabetes 1990, 39, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Hamel, F.G.; Upward, J.L.; Bennett, R.G. In vitro inhibition of insulin-degrading enzyme by long-chain fatty acids and their coenzyme A thioesters. Endocrinology 2003, 144, 2404–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhang, L.; Liu, S.; Wang, Z. Palmitic acid and docosahexaenoic acid opposingly regulate the expression of insulin-degrading enzyme in neurons. Die Pharm. 2010, 65, 231–232. [Google Scholar]
- Rosin, D.L.; Bond, J.S.; Bradley, S.G. A cysteine metalloproteinase from mouse liver cytosol. Proc. Soc. Exp. Biol. Med. 1984, 177, 112–119. [Google Scholar] [CrossRef]
- Hsu, M.C.; Bai, J.P. Investigation into the presence of insulin-degrading enzyme in cultured type II alveolar cells and the effects of enzyme inhibitors on pulmonary bioavailability of insulin in rats. J. Pharm. Pharmacol. 1998, 50, 507–514. [Google Scholar] [CrossRef]
- Bai, J.P.; Chang, L.L. Transepithelial transport of insulin: I. Insulin degradation by insulin-degrading enzyme in small intestinal epithelium. Pharm. Res. 1995, 12, 1171–1175. [Google Scholar] [CrossRef]
- Abdul-Hay, S.O.; Bannister, T.D.; Wang, H.; Cameron, M.D.; Caulfield, T.R.; Masson, A.; Bertrand, J.; Howard, E.A.; McGuire, M.P.; Crisafulli, U.; et al. Selective Targeting of Extracellular Insulin-Degrading Enzyme by Quasi-Irreversible Thiol-Modifying Inhibitors. ACS Chem. Biol. 2015, 10, 2716–2724. [Google Scholar] [CrossRef]
- Ding, L.; Becker, A.B.; Suzuki, A.; Roth, R.A. Comparison of the enzymatic and biochemical properties of human insulin-degrading enzyme and Escherichia coli protease III. J. Biol. Chem. 1992, 267, 2414–2420. [Google Scholar] [CrossRef]
- Leissring, M.A.; Malito, E.; Hedouin, S.; Reinstatler, L.; Sahara, T.; Abdul-Hay, S.O.; Choudhry, S.; Maharvi, G.M.; Fauq, A.H.; Huzarska, M.; et al. Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS ONE 2010, 5, e10504. [Google Scholar] [CrossRef]
- Leroux, F.; Bosc, D.; Beghyn, T.; Hermant, P.; Warenghem, S.; Landry, V.; Pottiez, V.; Guillaume, V.; Charton, J.; Herledan, A.; et al. Identification of ebselen as a potent inhibitor of insulin degrading enzyme by a drug repurposing screening. Eur. J. Med. Chem. 2019, 179, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Meotti, F.C.; Stangherlin, E.C.; Zeni, G.; Nogueira, C.W.; Rocha, J.B. Protective role of aryl and alkyl diselenides on lipid peroxidation. Environ. Res. 2004, 94, 276–282. [Google Scholar] [CrossRef]
- Chander, P.N.; Gealekman, O.; Brodsky, S.V.; Elitok, S.; Tojo, A.; Crabtree, M.; Gross, S.S.; Goligorsky, M.S. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: Prevention by chronic therapy with a peroxynitrite scavenger ebselen. J. Am. Soc. Nephrol. 2004, 15, 2391–2403. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, C.W.; Zeni, G.; Rocha, J.B. Organoselenium and organotellurium compounds: Toxicology and pharmacology. Chem. Rev. 2004, 104, 6255–6285. [Google Scholar] [CrossRef] [PubMed]
- Nikawa, T.; Schuch, G.; Wagner, G.; Sies, H. Interaction of ebselen with glutathione S-transferase and papain in vitro. Biochem. Pharmacol. 1994, 47, 1007–1012. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, Z.; Chen, N.; Shi, Q.; Tong, L.; Kong, F.; Cheng, X.; Chen, H.; Wang, C.; Tang, B. Target discovery of ebselen with a biotinylated probe. Chem. Commun. 2018, 54, 9506–9509. [Google Scholar] [CrossRef]
- Costa, M.D.; Gai, B.M.; Acker, C.I.; Souza, A.C.; Brandao, R.; Nogueira, C.W. Ebselen reduces hyperglycemia temporarily-induced by diazinon: A compound with insulin-mimetic properties. Chemico-Biol. Interact. 2012, 197, 80–86. [Google Scholar] [CrossRef]
- Wang, X.; Yun, J.W.; Lei, X.G. Glutathione peroxidase mimic ebselen improves glucose-stimulated insulin secretion in murine islets. Antioxid. Redox Signal. 2014, 20, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Kang, S.; Kim, D.S.; Shin, B.K.; Moon, N.R.; Daily, J.W., 3rd. Ebselen pretreatment attenuates ischemia/reperfusion injury and prevents hyperglycemia by improving hepatic insulin signaling and beta-cell survival in gerbils. Free Radic. Res. 2014, 48, 864–874. [Google Scholar] [CrossRef]
- Suire, C.N.; Nainar, S.; Fazio, M.; Kreutzer, A.G.; Paymozd-Yazdi, T.; Topper, C.L.; Thompson, C.R.; Leissring, M.A. Peptidic inhibitors of insulin-degrading enzyme with potential for dermatological applications discovered via phage display. PLoS ONE 2018, 13, e0193101. [Google Scholar] [CrossRef]
- Demidowich, A.P.; Levine, J.A.; Brady, S.M.; Johnson, C.D.; Soldin, S.J.; Yanovski, J.A. Bacitracin attenuates haemolysis-induced insulin degradation during insulin sensitivity testing: Repurposing an old drug for use in metabolic research. Diabetes Obes. Metab. 2020, 22, 1469–1473. [Google Scholar] [CrossRef] [PubMed]
- Rizos, C.V.; Liberopoulos, E.N.; Mikhailidis, D.P.; Elisaf, M.S. Pleiotropic effects of thiazolidinediones. Expert Opin. Pharmacother. 2008, 9, 1087–1108. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yamazaki, H.; Ikeda, T.; Watanabe, T.; Iwabuchi, H.; Nakajima, M.; Yokoi, T. Formation of a novel quinone epoxide metabolite of troglitazone with cytotoxicity to HepG2 cells. Drug Metab. Dispos. 2002, 30, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.O.; Delgado, T.C.; Viegas, J.; Gaspar, J.M.; Scott, D.K.; O’Doherty, R.M.; Macedo, M.P.; Jones, J.G. Mechanisms by which the thiazolidinedione troglitazone protects against sucrose-induced hepatic fat accumulation and hyperinsulinaemia. Br. J. Pharmacol. 2016, 173, 267–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneses, M.J.; Borges, D.O.; Dias, T.R.; Martins, F.O.; Oliveira, P.F.; Macedo, M.P.; Alves, M.G. Knockout of insulin-degrading enzyme leads to mice testicular morphological changes and impaired sperm quality. Mol. Cell. Endocrinol. 2019, 486, 11–17. [Google Scholar] [CrossRef]
- Agbaje, I.M.; Rogers, D.A.; McVicar, C.M.; McClure, N.; Atkinson, A.B.; Mallidis, C.; Lewis, S.E. Insulin dependant diabetes mellitus: Implications for male reproductive function. Hum. Reprod. 2007, 22, 1871–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez, O.; Ballester, B.; Romero, A.; Arnal, E.; Almansa, I.; Miranda, M.; Mesonero, J.E.; Terrado, J. Expression and regulation of insulin and the glucose transporter GLUT8 in the testes of diabetic rats. Horm. Metab. Res. 2009, 41, 343–349. [Google Scholar] [CrossRef]
- Mirsky, I.A. The role of insulinase and insulinase-inhibitors. Metab. Clin. Exp. 1956, 5, 138–143. [Google Scholar]
- Bennett, R.G.; Hamel, F.G.; Duckworth, W.C. An insulin-degrading enzyme inhibitor decreases amylin degradation, increases amylin-induced cytotoxicity, and increases amyloid formation in insulinoma cell cultures. Diabetes 2003, 52, 2315–2320. [Google Scholar] [CrossRef] [Green Version]
- Mirsky, I.A.; Perisutti, G. Effect of insulinase-inhibitor on hypoglycemic action of insulin. Science 1955, 122, 559–560. [Google Scholar] [CrossRef]
- Charton, J.; Gauriot, M.; Guo, Q.; Hennuyer, N.; Marechal, X.; Dumont, J.; Hamdane, M.; Pottiez, V.; Landry, V.; Sperandio, O.; et al. Imidazole-derived 2-[N-carbamoylmethyl-alkylamino]acetic acids, substrate-dependent modulators of insulin-degrading enzyme in amyloid-β hydrolysis. Eur. J. Med. Chem. 2014, 79, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durham, T.B.; Toth, J.L.; Klimkowski, V.J.; Cao, J.X.; Siesky, A.M.; Alexander-Chacko, J.; Wu, G.Y.; Dixon, J.T.; McGee, J.E.; Wang, Y.; et al. Dual Exosite-binding Inhibitors of Insulin-degrading Enzyme Challenge Its Role as the Primary Mediator of Insulin Clearance in Vivo. J. Biol. Chem. 2015, 290, 20044–20059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deprez-Poulain, R.; Hennuyer, N.; Bosc, D.; Liang, W.G.; Enée, E.; Marechal, X.; Charton, J.; Totobenazara, J.; Berte, G.; Jahklal, J.; et al. Catalytic site inhibition of insulin-degrading enzyme by a small molecule induces glucose intolerance in mice. Nat. Commun. 2015, 6, 8250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maianti, J.P.; Tan, G.A.; Vetere, A.; Welsh, A.J.; Wagner, B.K.; Seeliger, M.A.; Liu, D.R. Substrate-selective inhibitors that reprogram the activity of insulin-degrading enzyme. Nat. Chem. Biol. 2019, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Tomita, T. Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn. J. Basic Med. Sci. 2016, 16, 162–179. [Google Scholar] [CrossRef] [Green Version]
- Dor, Y.; Glaser, B. beta-cell dedifferentiation and type 2 diabetes. N. Engl. J. Med. 2013, 368, 572–573. [Google Scholar] [CrossRef]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Thorel, F.; Népote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Chera, S.; Baronnier, D.; Ghila, L.; Cigliola, V.; Jensen, J.N.; Gu, G.; Furuyama, K.; Thorel, F.; Gribble, F.M.; Reimann, F.; et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature 2014, 514, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Brereton, M.F.; Rohm, M.; Ashcroft, F.M. beta-Cell dysfunction in diabetes: A crisis of identity? Diabetes Obes. Metab. 2016, 18 (Suppl. 1), 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.S.; Stein, R.W. Evidence for Loss in Identity, De-Differentiation, and Trans-Differentiation of Islet β-Cells in Type 2 Diabetes. Front. Genet. 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Watanabe, R.M.; Valle, T.T.; Hauser, E.R.; Magnuson, V.L.; Langefeld, C.D.; Ally, D.S.; Mohlke, K.L.; Silander, K.; Kohtamäki, K.; et al. The Finland-United States investigation of non-insulin-dependent diabetes mellitus genetics (FUSION) study. I. An autosomal genome scan for genes that predispose to type 2 diabetes. Am. J. Hum. Genet. 2000, 67, 1174–1185. [Google Scholar] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, L.; Tura, A.; Patel, S.K.; Ibrahim, I.M.; Ferrannini, E.; Zeggini, E.; Weedon, M.N.; Mari, A.; Hattersley, A.T.; McCarthy, M.I.; et al. Common variants of the novel type 2 diabetes genes CDKAL1 and HHEX/IDE are associated with decreased pancreatic beta-cell function. Diabetes 2007, 56, 3101–3104. [Google Scholar] [CrossRef] [Green Version]
- Snehalatha, C.; Timothy, H.; Mohan, V.; Ramachandran, A.; Viswanathan, M. Immunoreactive insulin and insulin degrading enzymes in erythrocytes. A preliminary report. J. Assoc. Physicians India 1990, 38, 558–561. [Google Scholar]
- Standl, E.; Kolb, H.J. Insulin degrading enzyme activity and insulin binding of erythrocytes in normal subjects and Type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1984, 27, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Fawcett, J.; Sang, H.; Permana, P.A.; Levy, J.L.; Duckworth, W.C. Insulin metabolism in human adipocytes from subcutaneous and visceral depots. Biochem. Biophys. Res. Commun. 2010, 402, 762–766. [Google Scholar] [CrossRef]
- Pivovarova, O.; von Loeffelholz, C.; Ilkavets, I.; Sticht, C.; Zhuk, S.; Murahovschi, V.; Lukowski, S.; Döcke, S.; Kriebel, J.; de las Heras Gala, T.; et al. Modulation of insulin degrading enzyme activity and liver cell proliferation. Cell Cycle 2015, 14, 2293–2300. [Google Scholar] [CrossRef] [Green Version]
- Grarup, N.; Rose, C.S.; Andersson, E.A.; Andersen, G.; Nielsen, A.L.; Albrechtsen, A.; Clausen, J.O.; Rasmussen, S.S.; Jørgensen, T.; Sandbaek, A.; et al. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: Validation and extension of genome-wide association studies. Diabetes 2007, 56, 3105–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Hu, R.; Brissova, M.; Stein, R.W.; Powers, A.C.; Gu, G.; Kaverina, I. Microtubules Negatively Regulate Insulin Secretion in Pancreatic β Cells. Dev. Cell 2015, 34, 656–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnell, A.H.; Swenne, I.; Borg, L.A. Lysosomes and pancreatic islet function. A quantitative estimation of crinophagy in the mouse pancreatic B-cell. Cell Tissue Res. 1988, 252, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.; Ravazzola, M.; Amherdt, M.; Yanaihara, C.; Yanaihara, N.; Halban, P.; Renold, A.E.; Perrelet, A. Insulin, not C-peptide (proinsulin), is present in crinophagic bodies of the pancreatic B-cell. J. Cell Biol. 1984, 98, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchizono, Y.; Alarcón, C.; Wicksteed, B.L.; Marsh, B.J.; Rhodes, C.J. The balance between proinsulin biosynthesis and insulin secretion: Where can imbalance lead? Diabetes Obes. Metab. 2007, 9 (Suppl. 2), 56–66. [Google Scholar] [CrossRef]
- Lachaal, M.; Spangler, R.A.; Jung, C.Y. High Km of GLUT-2 glucose transporter does not explain its role in insulin secretion. Am. J. Physiol. 1993, 265, E914–E919. [Google Scholar] [CrossRef]
- Thorens, B.; Wu, Y.J.; Leahy, J.L.; Weir, G.C. The loss of GLUT2 expression by glucose-unresponsive beta cells of db/db mice is reversible and is induced by the diabetic environment. J. Clin. Investig. 1992, 90, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Unger, R.H. Diabetic hyperglycemia: Link to impaired glucose transport in pancreatic beta cells. Science 1991, 251, 1200–1205. [Google Scholar] [CrossRef]
- Maeda, Y.; Akazawa, S.; Akazawa, M.; Takao, Y.; Trocino, R.A.; Takino, H.; Kawasaki, E.; Yokota, A.; Okuno, S.; Nagataki, S. Glucose transporter gene expression in rat conceptus during early organogenesis and exposure to insulin-induced hypoglycemic serum. Acta Diabetol. 1993, 30, 73–78. [Google Scholar] [CrossRef]
- Guillam, M.T.; Hümmler, E.; Schaerer, E.; Yeh, J.I.; Birnbaum, M.J.; Beermann, F.; Schmidt, A.; Dériaz, N.; Thorens, B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef]
- Blum, B.; Hrvatin, S.; Schuetz, C.; Bonal, C.; Rezania, A.; Melton, D.A. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat. Biotechnol. 2012, 30, 261–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henquin, J.C.; Nenquin, M. Immaturity of insulin secretion by pancreatic islets isolated from one human neonate. J. Diabetes Investig. 2018, 9, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Roy, N.; Russ, H.A.; Leonhardt, L.; French, E.K.; Roy, R.; Bengtsson, H.; Scott, D.K.; Stewart, A.F.; Hebrok, M. Replication confers β cell immaturity. Nat. Commun. 2018, 9, 485. [Google Scholar] [CrossRef]
- Huang, C.; Walker, E.M.; Dadi, P.K.; Hu, R.; Xu, Y.; Zhang, W.; Sanavia, T.; Mun, J.; Liu, J.; Nair, G.G.; et al. Synaptotagmin 4 Regulates Pancreatic β Cell Maturation by Modulating the Ca(2+) Sensitivity of Insulin Secretion Vesicles. Dev. Cell 2018, 45, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Kropp, P.A.; Dunn, J.C.; Carboneau, B.A.; Stoffers, D.A.; Gannon, M. Cooperative function of Pdx1 and Oc1 in multipotent pancreatic progenitors impacts postnatal islet maturation and adaptability. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E308–E321. [Google Scholar] [CrossRef] [Green Version]
- Najjar, S.M. Regulation of insulin action by CEACAM1. Trends Endocrinol. Metab. 2002, 13, 240–245. [Google Scholar] [CrossRef]
- Bar, R.S.; Gorden, P.; Roth, J.; Kahn, C.R.; De Meyts, P. Fluctuations in the affinity and concentration of insulin receptors on circulating monocytes of obese patients: Effects of starvation, refeeding, and dieting. J. Clin. Investig. 1976, 58, 1123–1135. [Google Scholar] [CrossRef]
- Olefsky, J.M. The insulin receptor: Its role in insulin resistance of obesity and diabetes. Diabetes 1976, 25, 1154–1162. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Reaven, G.M. Decreased insulin binding to lymphocytes from diabetic subjects. J. Clin. Investig. 1974, 54, 1323–1328. [Google Scholar] [CrossRef]
- Beck-Nielsen, H. The pathogenic role of an insulin-receptor defect in diabetes mellitus of the obese. Diabetes 1978, 27, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Ronnett, G.V.; Knutson, V.P.; Lane, M.D. Insulin-induced down-regulation of insulin receptors in 3T3-L1 adipocytes. Altered rate of receptor inactivation. J. Biol. Chem. 1982, 257, 4285–4291. [Google Scholar] [CrossRef]
- Kono, T.; Barham, F.W. The relationship between the insulin-binding capacity of fat cells and the cellular response to insulin. Studies with intact and trypsin-treated fat cells. J. Biol. Chem. 1971, 246, 6210–6216. [Google Scholar] [CrossRef]
- Le Marchand-Brustel, Y.; Jeanrenaud, B.; Freychet, P. Insulin binding and effects in isolated soleus muscle of lean and obese mice. Am. J. Physiol. 1978, 234, E348–E358. [Google Scholar] [CrossRef] [PubMed]
- Hamel, F.G.; Peavy, D.E.; Ryan, M.P.; Duckworth, W.C. HPLC analysis of insulin degradation products from isolated hepatocytes. Effects of inhibitors suggest intracellular and extracellular pathways. Diabetes 1987, 36, 702–708. [Google Scholar] [CrossRef]
- Blackard, W.G.; Ludeman, C.; Stillman, J. Role of hepatocyte plasma membrane in insulin degradation. Am. J. Physiol. 1985, 248, E194–E202. [Google Scholar] [CrossRef]
- Hammons, G.T.; Smith, R.M.; Jarett, L. Inhibition by bacitracin of rat adipocyte plasma membrane degradation of 125I-insulin is associated with an increase in plasma membrane bound insulin and a potentiation of glucose oxidation by adipocytes. J. Biol. Chem. 1982, 257, 11563–11570. [Google Scholar] [CrossRef]
- Caro, J.F.; Muller, G.; Glennon, J.A. Insulin processing by the liver. J. Biol. Chem. 1982, 257, 8459–8466. [Google Scholar] [CrossRef]
- Marshall, S. Dual pathways for the intracellular processing of insulin. Relationship between retroendocytosis of intact hormone and the recycling of insulin receptors. J. Biol. Chem. 1985, 260, 13524–13531. [Google Scholar] [CrossRef]
- Suire, C.N.; Abdul-Hay, S.O.; Sahara, T.; Kang, D.; Brizuela, M.K.; Saftig, P.; Dickson, D.W.; Rosenberry, T.L.; Leissring, M.A. Cathepsin D regulates cerebral Aβ42/40 ratios via differential degradation of Aβ42 and Aβ40. Alzheimer’s Res. Ther. 2020, 12, 80. [Google Scholar] [CrossRef]
- Gliemann, J.; Sonne, O. Binding and receptor-mediated degradation of insulin in adipocytes. J. Biol. Chem. 1978, 253, 7857–7863. [Google Scholar] [PubMed]
- Gorden, P.J.; Carpentier, J.L.; Freychet, P.; LeCam, A.; Orci, L. Intracellular translocation of iodine-125-labeled insulin: Direct demonstration in isolated hepatocytes. Science 1978, 200, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Farris, W.; Chang, A.Y.; Walsh, D.M.; Wu, X.; Sun, X.; Frosch, M.P.; Selkoe, D.J. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 2003, 40, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; Depinho, R.A.; Accili, D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007, 6, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Leavens, K.F.; Easton, R.M.; Shulman, G.I.; Previs, S.F.; Birnbaum, M.J. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009, 10, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Wan, M.; Leavens, K.F.; Saleh, D.; Easton, R.M.; Guertin, D.A.; Peterson, T.R.; Kaestner, K.H.; Sabatini, D.M.; Birnbaum, M.J. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab. 2011, 14, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef]
- Garris, D.R.; Garris, B.L. Cytochemical analysis of pancreatic islet hypercytolipidemia following diabetes (db/db) and obese (ob/ob) mutation expression: Influence of genomic background. Pathobiology 2004, 71, 231–240. [Google Scholar] [CrossRef]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Cohen, S.E.; Kokkotou, E.; Biddinger, S.B.; Kondo, T.; Gebhardt, R.; Kratzsch, J.; Mantzoros, C.S.; Kahn, C.R. High circulating leptin receptors with normal leptin sensitivity in liver-specific insulin receptor knock-out (LIRKO) mice. J. Biol. Chem. 2007, 282, 23672–23678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, N.; Kubota, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Mineyama, T.; Ogata, H.; Tokuyama, K.; Ohsugi, M.; et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 2008, 8, 49–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sopasakis, V.R.; Liu, P.; Suzuki, R.; Kondo, T.; Winnay, J.; Tran, T.T.; Asano, T.; Smyth, G.; Sajan, M.P.; Farese, R.V.; et al. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010, 11, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, Y.; Ogawa, W.; Nishizawa, A.; Inoue, H.; Teshigawara, K.; Kinoshita, S.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; Sakaue, H.; et al. Restoration of glucokinase expression in the liver normalizes postprandial glucose disposal in mice with hepatic deficiency of PDK1. Diabetes 2007, 56, 1000–1009. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Li, L.; Qi, Y.; Zhu, X.; Gan, B.; DePinho, R.A.; Averitt, T.; Guo, S. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 2012, 153, 631–646. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Rüegg, M.A.; Hall, M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Ghadieh, H.E.; Russo, L.; Muturi, H.T.; Ghanem, S.S.; Manaserh, I.H.; Noh, H.L.; Suk, S.; Kim, J.K.; Hill, J.W.; Najjar, S.M. Hyperinsulinemia drives hepatic insulin resistance in male mice with liver-specific Ceacam1 deletion independently of lipolysis. Metab. Clin. Exp. 2019, 93, 33–43. [Google Scholar] [CrossRef]




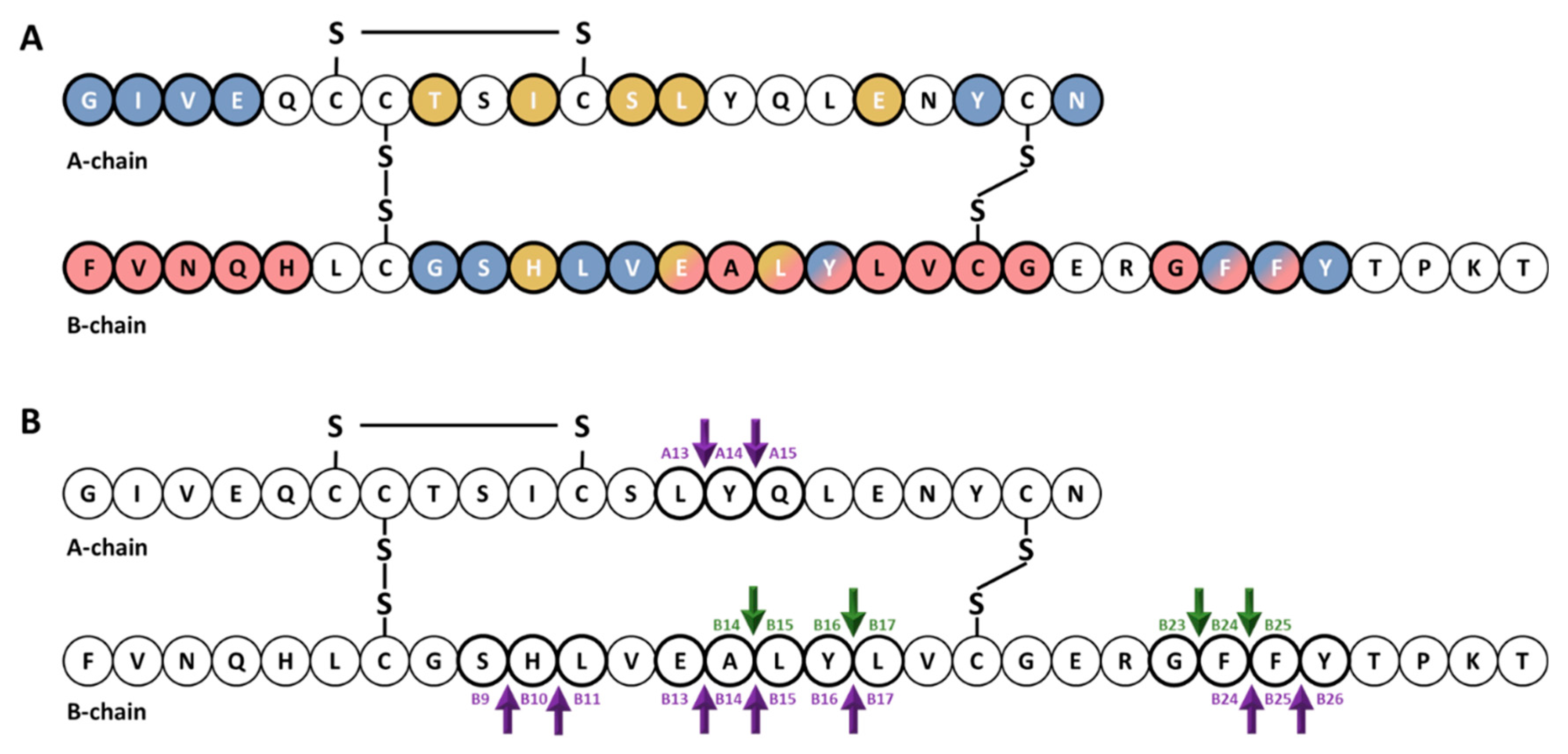{kind=link}
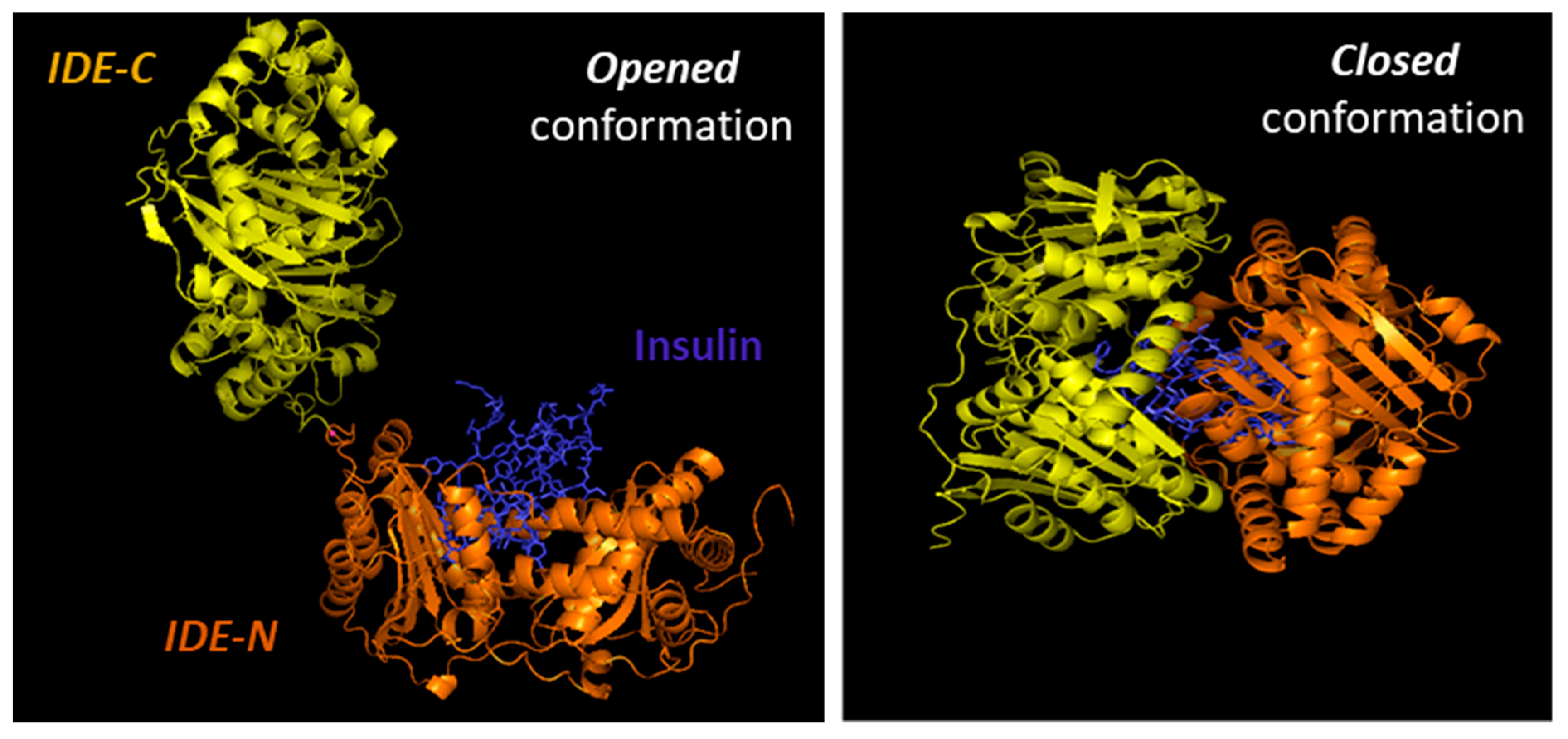{kind=link}
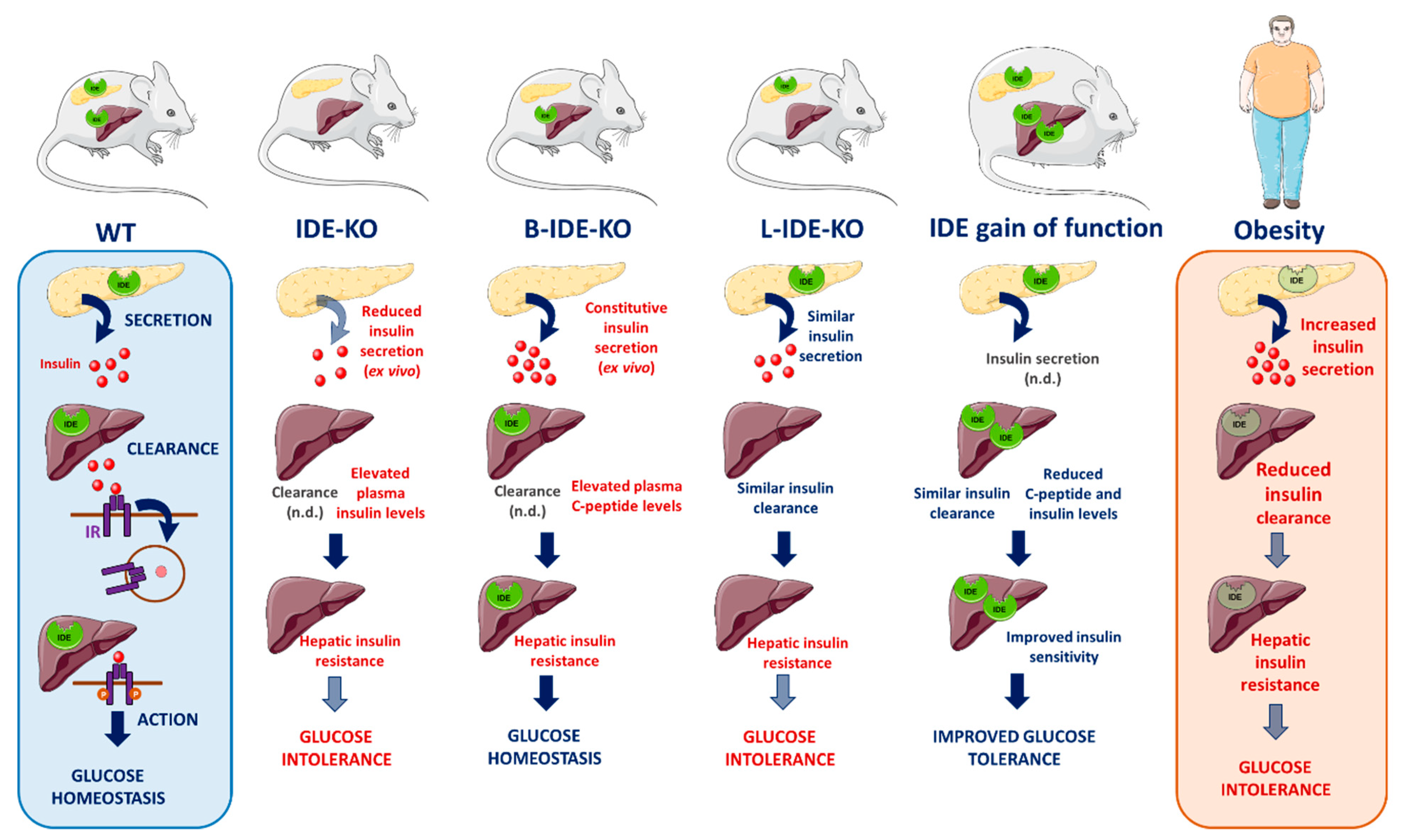{kind=link}
| UniProtKB Entry | Organism | Gene Name | Protein Name | Protein Length | E-Value * | Identity (%) * | Positives (%) * |
|---|---|---|---|---|---|---|---|
| Q5UPX9 (YL233_MIMIV) | Acanthamoeba polyphaga mimivirus (APMV) | MIMI_L233 | Putative zinc protease L233 | 440 | 1.3 × 10−5 | 20.9 | 40.4 |
| P31828 (PQQL_ECOLI) | Escherichia coli (strain K12) | pqqL | Probable zinc protease PqqL | 931 | 2.4 × 10−9 | 24.6 | 41.5 |
| P05458 (PTRA_ECOLI) | Escherichia coli (strain K12) | ptrA | Protease 3 (Pitrilysin) | 962 | 7.1 × 10−6 | 27.6 | 47.6 |
| O22941 (IDE1_ARATH) | Arabidopsis thaliana (Mouse-ear cress) | PXM16 | Insulin-degrading enzyme-like 1, peroxisomal | 970 | 0.0 | 38.5 | 57.1 |
| F4J3D9 (IDE2_ARATH) | Arabidopsis thaliana (Mouse-ear cress) | At3g57470 | Insulin-degrading enzyme-like 2 | 966 | 0.0 | 39.4 | 57.9 |
| Q06010 (STE23_YEAST) | Saccharomyces cerevisiae (strain ATCC 204508/S288c) (Baker’s yeast) | STE23 | A-factor-processing enzyme | 1027 | 0.0 | 38.4 | 60.2 |
| P40851 (AXL1_YEAST) | Saccharomyces cerevisiae (strain ATCC 204508/S288c) (Baker’s yeast) | AXL1 | Putative protease AXL1 | 1208 | 9.2 × 10−50 | 21.5 | 41.5 |
| P22817 (IDE_DROME) | Drosophila melanogaster (Fruit fly) | Ide | Insulin-degrading enzyme | 990 | 0.0 | 46.6 | 67.3 |
| Q9JHR7 (IDE_MOUSE) | Mus musculus (House mouse) | Ide | Insulin-degrading enzyme | 1019 | 0.0 | 95.0 | 97.4 |
| P35559 (IDE_RAT) | Rattus norvegicus (Norwegian rat) | Ide | Insulin-degrading enzyme | 1019 | 0.0 | 95.5 | 97.6 |
| Q24K02 (IDE_BOVIN) | Bos taurus (Bovine) | Ide | Insulin-degrading enzyme | 1019 | 0.0 | 98.8 | 99.2 |
| F7EFL5 (F7EFL5_MACMU) | Macaca mulatta (Rhesus macaque) | Ide | Insulin-degrading enzyme | 1019 | 0.0 | 99.5 | 99.7 |
| P14735 (IDE_HUMAN) | Homo sapiens (Human) | Ide | Insulin-degrading enzyme | 1019 | 0.0 | 100 | 100 |
| UniProtKB Entry | Organism | Gene Name | Protein Name | Protein Length | E-Value * | Identity (%) * | Positives (%) * |
|---|---|---|---|---|---|---|---|
| Q5JRX3 (PREP_HUMAN) | Homo sapiens (Human) | PITRM1 | Presequence protease, mitochondrial | 1037 | 7.6 × 10−1 | 28.9 | 51.3 |
| Q10713 (MPPA_HUMAN) | Homo sapiens (Human) | PMPCA | Mitochondrial-processing peptidase subunit alpha | 525 | 1.7 × 10−1 | 24.4 | 45.5 |
| P31930 (QCR1_HUMAN) | Homo sapiens (Human) | UQCRC1 | Cytochrome b-c1 complex subunit 1, mitochondrial | 480 | 1.3 × 10−2 | 21.7 | 40.1 |
| O75439 (MPPB_HUMAN) | Homo sapiens (Human) | PMPCB | Mitochondrial-processing peptidase subunit beta | 489 | 7.9 × 10−3 | 22.8 | 39.6 |
| O43847 (NRDC_HUMAN) | Homo sapiens (Human) | NRDC | Nardilysin | 1151 | 8 × 10−174 | 34.6 | 55.6 |
| P14735 (IDE_HUMAN) | Homo sapiens (Human) | Ide | Insulin-degrading enzyme | 1019 | 0.0 | 100 | 100 |
| Mouse Model | Genetic Background | Target Protein | Target Tissue | Metabolic Phenotype | Insulin Resistance | Insulin Clearance | Refs. |
|---|---|---|---|---|---|---|---|
| db/db | C57BLKs/J | OB-R | Spontaneous mutation of the leptin receptor | Hyperinsulinemia, hyperglycemia, higher body weight | + | n.d. | [288,289] |
| ob/ob | C57BLKs/J | Leptin | Recessive mutation of leptin | Hyperinsulinemia, hyperglycemia, higher body weight | + | n.d. | [289,290] |
| L-IDE-KO | C57BL/6J | IDE | Liver | Normoinsulinemia, higher glucose levels, similar body weight | + | = | [86,87] |
| LIRKO | Mixed genetic background | IR | Liver | Hyperinsulinemia, hyperglycemia, similar body weight | + | Lower | [291,292] |
| LIrs1KO | Mixed genetic background | IRS1 | Liver | Hyperinsulinemia, euglycemia, similar body weight | + (*) | n.d. | [293] |
| LIrs2KO | Mixed genetic background | IRS2 | Liver | Hyperinsulinemia, euglycemia, similar body weight | + ($) | n.d. | [293] |
| L-p110-αKO | Mixed genetic background | PI3K catalytic subunit p110-α | Liver | Hyperinsulinemia, hyperglycemia, increased fat mass | + | n.d. | [294] |
| L-p110βKO | Mixed genetic background | PI3K catalytic subunit p110-β | Liver | Hyperinsulinemia, similar blood glucose levels | + | n.d. | [295] |
| L-Pdk1KO | Mixed genetic background | PDK1 | Liver | Hyperinsulinemia, hyperglycemia, similar body weight | + | n.d. | [296] |
| L-Akt2 | C57BL/6J | AKT2 | Liver | Normoinsulinemia, euglycemia, similar body weight | - | n.d. | [286,287] |
| L1KO(l-FoxO1) | C57BL/6J | FoxO1 | Liver | Normoinsulinemia, lower plasma glucose, similar body weight | - | n.d. | [285,297] |
| LiRiKO | C57BL/6J | Rictor | Liver | Hyperinsulinemia, hyperglycemia, similar body weight | + | n.d. | [298] |
| L-SACC1 (AlbCre + Cc1fl/fl) | C57BL/6J | CEACAM1 | Liver | Hyperinsulinemia, hyperglycemia, increased fat mass | + | Lower | [299] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Casimiro, C.M.; Merino, B.; Casanueva-Álvarez, E.; Postigo-Casado, T.; Cámara-Torres, P.; Fernández-Díaz, C.M.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines 2021, 9, 86. https://doi.org/10.3390/biomedicines9010086
González-Casimiro CM, Merino B, Casanueva-Álvarez E, Postigo-Casado T, Cámara-Torres P, Fernández-Díaz CM, Leissring MA, Cózar-Castellano I, Perdomo G. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines. 2021; 9(1):86. https://doi.org/10.3390/biomedicines9010086
Chicago/Turabian StyleGonzález-Casimiro, Carlos M., Beatriz Merino, Elena Casanueva-Álvarez, Tamara Postigo-Casado, Patricia Cámara-Torres, Cristina M. Fernández-Díaz, Malcolm A. Leissring, Irene Cózar-Castellano, and Germán Perdomo. 2021. "Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme" Biomedicines 9, no. 1: 86. https://doi.org/10.3390/biomedicines9010086