RNA Sequencing in Comparison to Immunohistochemistry for Measuring Cancer Biomarkers in Breast Cancer and Lung Cancer Specimens

Abstract
:1. Introduction
2. Materials and Methods
2.1. BC Biosamples
2.2. LC Biosamples
2.3. Preparation of Libraries and RNA Sequencing
2.4. Processing of RNA Sequencing Data
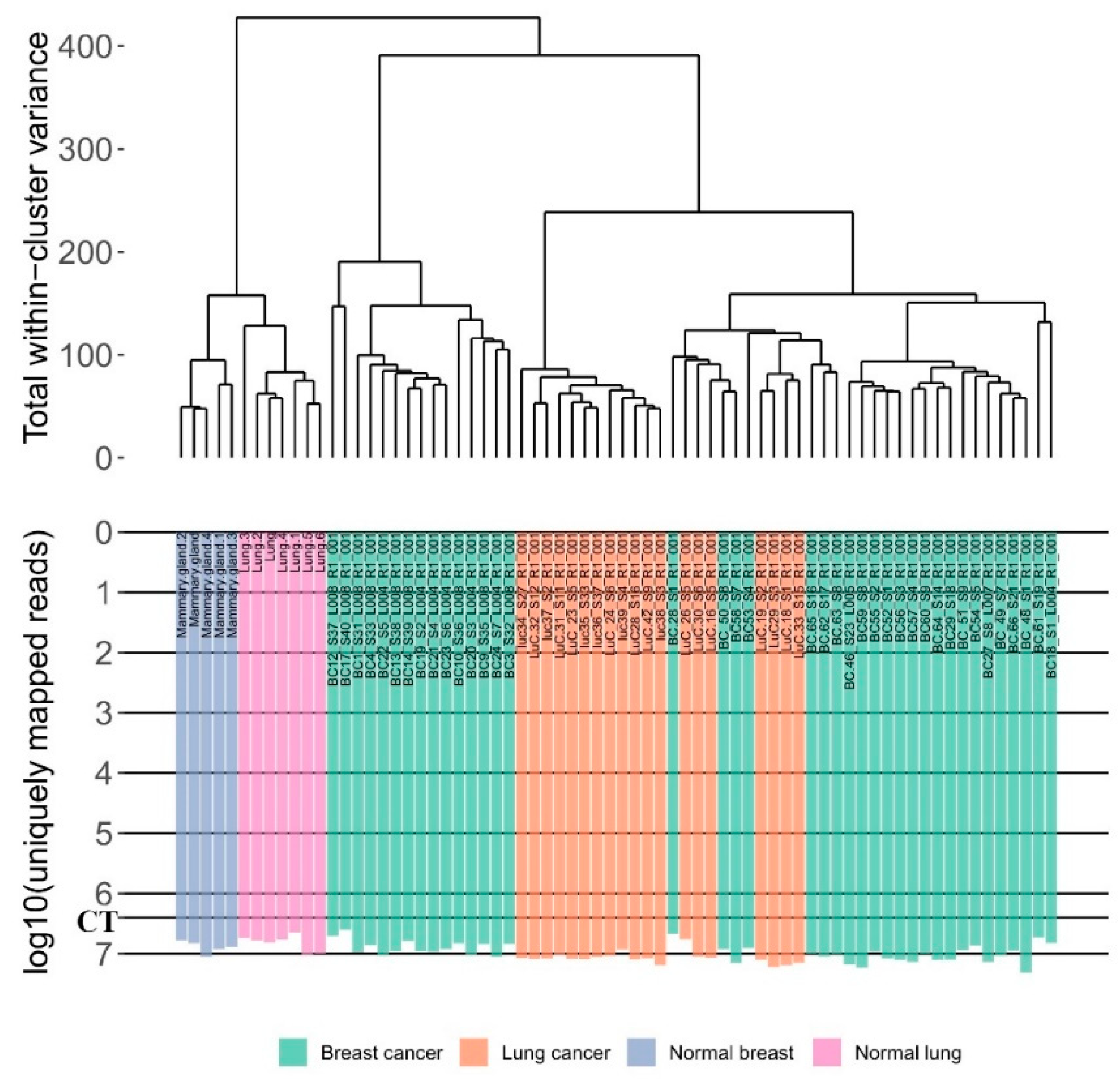
2.5. Data Clustering
2.6. Immunohistochemistry
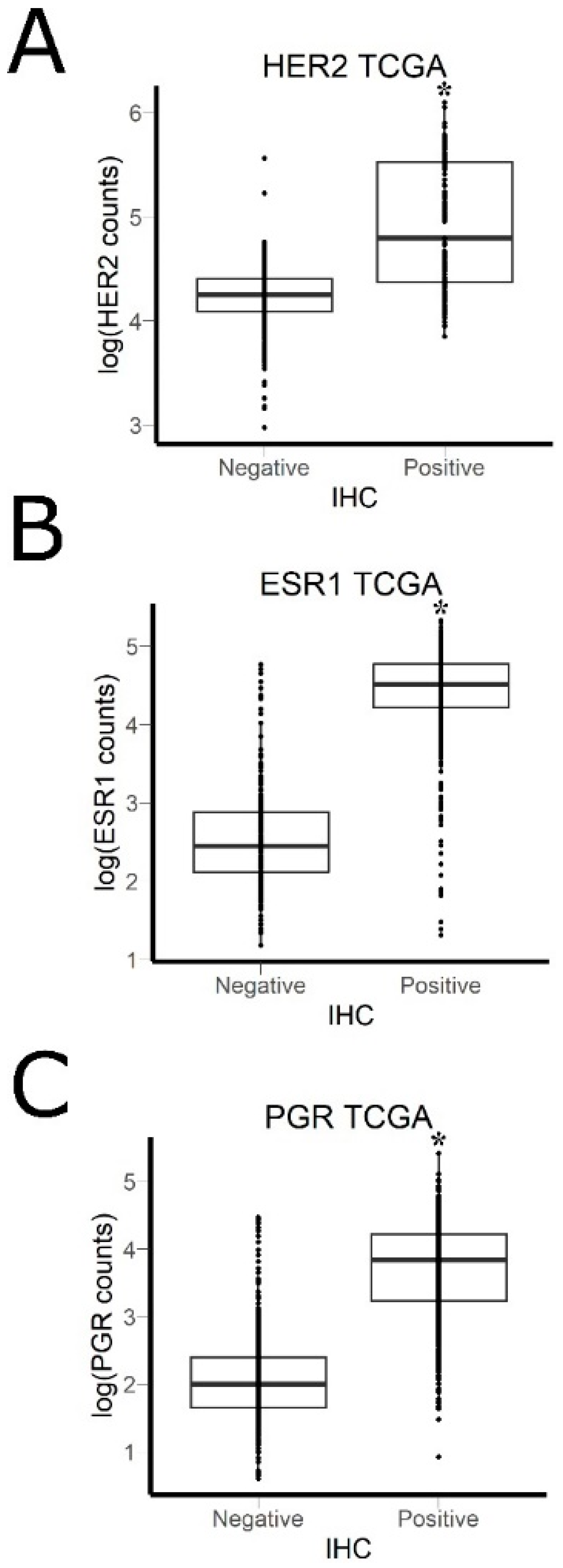
2.7. Literature Gene Expression Data
2.8. Statistical Analysis
3. Results
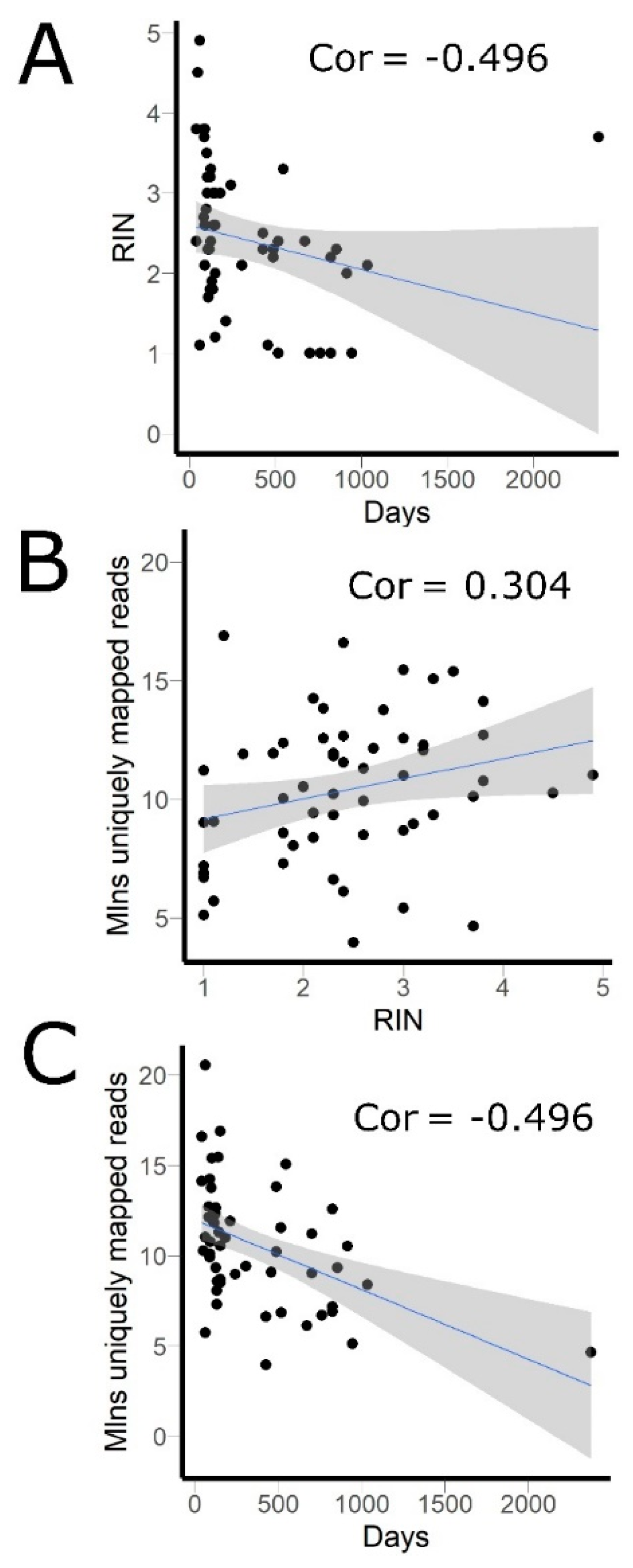
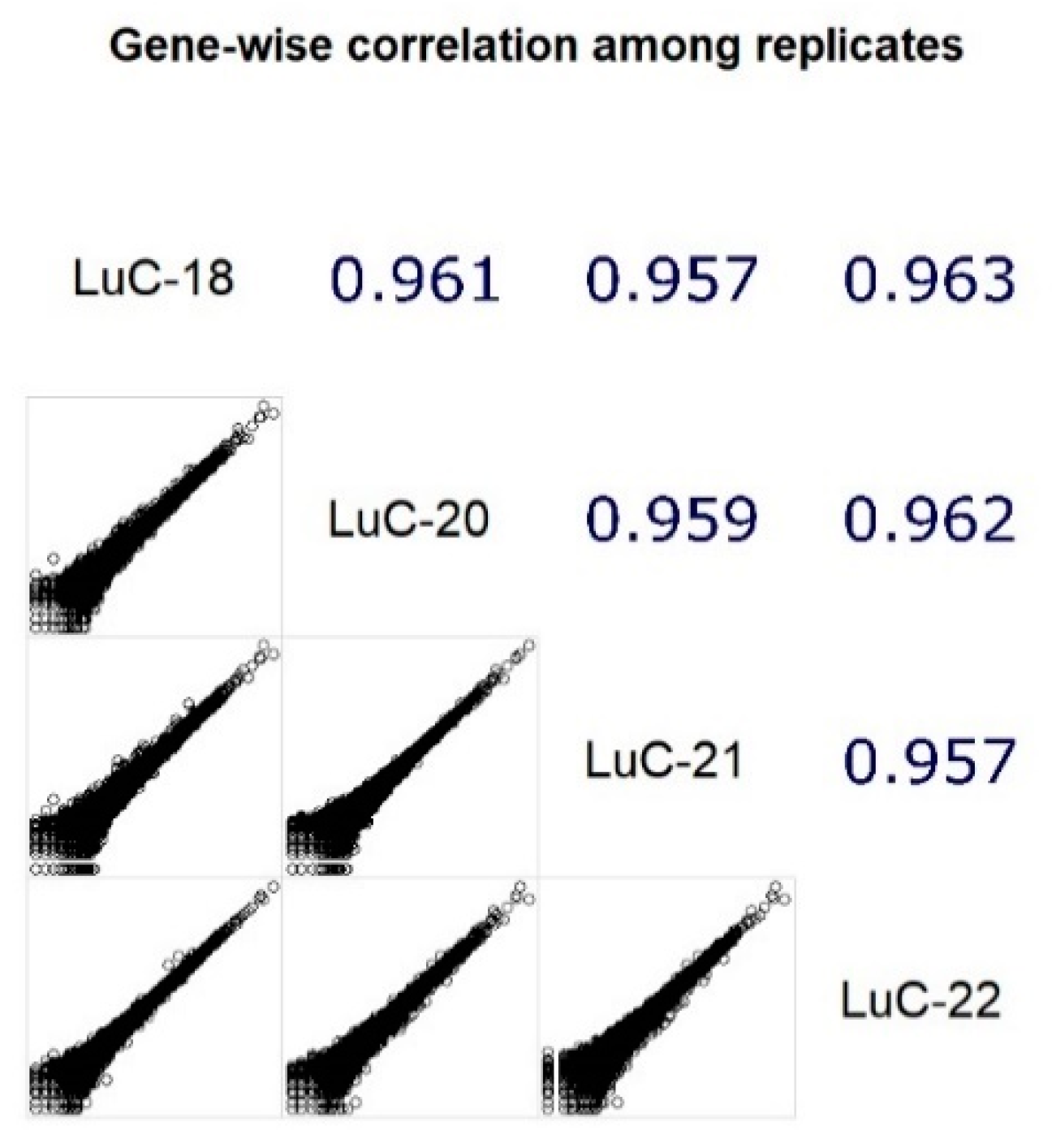
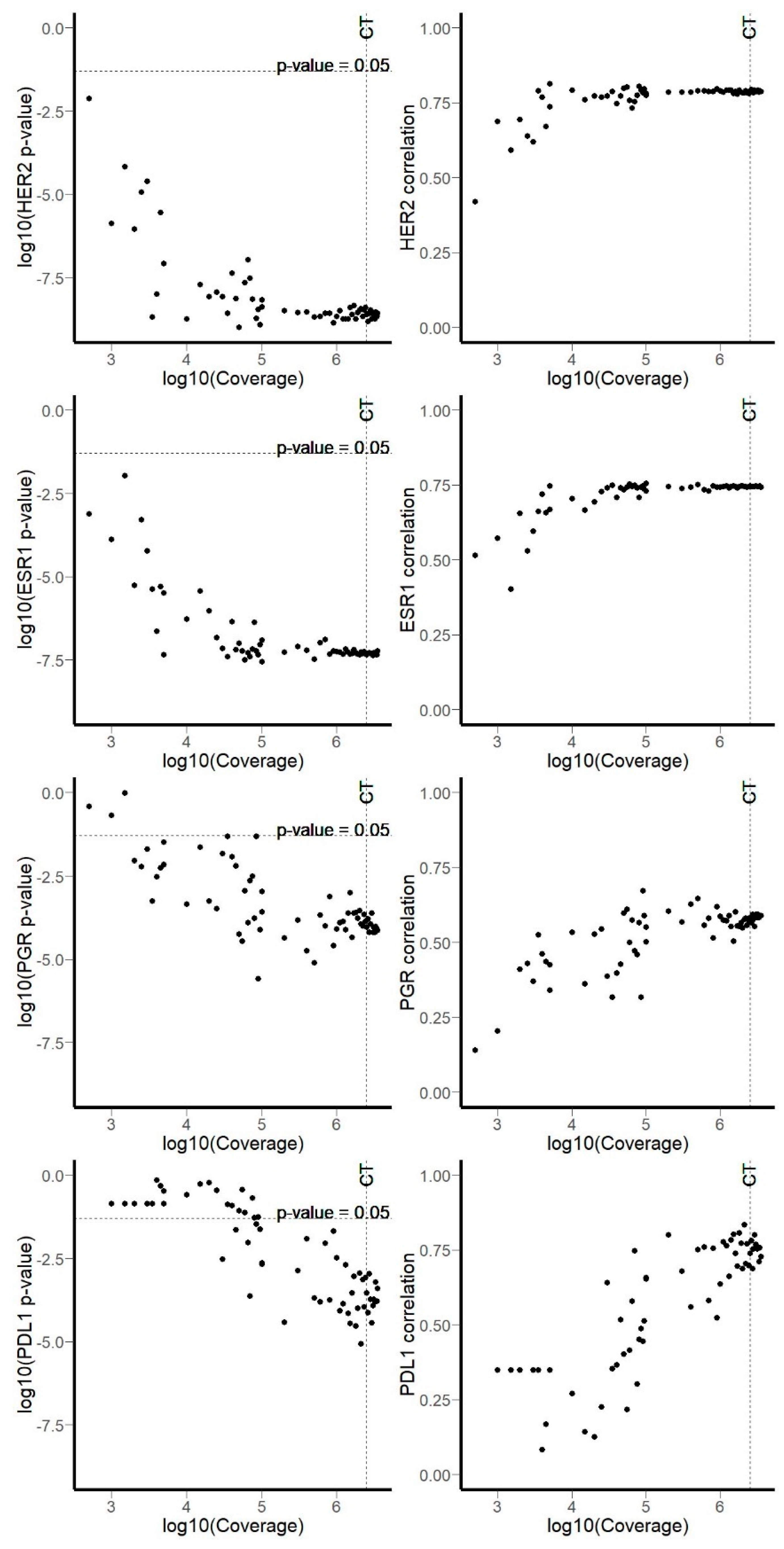
3.1. RNA Sequencing Data
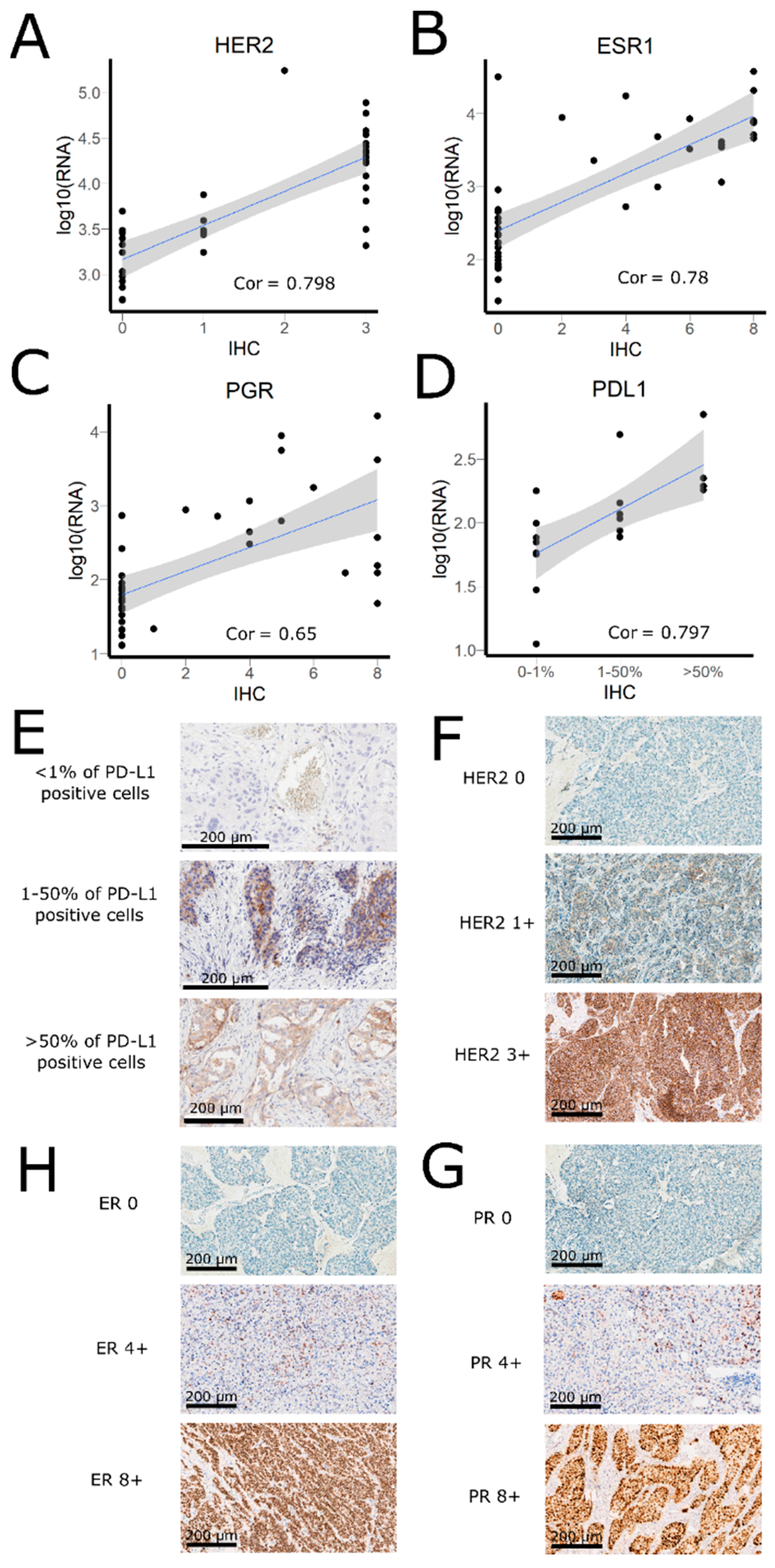
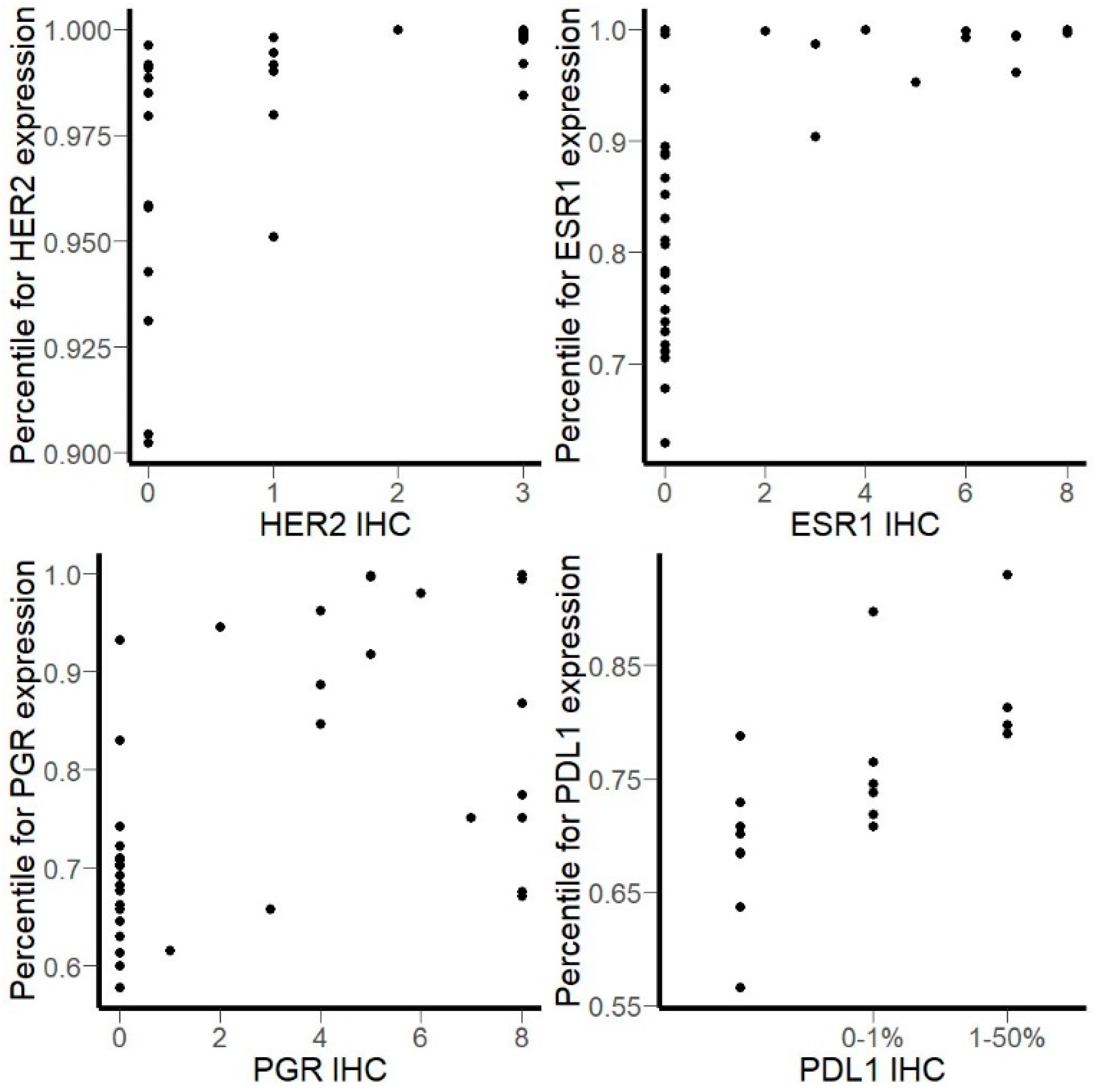
3.2. Comparison of RNA Sequencing and Immunohistochemistry Staining Results
3.3. Correlation of HER2, ER, and PGR Statuses Measured by RNA Sequencing and IHC for Freshly Frozen Tumor Samples
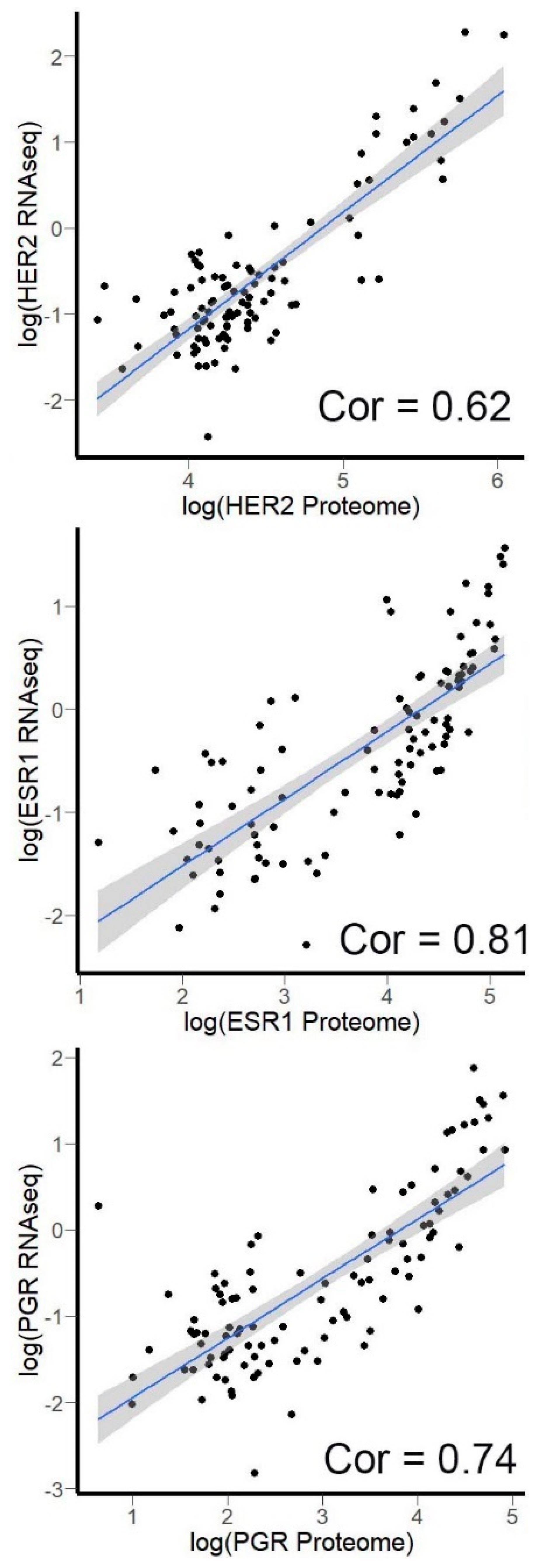
3.4. Correlation of HER2, ER, and PGR Expression Measured by RNA Sequencing versus Quantitative Proteomics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ruggles, K.V.; Krug, K.; Wang, X.; Clauser, K.R.; Wang, J.; Payne, S.H.; Fenyö, D.; Zhang, B.; Mani, D.R. Methods, Tools and Current Perspectives in Proteogenomics. Mol. Cell. Proteom. 2017, 16, 959–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.J.; Biggin, M.D. Gene expression. Statistics requantitates the central dogma. Science 2015, 347, 1066–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Klerk, E.; Fokkema, I.F.A.C.; Thiadens, K.A.M.H.; Goeman, J.J.; Palmblad, M.; den Dunnen, J.T.; von Lindern, M.; ‘t Hoen, P.A.C. Assessing the translational landscape of myogenic differentiation by ribosome profiling. Nucleic Acids Res. 2015, 43, 4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, K.C.; Ingolia, N.T.; Vance, R.E. Global analysis of gene expression reveals mRNA superinduction is required for the inducible immune response to a bacterial pathogen. Elife 2017, 6. [Google Scholar] [CrossRef]
- Dunn, J.G.; Foo, C.K.; Belletier, N.G.; Gavis, E.R.; Weissman, J.S. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. Elife 2013, 2, e01179. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Piehowski, P.D.; Kelly, R.T.; Qian, W.-J. Nanoproteomics comes of age. Expert Rev. Proteom. 2018, 15, 865–871. [Google Scholar] [CrossRef]
- Painter, J.T.; Clayton, N.P.; Herbert, R.A. Useful immunohistochemical markers of tumor differentiation. Toxicol. Pathol. 2010, 38, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Liang, Z.; Zhou, H.; Qu, L. Applications of RNA Indexes for Precision Oncology in Breast Cancer. Genomics Proteom. Bioinform. 2018, 16, 108–119. [Google Scholar] [CrossRef]
- Bossel Ben-Moshe, N.; Gilad, S.; Perry, G.; Benjamin, S.; Balint-Lahat, N.; Pavlovsky, A.; Halperin, S.; Markus, B.; Yosepovich, A.; Barshack, I.; et al. mRNA-seq whole transcriptome profiling of fresh frozen versus archived fixed tissues. BMC Genom. 2018, 19, 419. [Google Scholar] [CrossRef]
- Nault, R.; Fader, K.A.; Zacharewski, T. RNA-Seq versus oligonucleotide array assessment of dose-dependent TCDD-elicited hepatic gene expression in mice. BMC Genom. 2015, 16, 373. [Google Scholar] [CrossRef] [Green Version]
- SEQC/MAQC-III Consortium. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014, 32, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Lahens, N.F.; Ricciotti, E.; Smirnova, O.; Toorens, E.; Kim, E.J.; Baruzzo, G.; Hayer, K.E.; Ganguly, T.; Schug, J.; Grant, G.R. A comparison of Illumina and Ion Torrent sequencing platforms in the context of differential gene expression. BMC Genom. 2017, 18, 602. [Google Scholar] [CrossRef] [PubMed]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, N.; Suntsova, M.; Sorokin, M.; Garazha, A.; Kovalchuk, O.; Aliper, A.; Ilnitskaya, E.; Lezhnina, K.; Korzinkin, M.; Tkachev, V.; et al. Data aggregation at the level of molecular pathways improves stability of experimental transcriptomic and proteomic data. Cell Cycle 2017, 16. [Google Scholar] [CrossRef] [Green Version]
- Buzdin, A.A.; Zhavoronkov, A.A.; Korzinkin, M.B.; Roumiantsev, S.A.; Aliper, A.M.; Venkova, L.S.; Smirnov, P.Y.; Borisov, N.M. The OncoFinder algorithm for minimizing the errors introduced by the high-throughput methods of transcriptome analysis. Front. Mol. Biosci. 2014, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Borisov, N.; Shabalina, I.; Tkachev, V.; Sorokin, M.; Garazha, A.; Pulin, A.; Eremin, I.I.; Buzdin, A. Shambhala: A platform-agnostic data harmonizer for gene expression data. BMC Bioinform. 2019, 20, 66. [Google Scholar] [CrossRef] [Green Version]
- Buzdin, A.; Sorokin, M.; Poddubskaya, E.; Borisov, N. High-Throughput Mutation Data Now Complement Transcriptomic Profiling: Advances in Molecular Pathway Activation Analysis Approach in Cancer Biology. Cancer Inform. 2019, 18, 1176935119838844. [Google Scholar] [CrossRef] [Green Version]
- Tkachev, V.; Sorokin, M.; Mescheryakov, A.; Simonov, A.; Garazha, A.; Buzdin, A.; Muchnik, I.; Borisov, N. FLOating-Window Projective Separator (FloWPS): A Data Trimming Tool for Support Vector Machines (SVM) to Improve Robustness of the Classifier. Front. Genet. 2019, 9, 717. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Sekacheva, M.; Kim, E.; Zhukov, N.; Wang, Y.; Li, X.; Kar, S.; Hartmann, C.; et al. Molecular pathway activation—New type of biomarkers for tumor morphology and personalized selection of target drugs. Semin. Cancer Biol. 2018, 53. [Google Scholar] [CrossRef] [PubMed]
- Aliper, A.M.; Korzinkin, M.B.; Kuzmina, N.B.; Zenin, A.A.; Venkova, L.S.; Smirnov, P.Y.; Zhavoronkov, A.A.; Buzdin, A.A.; Borisov, N.M. Mathematical Justification of Expression-Based Pathway Activation Scoring (PAS). Methods Mol. Biol. 2017, 1613, 31–51. [Google Scholar] [PubMed]
- Buzdin, A.A.; Prassolov, V.; Zhavoronkov, A.A.; Borisov, N.M. Bioinformatics Meets Biomedicine: OncoFinder, a Quantitative Approach for Interrogating Molecular Pathways Using Gene Expression Data. Methods Mol. Biol. 2017, 1613, 53–83. [Google Scholar] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, T.O.; Yang, S.; Verma, R.X.; Pillalamarri, V.; Arking, D.E.; Rosenberg, A.Z.; McCall, M.N.; Halushka, M.K. Basal Contamination of Sequencing: Lessons from the GTEx dataset. bioRxiv 2020, 602367. [Google Scholar]
- Cancer Genome Atlas Research Network, J.N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Stern, D.F.; Zhao, H. Transcriptional Profiles from Paired Normal Samples Offer Complementary Information on Cancer Patient Survival—Evidence from TCGA Pan-Cancer Data. Sci. Rep. 2016, 6, 20567. [Google Scholar] [CrossRef] [Green Version]
- Casbas-Hernandez, P.; Sun, X.; Roman-Perez, E.; D’Arcy, M.; Sandhu, R.; Hishida, A.; McNaughton, K.K.; Yang, X.R.; Makowski, L.; Sherman, M.E.; et al. Tumor Intrinsic Subtype Is Reflected in Cancer-Adjacent Tissue. Cancer Epidemiol. Biomark. Prev. 2015, 24, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yu, P.; Wu, R.; Ge, Y.; Wu, J.; Zhu, J.; Jia, R. Renal cell carcinoma-adjacent tissues enhance mobilization and recruitment of endothelial progenitor cells to promote the invasion of the neoplasm. Biomed. Pharmacother. 2013, 67, 643–649. [Google Scholar] [CrossRef]
- Jones, A.C.; Antillon, K.S.; Jenkins, S.M.; Janos, S.N.; Overton, H.N.; Shoshan, D.S.; Fischer, E.G.; Trujillo, K.A.; Bisoffi, M. Prostate Field Cancerization: Deregulated Expression of Macrophage Inhibitory Cytokine 1 (MIC-1) and Platelet Derived Growth Factor A (PDGF-A) in Tumor Adjacent Tissue. PLoS ONE 2015, 10, e0119314. [Google Scholar] [CrossRef] [PubMed]
- Suntsova, M.; Gaifullin, N.; Allina, D.; Reshetun, A.; Li, X.; Mendeleeva, L.; Surin, V.; Sergeeva, A.; Spirin, P.; Prassolov, V.; et al. Atlas of RNA sequencing profiles for normal human tissues. Sci. Data 2019, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Mehta, M.; Walton, A.; Talsania, K.; Levin, Y.; Shetty, J.; Gillanders, E.M.; Tran, B.; Carrick, D.M. Robustness of RNA sequencing on older formalin-fixed paraffin-embedded tissue from high-grade ovarian serous adenocarcinomas. PLoS ONE 2019, 14, e0216050. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Codina, A.; Arpi, O.; Martinez-García, M.; Pineda, E.; Mallo, M.; Gut, M.; Carrato, C.; Rovira, A.; Lopez, R.; Tortosa, A.; et al. A Comparison of RNA-Seq Results from Paired Formalin-Fixed Paraffin-Embedded and Fresh-Frozen Glioblastoma Tissue Samples. PLoS ONE 2017, 12, e0170632. [Google Scholar] [CrossRef] [Green Version]
- Scolnick, J.A.; Dimon, M.; Wang, I.-C.; Huelga, S.C.; Amorese, D.A. An Efficient Method for Identifying Gene Fusions by Targeted RNA Sequencing from Fresh Frozen and FFPE Samples. PLoS ONE 2015, 10, e0128916. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen Receptor Status by Immunohistochemistry Is Superior to the Ligand-Binding Assay for Predicting Response to Adjuvant Endocrine Therapy in Breast Cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L.; Ni, R.; Zhou, W. Advances in HER2 testing. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; Volume 91, pp. 123–162. [Google Scholar]
- Verocq, C.; Decaestecker, C.; Rocq, L.; Clercq, S.D.; Verrellen, A.; Mekinda, Z.; Ocak, S.; Compère, C.; Stanciu-Pop, C.; Salmon, I.; et al. The daily practice reality of PD-L1 (CD274) evaluation in non-small cell lung cancer: A retrospective study. Oncol. Lett. 2020, 19, 3400. [Google Scholar] [CrossRef]
- Green, D.; Swets, J. Signal Detection Theory and Psychophysics; Wiley: New York, NY, USA, 1966. [Google Scholar]
- Chen, L.; Zhou, Y.; Tang, X.; Yang, C.; Tian, Y.; Xie, R.; Chen, T.; Yang, J.; Jing, M.; Chen, F.; et al. EGFR mutation decreases FDG uptake in non-small cell lung cancer via the NOX4/ROS/GLUT1 axis. Int. J. Oncol. 2019, 54, 370–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Cheng, G.; Kang, X.; Xi, Y.; Zhu, Y.; Wang, K.; Sun, C.; Ye, J.; Li, P.; Yin, H. Noninvasively evaluating the grading and IDH1 mutation status of diffuse gliomas by three-dimensional pseudo-continuous arterial spin labeling and diffusion-weighted imaging. Neuroradiology 2018, 60, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Tanioka, M.; Fan, C.; Parker, J.S.; Hoadley, K.A.; Hu, Z.; Li, Y.; Hyslop, T.M.; Pitcher, B.N.; Soloway, M.G.; Spears, P.A.; et al. Integrated Analysis of RNA and DNA from the Phase III Trial CALGB 40601 Identifies Predictors of Response to Trastuzumab-Based Neoadjuvant Chemotherapy in HER2-Positive Breast Cancer. Clin. Cancer Res. 2018, 24, 5292–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolotovskaia, M.A.; Sorokin, M.I.; Roumiantsev, S.A.; Borisov, N.M.; Buzdin, A.A. Pathway Instability Is an Effective New Mutation-Based Type of Cancer Biomarkers. Front. Oncol. 2018, 8, 658. [Google Scholar] [CrossRef]
- Lezhnina, K.; Kovalchuk, O.; Zhavoronkov, A.A.; Korzinkin, M.B.; Zabolotneva, A.A.; Shegay, P.V.; Sokov, D.G.; Gaifullin, N.M.; Rusakov, I.G.; Aliper, A.M.; et al. Novel robust biomarkers for human bladder cancer based on activation of intracellular signaling pathways. Oncotarget 2014, 5, 9022–9032. [Google Scholar] [CrossRef]
- Borisov, N.M.; Terekhanova, N.V.; Aliper, A.M.; Venkova, L.S.; Smirnov, P.Y.; Roumiantsev, S.; Korzinkin, M.B.; Zhavoronkov, A.A.; Buzdin, A.A. Signaling pathways activation profiles make better markers of cancer than expression of individual genes. Oncotarget 2014, 5, 10198–10205. [Google Scholar] [CrossRef]
- Boyd, J.C. Mathematical tools for demonstrating the clinical usefulness of biochemical markers. Scand. J. Clin. Lab. Invest. Suppl. 1997, 227, 46–63. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.-V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network, G.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef] [Green Version]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef]
- Kim, S.-J.; Kim, S.; Kim, D.-W.; Kim, M.; Keam, B.; Kim, T.M.; Lee, Y.; Koh, J.; Jeon, Y.K.; Heo, D.S. Alterations in PD-L1 Expression Associated with Acquisition of Resistance to ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Res. Treat. 2018. [Google Scholar] [CrossRef]
- Smith, N.R.; Womack, C. A matrix approach to guide IHC-based tissue biomarker development in oncology drug discovery. J. Pathol. 2014, 232, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Le Stang, N.; Rouquette, I.; Cazes, A.; Badoual, C.; Pinot-Roussel, H.; Tixier, L.; Danel, C.; Damiola, F.; Damotte, D.; et al. Multicenter harmonization study for PD-L1 IHC testing in non-small-cell lung cancer. Ann. Oncol. 2018, 29, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Conroy, J.M.; Pabla, S.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Andreas, J.; Giamo, V.; Wang, Y.; Lenzo, F.L.; et al. Next generation sequencing of PD-L1 for predicting response to immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 18. [Google Scholar] [CrossRef]
- Paluch, B.E.; Glenn, S.T.; Conroy, J.M.; Papanicolau-Sengos, A.; Bshara, W.; Omilian, A.R.; Brese, E.; Nesline, M.; Burgher, B.; Andreas, J.; et al. Robust detection of immune transcripts in FFPE samples using targeted RNA sequencing. Oncotarget 2017, 8, 3197–3205. [Google Scholar] [CrossRef] [PubMed]
- Tsimafeyeu, I.; Imyanitov, E.; Zavalishina, L.; Raskin, G.; Povilaitite, P.; Savelov, N.; Kharitonova, E.; Rumyantsev, A.; Pugach, I.; Andreeva, Y.; et al. Agreement between PDL1 immunohistochemistry assays and polymerase chain reaction in non-small cell lung cancer: CLOVER comparison study. Sci. Rep. 2020, 10, 3928. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, K.; Krazinski, B.E.; Kowalczyk, A.E.; Dworakowska, B.; Jakiela, S.; Stobiecka, M. Hairpin-Hairpin Molecular Beacon Interactions for Detection of Survivin mRNA in Malignant SW480 Cells. ACS Appl. Mater. Interfaces 2018, 10, 17028–17039. [Google Scholar] [CrossRef]
- Stobiecka, M.; Ratajczak, K.; Jakiela, S. Toward early cancer detection: Focus on biosensing systems and biosensors for an anti-apoptotic protein survivin and survivin mRNA. Biosens. Bioelectron. 2019, 137, 58–71. [Google Scholar] [CrossRef]
- Stobiecka, M.; Dworakowska, B.; Jakiela, S.; Lukasiak, A.; Chalupa, A.; Zembrzycki, K. Sensing of survivin mRNA in malignant astrocytes using graphene oxide nanocarrier-supported oligonucleotide molecular beacons. Sens. Actuators B Chem. 2016, 235, 136–145. [Google Scholar] [CrossRef]
- Brueffer, C.; Vallon-Christersson, J.; Grabau†, D.; Ehinger, A.; Häkkinen, J.; Hegardt, C.; Malina, J.; Chen, Y.; Bendahl, P.-O.; Manjer, J.; et al. Clinical Value of RNA Sequencing–Based Classifiers for Prediction of the Five Conventional Breast Cancer Biomarkers: A Report From the Population-Based Multicenter Sweden Cancerome Analysis Network—Breast Initiative. JCO Precis. Oncol. 2018, 1–18. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Passiglia, F.; Galvano, A.; Rizzo, S.; Incorvaia, L.; Listì, A.; Bazan, V.; Russo, A. Looking for the best immune-checkpoint inhibitor in pre-treated NSCLC patients: An indirect comparison between nivolumab, pembrolizumab and atezolizumab. Int. J. cancer 2018, 142, 1277–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, V.; Rubin, E.; Depil, S.; Pawitan, Y.; Martini, J.-F.; Gomez-Navarro, J.; Yver, A.; Kan, Z.; Dry, J.R.; Kehren, J.; et al. A simplified interventional mapping system (SIMS) for the selection of combinations of targeted treatments in non-small cell lung cancer. Oncotarget 2015, 6, 14139–14152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzdin, A.; Sorokin, M.; Glusker, A.; Garazha, A.; Poddubskaya, E.; Shirokorad, V.; Naskhletashvili, D.; Kashintsev, K.; Sokov, D.; Suntsova, M.; et al. Activation of intracellular signaling pathways as a new type of biomarkers for selection of target anticancer drugs. J. Clin. Oncol. 2017, 35, e23142. [Google Scholar] [CrossRef]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Glusker, A.; Aleshin, A.; Poddubskaya, E.; Sekacheva, M.; Kim, E.; Gaifullin, N.; Giese, A.; et al. RNA sequencing for research and diagnostics in clinical oncology. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Poddubskaya, E.; Buzdin, A.; Garazha, A.; Sorokin, M.; Glusker, A.; Aleshin, A.; Allina, D.; Moiseev, A.; Sekacheva, M.; Suntsova, M.; et al. Oncobox, gene expression-based second opinion system for predicting response to treatment in advanced solid tumors. J. Clin. Oncol. 2019, 37, e13143. [Google Scholar] [CrossRef]
- Poddubskaya, E.; Bondarenko, A.; Boroda, A.; Zotova, E.; Glusker, A.; Sletina, S.; Makovskaia, L.; Kopylov, P.; Sekacheva, M.; Moisseev, A.; et al. Transcriptomics-Guided Personalized Prescription of Targeted Therapeutics for Metastatic ALK-Positive Lung Cancer Case Following Recurrence on ALK Inhibitors. Front. Oncol. 2019, 9, 1026. [Google Scholar] [CrossRef] [Green Version]
- Poddubskaya, E.V.; Baranova, M.P.; Allina, D.O.; Sekacheva, M.I.; Makovskaia, L.A.; Kamashev, D.E.; Suntsova, M.V.; Barbara, V.S.; Kochergina-Nikitskaya, I.N.; Aleshin, A.A. Personalized prescription of imatinib in recurrent granulosa cell tumor of the ovary: Case report. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003434. [Google Scholar] [CrossRef]
- Poddubskaya, E.V.; Baranova, M.P.; Allina, D.O.; Smirnov, P.Y.; Albert, E.A.; Kirilchev, A.P.; Aleshin, A.A.; Sekacheva, M.I.; Suntsova, M. V Personalized prescription of tyrosine kinase inhibitors in unresectable metastatic cholangiocarcinoma. Exp. Hematol. Oncol. 2018, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Moisseev, A.; Albert, E.; Lubarsky, D.; Schroeder, D.; Clark, J. Transcriptomic and Genomic Testing to Guide Individualized Treatment in Chemoresistant Gastric Cancer Case. Biomedicines 2020, 8, 67. [Google Scholar] [CrossRef] [Green Version]
- BGI. Available online: https://www.bgi.com/ (accessed on 29 March 2020).
- Loong, H.H.; Wong, C.K.H.; Leung, L.K.S.; Dhankhar, P.; Insinga, R.P.; Chandwani, S.; Hsu, D.C.; Lee, M.Y.K.; Huang, M.; Pellissier, J.; et al. Cost Effectiveness of PD-L1-Based Test-and-Treat Strategy with Pembrolizumab as the First-Line Treatment for Metastatic NSCLC in Hong Kong. PharmacoEconomics Open 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Primary Tumor or Metastasis | Age | Stage | HER2 Score | ER Score | PR Score | Coverage (mln Mapped Reads) | RIN |
|---|---|---|---|---|---|---|---|---|
| BC-1 | primary | 39 | T2N3aM0, IIIC | 3 | 0 | 0 | 9.42 | 2.1 |
| BC-10 | primary | 48 | T2N0M0, II | 3 | 0 | 0 | 6.70 | 1 |
| BC-12 | primary | 60 | T2N0M0, IIA | 3 | 0 | 0 | 5.12 | 1 |
| BC-13 | primary | 69 | T2N3M0, III C | 3 | 8 | 4 | 9.03 | 1 |
| BC-14 | primary | 49 | T2N2M0, IIIA | 3 | 0 | 0 | 6.11 | 2.4 |
| BC-17 | primary | 59 | T4N2M0 | 3 | 7 | 2 | 3.96 | 2.5 |
| BC-18 | lymph node metastasis | 47 | T3N1M0, IIIA | 3 | 0 | 0 | 6.62 | 2.3 |
| BC-19 | primary | 48 | T1N0M0, I | 3 | 5 | 5 | 9.07 | 1.1 |
| BC-20 | lymph node metastasis | 51 | T2N0M0, II | 3 | 0 | 0 | 10.22 | 2.3 |
| BC-21 | primary | 49 | T1N3M0, IIIC | 3 | 0 | 0 | 9.34 | 2.3 |
| BC-22 | primary | 47 | T2N0M0, II | 3 | 6 | 5 | 10.52 | 2 |
| BC-23 | primary | 46 | T2N2M0, IIIA | 3 | 7 | 6 | 8.39 | 2.1 |
| BC-24 | primary | 57 | T2N0M0, IIA | 3 | 6 | 4 | 11.21 | 1 |
| BC-27 | primary | 44 | T2N0M0 | 3 | 0 | 0 | 13.82 | 2.2 |
| BC-28 | ovary metastasis | 53 | T2N0M0, IIA | 0 | 7 | 4 | 4.65 | 3.7 |
| BC-29 | primary | 65 | T4N3M1,IV | 3 | 0 | 0 | 12.56 | 2.2 |
| BC-3 | primary | 55 | T2N1M0, IIIa | 3 | 0 | 0 | 6.84 | 1 |
| BC-4 | primary | 58 | T2N1M0, IIB | 3 | 0 | 0 | 7.17 | 1 |
| BC-46 | liver metastasis | 27 | T2N2M0 | 0 | 8 | 8 | 15.07 | 3.3 |
| BC-48 | relapse in the scar | 36 | T3N1M0 | 1 | 0 | 0 | 20.54 | NA |
| BC-49 | primary | 54 | T1N2M0 | 0 | 2 | 8 | 10.54 | 2 |
| BC-50 | primary | 51 | T2N0M0 | 0 | 0 | 0 | 8.49 | 2.6 |
| BC-51 | primary | 38 | T2N1M0 | 0 | 0 | 0 | 8.68 | 3 |
| BC-52 | primary | 78 | T1N2M0 | 1 | 4 | 8 | 11.92 | 1.7 |
| BC-53 | primary | 50 | T2N0M0 | 1 | 0 | 8 | 8.06 | 1.9 |
| BC-54 | primary | 50 | T2N0M0 | 0 | 0 | 0 | 7.30 | 1.8 |
| BC-55 | primary | 71 | T2N3M0 | 1 | 8 | 8 | 9.32 | 3.3 |
| BC-56 | primary | 60 | T1N1M1 | 0 | 0 | 8 | 12.66 | 2.4 |
| BC-57 | primary | 55 | T3N2M0 | 1 | 0 | 0 | 13.77 | 2.8 |
| BC-58 | lymph node metastasis | 55 | T1N0M0 | 0 | 7 | 7 | 14.24 | 2.1 |
| BC-59 | scar metastasis | 61 | T1N1M0 | 0 | 3 | 1 | 16.88 | 1.2 |
| BC-60 | primary | 33 | T2N1M0 | 2 | 0 | 0 | 10.03 | 1.8 |
| BC-61 | liver metastasis | 38 | T2N2M0 | 0 | 8 | 8 | 5.42 | 3 |
| BC-62 | brain metastasis | 44 | T2N0M0 | 0 | 0 | 0 | 10.99 | 3 |
| BC-63 | primary | 66 | T4N2M0 | 0 | 0 | 0 | 10.11 | 3.7 |
| BC-64 | primary | 60 | T3N3M0 | 1 | 0 | 0 | 12.71 | 3.8 |
| BC-65 | primary | 42 | T2N0M0 | 0 | 0 | 0 | 9.92 | 2.6 |
| BC-66 | primary | 55 | T3N1M0 | 3 | 3 | 3 | 8.96 | 3.1 |
| BC-9 | primary | 57 | T1N1M0, IIB | 3 | 8 | 5 | 6.88 | 1 |
| ID | Histology | Age | Stage | Sex | Percent of PDL1 Positive Cells | Coverage (mln Mapped Reads) | RIN |
|---|---|---|---|---|---|---|---|
| LuC_16 | squamous cell carcinoma | 75 | T3N2M1, IV | male | 1%–50% | 11.54 | 2.4 |
| LuC_18 | squamous cell carcinoma | 63 | T2N1M0 | male | 0 | 15.45 | 3 |
| LuC_19 | squamous cell carcinoma | 65 | T2N0M0 | male | >50% | 12.57 | 3 |
| LuC_30 | Unidentified | 79 | T2NXM0 | male | >50% | 11.01 | 4.9 |
| LuC_31 | adenocarcinoma | 66 | T3N2M0 | male | 1%–50% | 10.27 | 4.5 |
| LuC_32 | adeno-squamous cell carcinoma | 70 | T2N1M0 | male | >50% | 12.14 | 2.7 |
| LuC_33 | squamous cell carcinoma | 57 | T3N0M0 | male | 0 | 14.12 | 3.8 |
| LuC_42 | adenocarcinoma | 67 | T1N1M0 | male | 1%–50% | 11.9 | 1.4 |
| LuC_23 | adenocarcinoma | 60 | T2N0M0 | male | 0 | 12.06 | 3.2 |
| LuC_24 | adenocarcinoma | 67 | T2N0M0 | male | 0 | 10.77 | 3.8 |
| LuC_26 | small cell carcinoma | 65 | T3N2M0, IIIa | male | 1%–50% | 5.71 | 1.1 |
| LuC_28 | adenocarcinoma | 76 | T2N0M0 | male | 0 | 12.37 | 1.8 |
| LuC_29 | squamous cell carcinoma | 65 | T2N0M0 | male | 0 | 16.58 | 2.4 |
| LuC_34 | adenocarcinoma | 62 | pT1bN0M0 | female | 0 | 11.82 | 2.3 |
| LuC_35 | squamous cell carcinoma | 75 | T3N0M0 | male | >50% | 12.28 | 3.2 |
| LuC_36 | adenocarcinoma | 57 | pT2N0M0 | male | 1%–50% | 11.3 | 2.6 |
| LuC_37 | squamous cell carcinoma | 68 | T3N1M0 | male | 0 | 11.93 | 2.3 |
| LuC_38 | adenocarcinoma | 68 | pT2aN2M0 | male | 1%–50% | 15.38 | 3.5 |
| LuC_39 | adenocarcinoma | 68 | pT2pNXpM1 | female | 0 | 8.58 |
| Protein | Experimental Dataset | The Cancer Genome Atlas |
|---|---|---|
| HER2 | 0.963 | 0.818 |
| ESR1 | 0.921 | 0.959 |
| PGR | 0.912 | 0.923 |
| PDL1 | 0.922 | Not available |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorokin, M.; Ignatev, K.; Poddubskaya, E.; Vladimirova, U.; Gaifullin, N.; Lantsov, D.; Garazha, A.; Allina, D.; Suntsova, M.; Barbara, V.; et al. RNA Sequencing in Comparison to Immunohistochemistry for Measuring Cancer Biomarkers in Breast Cancer and Lung Cancer Specimens. Biomedicines 2020, 8, 114. https://doi.org/10.3390/biomedicines8050114
Sorokin M, Ignatev K, Poddubskaya E, Vladimirova U, Gaifullin N, Lantsov D, Garazha A, Allina D, Suntsova M, Barbara V, et al. RNA Sequencing in Comparison to Immunohistochemistry for Measuring Cancer Biomarkers in Breast Cancer and Lung Cancer Specimens. Biomedicines. 2020; 8(5):114. https://doi.org/10.3390/biomedicines8050114
Chicago/Turabian StyleSorokin, Maxim, Kirill Ignatev, Elena Poddubskaya, Uliana Vladimirova, Nurshat Gaifullin, Dmitriy Lantsov, Andrew Garazha, Daria Allina, Maria Suntsova, Victoria Barbara, and et al. 2020. "RNA Sequencing in Comparison to Immunohistochemistry for Measuring Cancer Biomarkers in Breast Cancer and Lung Cancer Specimens" Biomedicines 8, no. 5: 114. https://doi.org/10.3390/biomedicines8050114