Familial Combined Hyperlipidemia (FCH) Patients with High Triglyceride Levels Present with Worse Lipoprotein Function Than FCH Patients with Isolated Hypercholesterolemia

 and
and
Abstract
:1. Introduction
2. Experimental Section
2.1. Study Population
2.2. Plasma Determinations
2.3. Lipoprotein Isolation and Composition
2.4. LDL Functional Assays
2.5. HDL Function Assays
2.5.1. Antioxidant Capacity of HDL
2.5.2. Cholesterol Efflux
2.5.3. Anti-Inflammatory Activity of HDL
2.6. Statistical Analysis
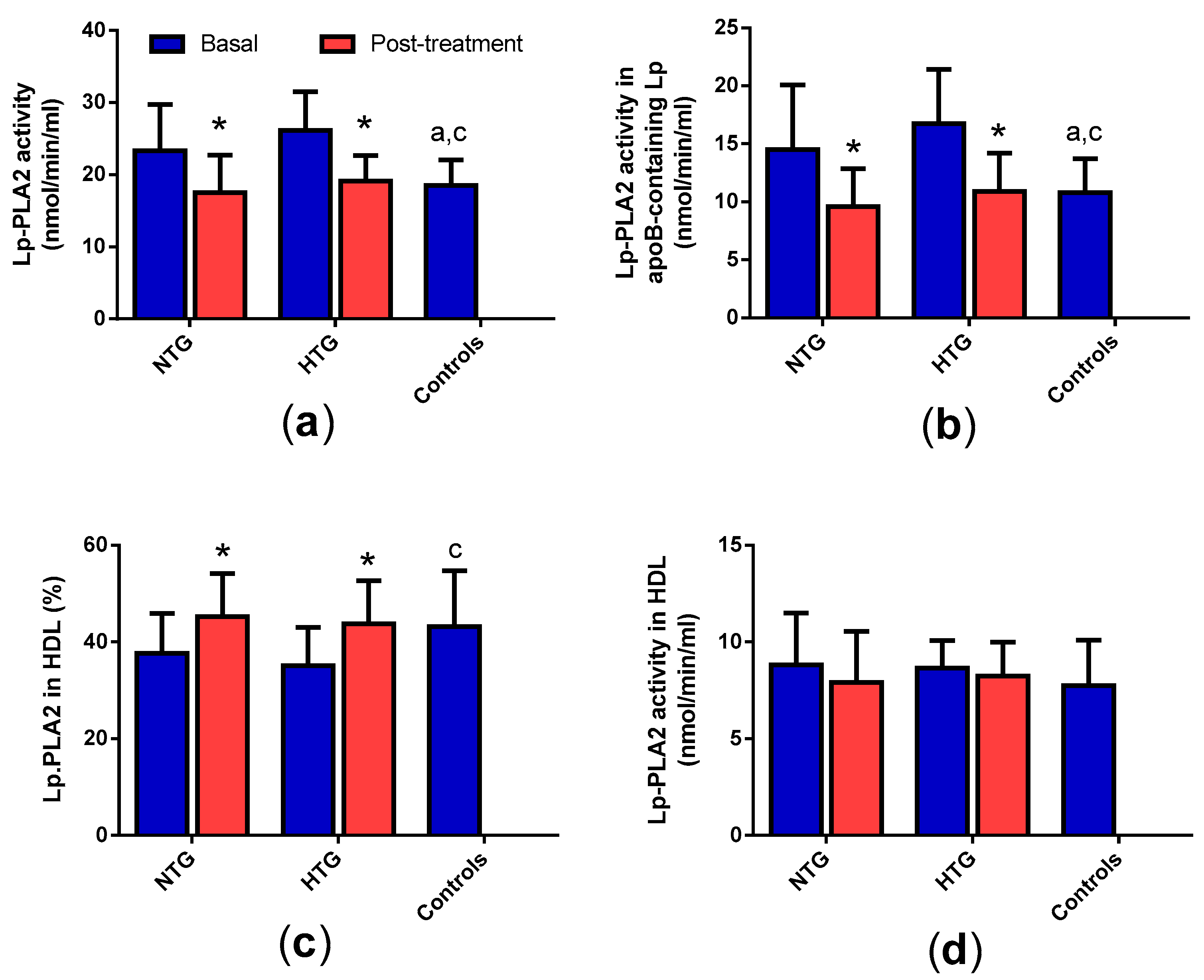
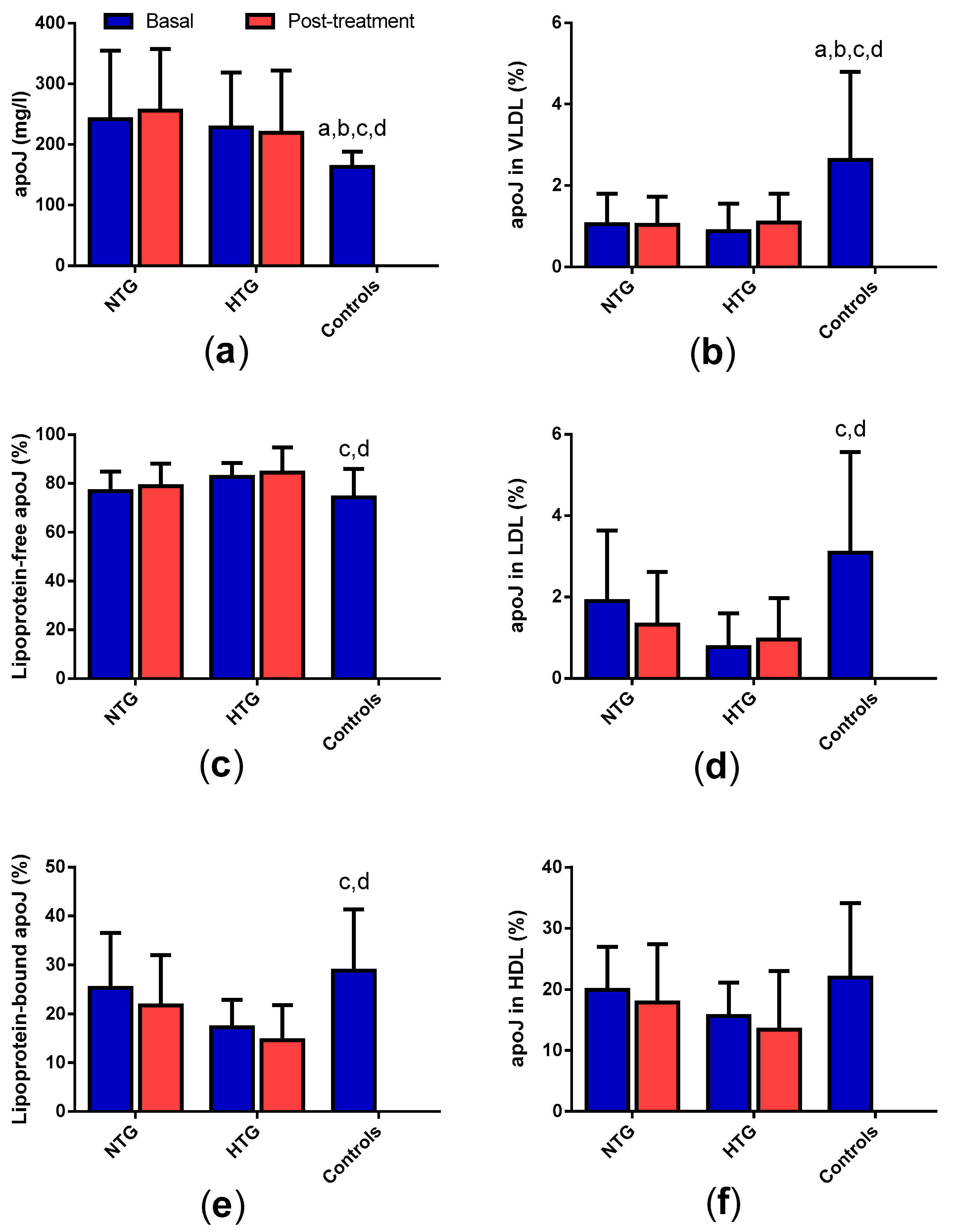
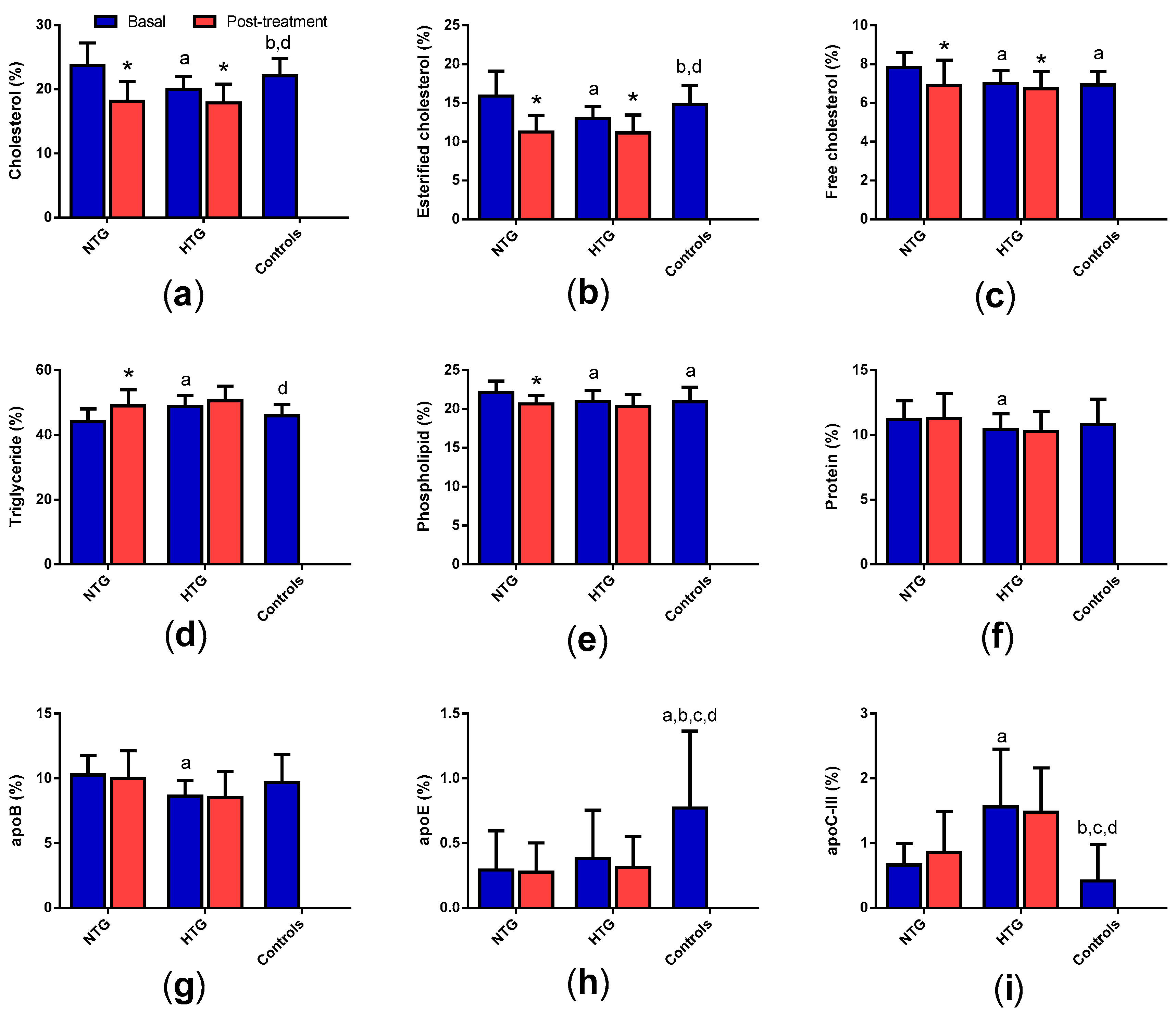
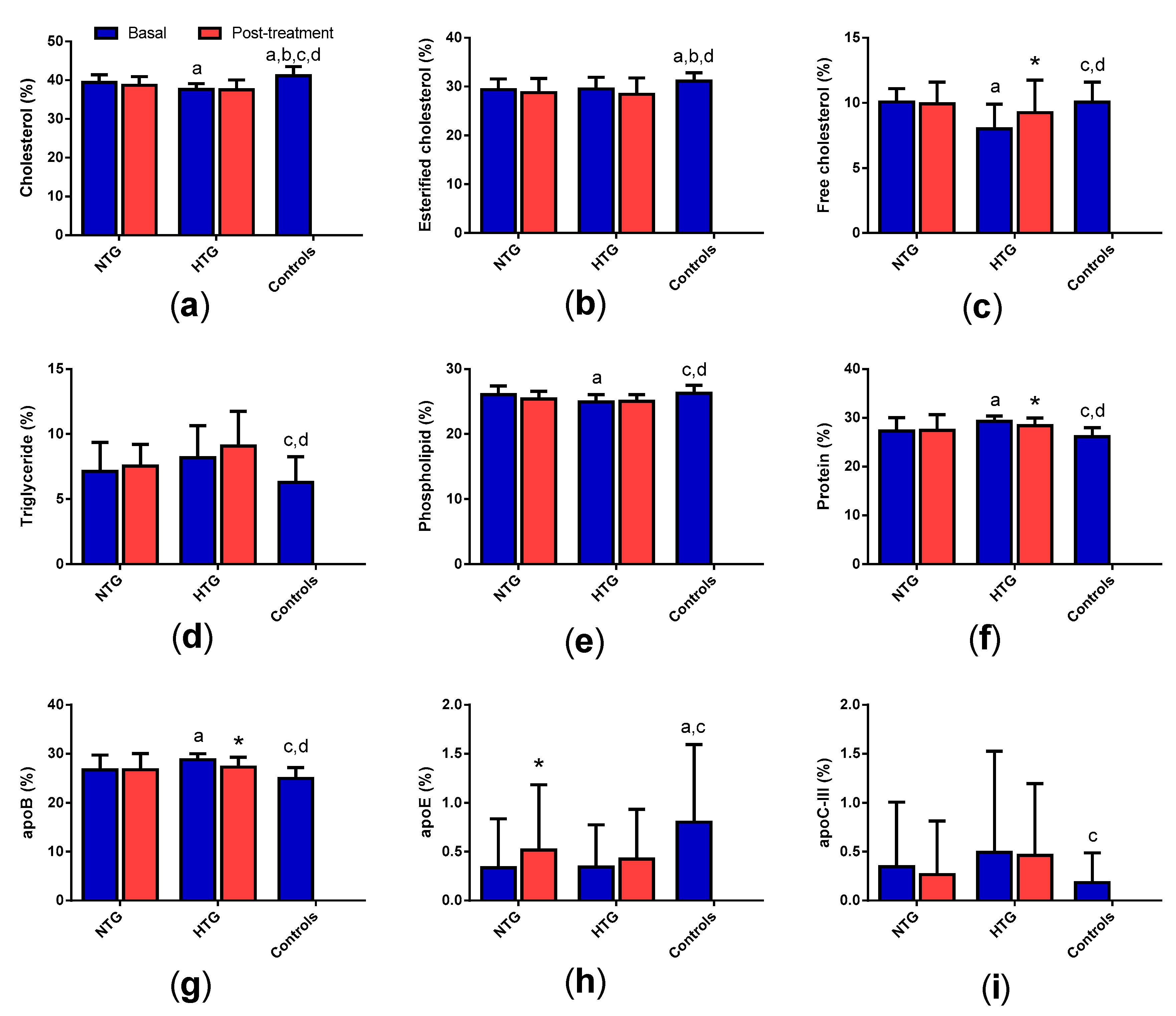
3. Results
3.1. Lipid Profile and Inflammation Markers
3.2. Distribution of Lp-PLA2 Activity in Plasma
3.3. Concentration and Distribution of Apolipoprotein J in Plasma
3.4. Very Low-Density Lipoprotein (VLDL) Composition
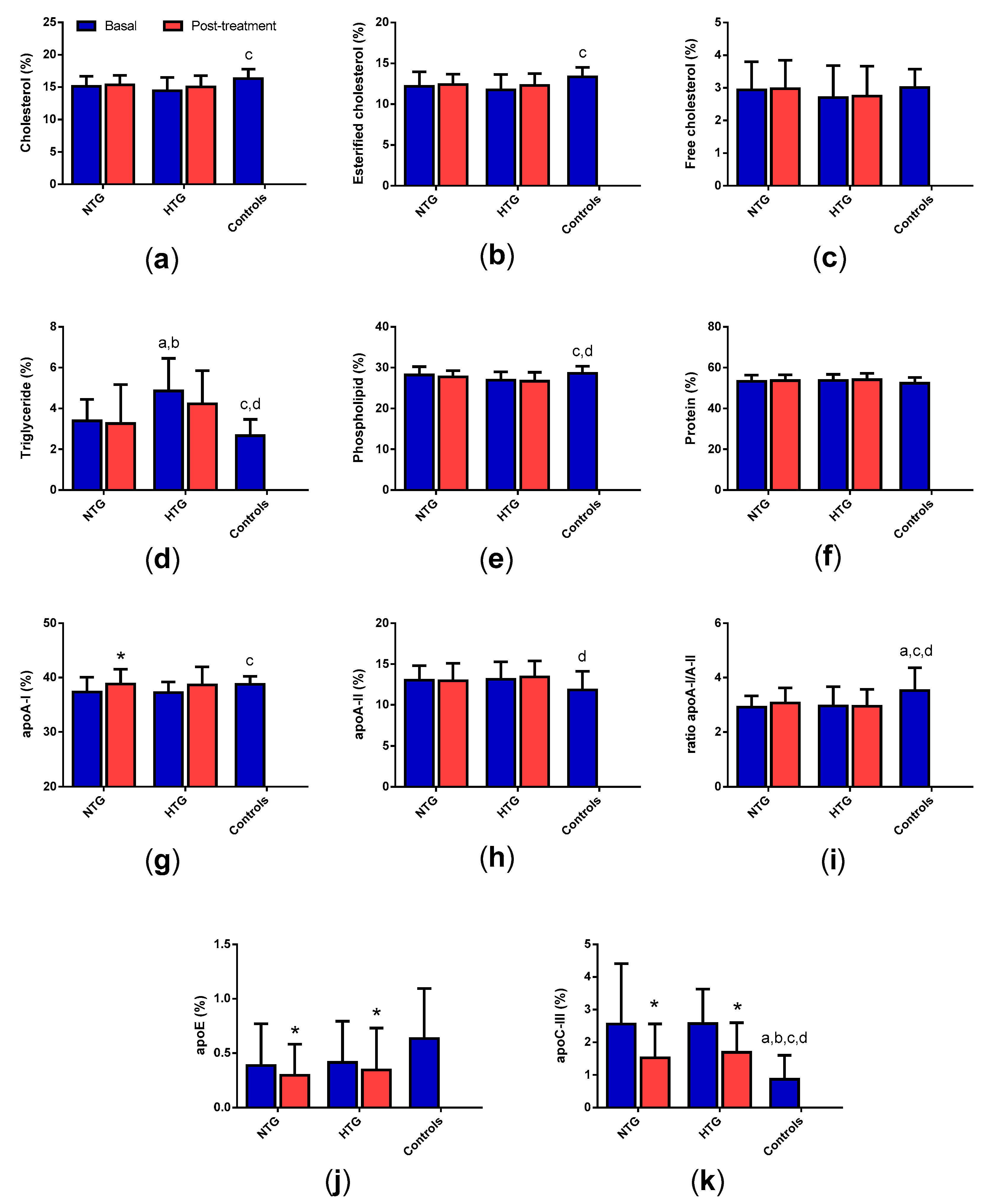
3.5. Low-Density Lipoprotein (LDL) Composition
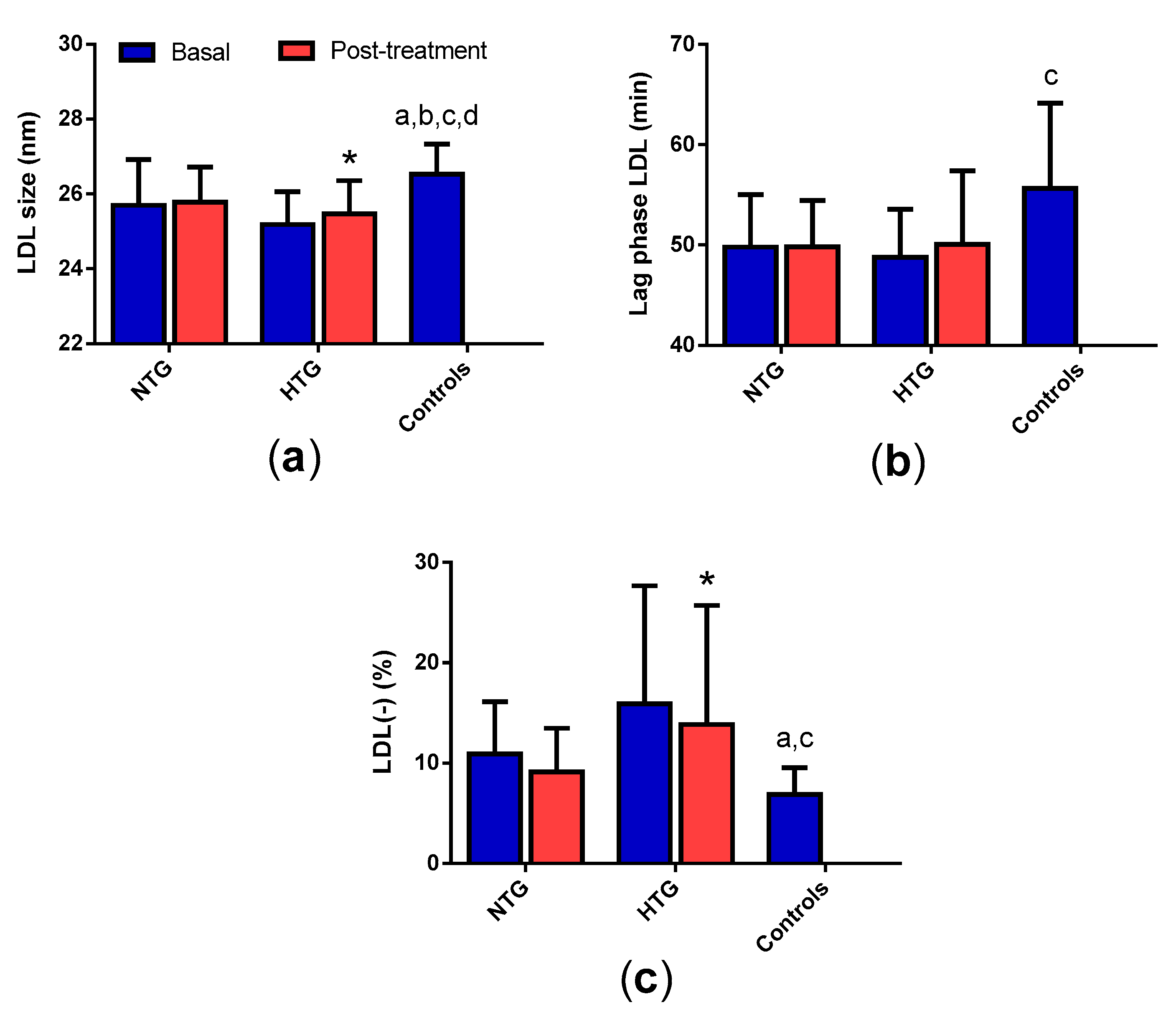
3.6. LDL Qualitative Properties
3.7. High-Density Lipoprotein (HDL) Composition
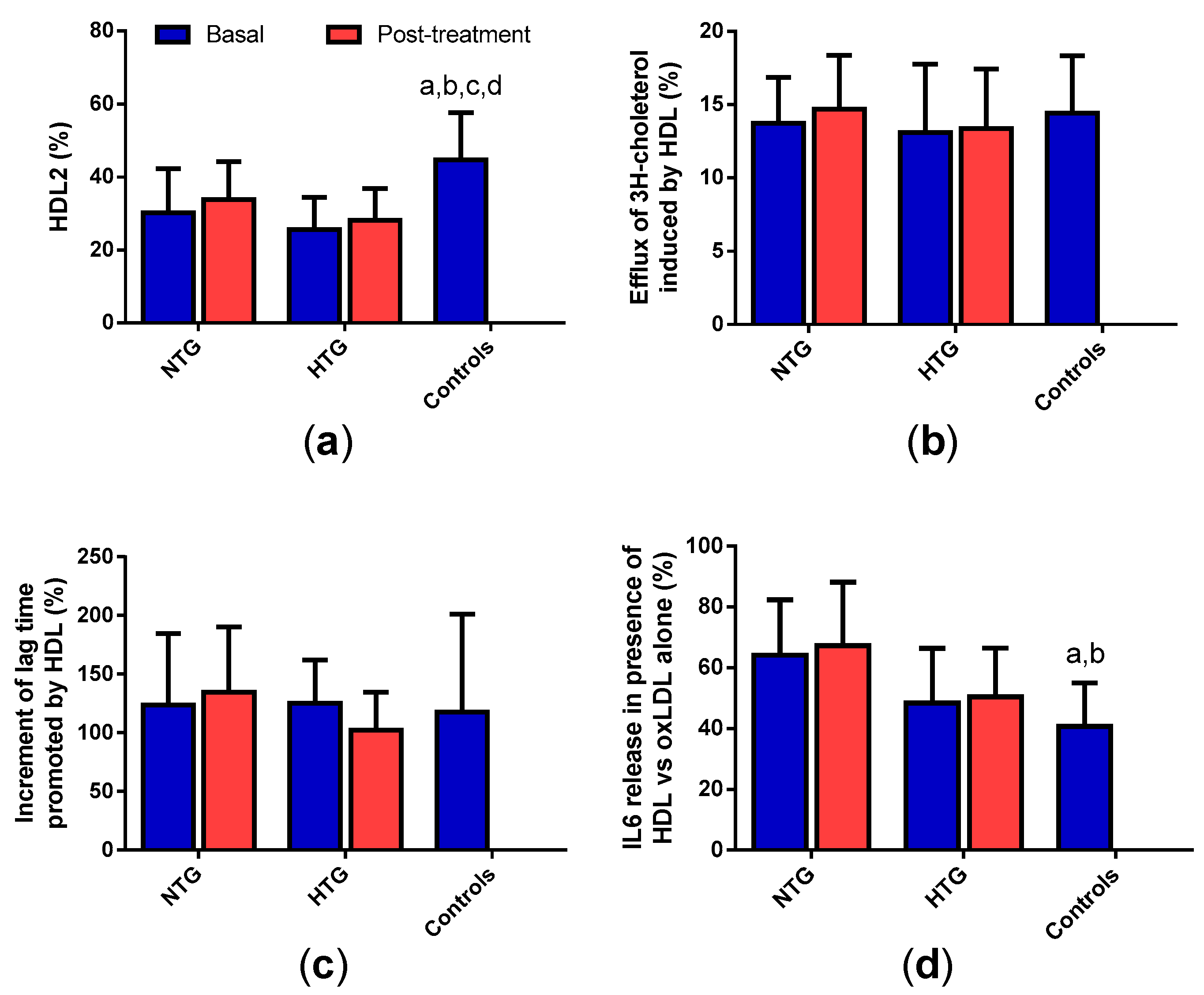
3.8. HDL Qualitative Properties
3.9. Correlation of Triglyceride Plasma Levels with the Qualitative Properties of Lipoproteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Diffenderfer, M.R.; Schaefer, E.J. The composition and metabolism of large and small LDL. Curr. Opin. Lipidol. 2014, 25, 221–226. [Google Scholar] [CrossRef]
- Rivas-Urbina, A.; Rull, A.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Electronegative LDL: An active player in atherogenesis or a by-product of atherosclerosis? Curr. Med. Chem. 2019, 26, 1665–1679. [Google Scholar] [CrossRef]
- Tsimikas, S.; Miller, Y.I. Oxidative modification of lipoproteins: Mechanisms, role in inflammation and potential clinical applications in cardiovascular disease. Curr. Pharm. Des. 2011, 17, 27–37. [Google Scholar] [CrossRef]
- Feng, M.; Rached, F.; Kontush, A.; Chapman, C.M. Impact of Lipoproteins on Atherobiology: Emerging Insights. Cardiol. Clin. 2018, 36, 193–201. [Google Scholar] [CrossRef]
- Navab, M.; Reddy, S.T.; Van Lenten, B.J.; Fogelman, A.M. HDL and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 2011, 8, 222–232. [Google Scholar] [CrossRef]
- Pirillo, A.; Catapano, A.L. Increasing high-density lipoprotein cholesterol levels for cardiovascular benefit: The end of a dream? Eur. J. Prev. Cardiol. 2019, 26, 531–532. [Google Scholar] [CrossRef]
- van der Stoep, M.; Korporaal, S.J.; Van Eck, M. High-density lipoprotein as a modulator of platelet and coagulation responses. Cardiovasc. Res. 2014, 103, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Camont, L.; Lhomme, M.; Rached, F.; Le Goff, W.; Negre-Salvayre, A.; Salvayre, R.; Calzada, C.; Lagarde, M.; Chapman, M.J.; Kontush, A. Small, dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: Relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2715–2723. [Google Scholar] [CrossRef] [Green Version]
- Riwanto, M.; Rohrer, L.; Roschitzki, B.; Besler, C.; Mocharla, P.; Mueller, M.; Perisa, D.; Heinrich, K.; Altwegg, L.; von Eckardstein, A.; et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: Role of high-density lipoprotein-proteome remodeling. Circulation 2013, 127, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef]
- Hussein, H.; Saheb, S.; Couturier, M.; Atassi, M.; Orsoni, A.; Carrie, A.; Therond, P.; Chantepie, S.; Robillard, P.; Bruckert, E.; et al. Small, dense high-density lipoprotein 3 particles exhibit defective antioxidative and anti-inflammatory function in familial hypercholesterolemia: Partial correction by low-density lipoprotein apheresis. J. Clin. Lipidol. 2016, 10, 124–133. [Google Scholar] [CrossRef]
- Brahm, A.J.; Hegele, R.A. Combined hyperlipidemia: Familial but not (usually) monogenic. Curr. Opin. Lipidol. 2016, 27, 131–140. [Google Scholar] [CrossRef]
- Brouwers, M.C.; van Greevenbroek, M.M.; Stehouwer, C.D.; de Graaf, J.; Stalenhoef, A.F. The genetics of familial combined hyperlipidaemia. Nat. Rev. Endocrinol. 2012, 8, 352–362. [Google Scholar] [CrossRef]
- Bello-Chavolla, O.Y.; Kuri-García, A.; Ríos-Ríos, M.; Vargas-Vázquez, A.; Cortés-Arroyo, J.E.; Tapia-González, G.; Cruz-Bautista, I.; Aguilar-Salinas, C.A. Familial combined hyperlipidemia: Current knowledge, perspectives, and Controversies. Rev. Investig. Clin. 2018, 70, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Ruiz, M.; Martinez-Triguero, M.L.; Lopez-Ruiz, A.; Fernandez-de la Cruz, F.; Banuls, C.; Hernandez-Mijares, A. Metabolic disorders and inflammation are associated with familial combined hyperlipemia. Clin. Chim. Acta 2019, 490, 194–199. [Google Scholar] [CrossRef]
- Baila-Rueda, L.; Mateo-Gallego, R.; Jarauta, E.; de Castro-Oros, I.; Bea, A.M.; Cenarro, A.; Civeira, F. Bile acid synthesis precursors in familial combined hyperlipidemia: The oxysterols 24S-hydroxycholesterol and 27-hydroxycholesterol. Biochem. Biophys. Res. Commun. 2014, 446, 731–735. [Google Scholar] [CrossRef]
- van Greevenbroek, M.M.; Stalenhoef, A.F.; de Graaf, J.; Brouwers, M.C. Familial combined hyperlipidemia: From molecular insights to tailored therapy. Curr. Opin. Lipidol. 2014, 25, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Bautista, I.; Mehta, R.; Cabiedes, J.; Garci´a-Ulloa, C.; Guillen-Pineda, L.E.; Almeda-Valdes, P.; Cuevas-Ramos, D.; Aguilar-Salinas, C.A. Determinants of VLDL composition and apo B-containing particles in familial combined hyperlipidemia. Clin. Chim. Acta 2015, 438, 160–165. [Google Scholar] [CrossRef]
- Liu, M.L.; Ylitalo, K.; Vakkilainen, J.; Nuotio, I.; Valkonen, M.; Lahdenpera, S.; Viikari, J.; Taskinen, M.R. Susceptibility of LDL to oxidation in vitro and antioxidant capacity in familial combined hyperlipidemia: Comparison of patients with different lipid phenotypes. Ann. Med. 2002, 34, 48–54. [Google Scholar] [CrossRef]
- Pauciullo, P.; Gentile, M.; Marotta, G.; Baiano, A.; Ubaldi, S.; Jossa, F.; Iannuzzo, G.; Faccenda, F.; Panico, S.; Rubba, P. Small dense low-density lipoprotein in familial combined hyperlipidemia: Independent of metabolic syndrome and related to history of cardiovascular events. Atherosclerosis 2009, 203, 320–324. [Google Scholar] [CrossRef]
- Jarauta, E.; Mateo-gallego, R.; Gilabert, R.; Plana, N.; Junyent, M.; de Groot, E.; Cenarro, A.; Masana, L.; Ros, E.; Civeira, F. Carotid atherosclerosis and lipoprotein particle subclasses in familial hypercholesterolaemia and familial combined hyperlipidaemia. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 591–597. [Google Scholar] [CrossRef]
- Georgieva, A.M.; Van Greevenbroek, M.M.; Krauss, R.M.; Brouwers, M.C.G.; Vermeulen, V.M.; Robertus-Teunissen, M.G.; van der Kallen, C.J.H.; de Bruin, T.W.A. Subclasses of Low-Density Lipoprotein and Very Low-Density Lipoprotein in Familial Combined Hyperlipidemia: Relationship to Multiple Lipoprotein Phenotype. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 744–749. [Google Scholar] [CrossRef] [Green Version]
- de Rijke, Y.B.; Bredie, S.J.; Demacker, P.N.; Vogelaar, J.M.; Hak-Lemmers, H.L.; Stalenhoef, A.F. The redox status of coenzyme Q10 in total LDL as an indicator of in vivo oxidative modification. Studies on subjects with familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Tavridou, A.; Efthimiadis, A.; Efthimiadis, I.; Manolopoulos, V.G. Simvastatin-induced changes in circulating oxidized low-density lipoprotein in different types of dyslipidemia. Heart Vessel. 2010, 25, 288–293. [Google Scholar] [CrossRef]
- Liu, M.L.; Ylitalo, K.; Salonen, R.; Salonen, J.T.; Taskinen, M.R. Circulating oxidized low-density lipoprotein and its association with carotid intima-media thickness in asymptomatic members of familial combined hyperlipidemia families. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1492–1497. [Google Scholar] [CrossRef] [Green Version]
- Reiner, Z. Hypertriglyceridaemia and risk of coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 401–411. [Google Scholar] [CrossRef]
- Mata, P.; Alonso, R.; Ruiz-Garcia, A.; Diaz-Diaz, J.L.; Gonzalez, N.; Gijon-Conde, T.; Martinez-Faedo, C.; Morón, I.; Arranz, E.; Aguado, R.; et al. Familial combined hyperlipidemia: Consensus document. Aten. Primaria. 2014, 46, 440–446. [Google Scholar] [CrossRef] [Green Version]
- National Cholesterol Education Program (US); Expert Panel on Detection and Treatment of High Blood Cholesterol in Adults. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002, 106, 3143–3421. [Google Scholar] [CrossRef]
- Sanchez-Quesada, J.L.; Benitez, S.; Otal, C.; Franco, M.; Blanco-Vaca, F.; Ordóñez-Llanos, J. Density distribution of electronegative LDL in normolipemic and hyperlipemic subjects. J. Lipid Res. 2002, 43, 699–705. [Google Scholar]
- Benitez, S.; Sanchez-Quesada, J.L.; Ribas, V.; Jorba, O.; Blanco-Vaca, F.; González-Sastre, F.; Ordóñez-Llanos, J. Platelet-activating factor acetylhydrolase is mainly associated with electronegative low-density lipoprotein subfraction. Circulation 2003, 108, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Quesada, J.L.; Vinagre, I.; De Juan-Franco, E.; Sanchez-Hernandez, J.; Bonet-Marques, R.; Blanco-Vaca, F.; Ordóñez-Llanos, J.; Pérez, A. Impact of the LDL subfraction phenotype on Lp-PLA2 distribution, LDL modification and HDL composition in type 2 diabetes. Cardiovasc. Diabetol. 2013, 12, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Quesada, J.L.; Ortega, H.; Payes-Romero, A.; Serrat-Serrat, J.; Gonzalez-Sastre, F.; Lasuncion, M.A.; Ordonez-Llanos, J. LDL from aerobically-trained subjects shows higher resistance to oxidative modification than LDL from sedentary subjects. Atherosclerosis 1997, 132, 207–213. [Google Scholar] [CrossRef]
- Benitez, S.; Sanchez-Quesada, J.L.; Lucero, L.; Arcelus, R.; Ribas, V.; Jorba, O.; Castellví, A.; Alonso, E.; Blanco-Vaca, F.; Ordóñez-Llanos, J. Changes in low-density lipoprotein electronegativity and oxidizability after aerobic exercise are related to the increase in associated non-esterified fatty acids. Atherosclerosis 2002, 160, 223–232. [Google Scholar] [CrossRef]
- De Juan-Franco, E.; Perez, A.; Ribas, V.; Sanchez-Hernandez, J.A.; Blanco-Vaca, F.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. Standardization of a method to evaluate the antioxidant capacity of high-density lipoproteins. Int. J. Biomed. Sci. 2009, 5, 402–410. [Google Scholar]
- Escola-Gil, J.C.; Lee-Rueckert, M.; Santos, D.; Cedo, L.; Blanco-Vaca, F.; Julve, J. Quantification of in vitro macrophage cholesterol efflux and in vivo macrophage-specific reverse cholesterol transport. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2015; pp. 211–233. [Google Scholar]
- Estruch, M.; Minambres, I.; Sanchez-Quesada, J.L.; Soler, M.; Perez, A.; Ordonez-Llanos, J.; Benitez, S. Increased inflammatory effect of electronegative LDL and decreased protection by HDL in type 2 diabetic patients. Atherosclerosis 2017, 265, 292–298. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef] [Green Version]
- Rull, A.; Martinez-Bujidos, M.; Perez-Cuellar, M.; Perez, A.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Increased concentration of clusterin/apolipoprotein J (apoJ) in hyperlipemic serum is paradoxically associated with decreased apoJ content in lipoproteins. Atherosclerosis 2015, 241, 463–470. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Wu, M.; Gosmanova, A.K.; Becker, J.O.; Wijsman, E.M.; Brunzell, J.D.; Kahn, S.E.; Knopp, R.H.; Lyons, T.J.; Heinecke, J.W. Low clusterin levels in high-density lipoprotein associate with insulin resistance, obesity, and dyslipoproteinemia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2528–2534. [Google Scholar] [CrossRef] [Green Version]
- Rull, A.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. The role of LDL-bound apoJ in the development of atherosclerosis. Clin. Lipidol. 2015, 10, 321–328. [Google Scholar] [CrossRef]
- Schwarz, M.; Spath, L.; Lux, C.A.; Paprotka, K.; Torzewski, M.; Dersch, K.; Koch-Brandt, C.; Husmann, M.; Bhakdi, S. Potential protective role of apoprotein J (clusterin) in atherogenesis: Binding to enzymatically modified low-density lipoprotein reduces fatty acid-mediated cytotoxicity. Thromb. Haemost. 2008, 100, 110–118. [Google Scholar]
- Martinez-Bujidos, M.; Rull, A.; Gonzalez-Cura, B.; Perez-Cuellar, M.; Montoliu-Gaya, L.; Villegas, S.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2015, 29, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.L.; Guo, Y.L.; Li, J.J. Apoprotein C-III: A review of its clinical implications. Clin. Chim. Acta 2016, 460, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Dergunov, A.D.; Rosseneu, M. The significance of apolipoprotein E structure to the metabolism of plasma triglyceride-rich lipoproteins. Biol. Chem. Hoppe Seyler 1994, 375, 485–495. [Google Scholar] [PubMed]
- Krauss, R.M.; Siri, P.W. Metabolic abnormalities: Triglyceride and low-density lipoprotein. Endocrinol. Metab. Clin. N. Am. 2004, 33, 405–415. [Google Scholar] [CrossRef]
- Musliner, T.A.; Krauss, R.M. Lipoprotein subspecies and risk of coronary disease. Clin. Chem. 1988, 34, B78–B83. [Google Scholar]
- Sanchez-Quesada, J.L.; Villegas, S.; Ordoñez-Llanos, J. Electronegative low-density lipoprotein. A link between apolipoprotein B misfolding, lipoprotein aggregation and proteoglycan binding. Curr. Opin. Lipidol. 2012, 23, 479–486. [Google Scholar] [CrossRef]
- Estruch, M.; Sanchez-Quesada, J.L.; Ordoñez Llanos, J.; Benitez, S. Electronegative LDL: A circulating modified LDL with a role in inflammation. Mediat. Inflamm. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sacks, F.M.; Jensen, M.K. From HDL cholesterol to measurements of function: Prospects for the development of tests for HDL functionality in CVD. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.K.; Aroner, S.A.; Mukamal, K.J.; Furtado, J.D.; Post, W.S.; Tsai, M.Y.; Tjønneland, A.; Polak, J.F.; Rimm, E.B.; Overvad, K.; et al. High-Density Lipoprotein Subspecies Defined by Presence of Apolipoprotein C-III and Incident Coronary Heart Disease in Four Cohorts. Circulation 2018, 137, 1364–1373. [Google Scholar] [CrossRef]
- Yingchun, H.; Yahong, M.; Jiangping, W.; Xiaokui, H.; Xiaohong, Z. Increased inflammation, endoplasmic reticulum stress and oxidative stress in endothelial and macrophage cells exacerbate atherosclerosis in ApoCIII transgenic mice. Lipids Health Dis. 2018, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Botteri, G.; Montori, M.; Guma, A.; Pizarro, J.; Cedo, L.; Li, D.; Barroso, E.; Palomer, X.; Kohan, A.B.; Vázquez-Carrera, M. VLDL and apolipoprotein CIII induce ER stress and inflammation and attenuate insulin signalling via Toll-like receptor 2 in mouse skeletal muscle cells. Diabetologia 2017, 60, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Azcutia, V.; Aikawa, E.; Figueiredo, J.L.; Croce, K.; Sonoki, H.; Sacks, F.M.; Luscinskas, F.W.; Aikawa, M. Statins suppress apolipoprotein CIII-induced vascular endothelial cell activation and monocyte adhesion. Eur. Heart J. 2013, 34, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NTG Group (n = 13) | HTG Group (n = 12) | Control Group (n = 15) | |||
|---|---|---|---|---|---|
| Age (years) | 48.6 ± 10.6 | 51.1 ± 6.6 | 48.4 ± 6.0 | ||
| Gender (M/F) | 7/5 | 8/4 | 9/6 | ||
| Baseline | After Therapy | Baseline | After Therapy | ||
| BMI (kg/m2) | 27.1 ± 4.3 | 26.5 ± 4.4 | 27.1 ± 4.7 | 27.3 ± 4.6 | 24.7 ± 4.3 |
| Cholesterol (mmol/L) | 7.18 ± 0.89 | 4.71 ± 0.66 * | 7.44 ± 0.81 | 5.04 ± 0.91 * | 5.44 ± 0.99 a,c |
| Triglycerides (mmol/L) | 1.84 ± 0.44 | 1.40 ± 0.54 * | 3.61 ± 0.79 a | 2.35 ± 0.73 *,a,b | 0.93 ± 0.28 a,c,d |
| VLDL-c (mmol/L) | 0.87 ± 0.20 | 0.59 ± 0.20 * | 1.51 ± 0.47 a | 1.02 ± 0.25 *,a,b | 0.43 ± 0.13 a,c,d |
| LDL-c (mmol/L) | 5.02 ± 0.78 | 2.73 ± 0.54 * | 4.95 ± 0.75 | 3.03 ± 0.89 * | 3.31 ± 0.96 a,c |
| HDL-c (mmol/L) | 1.29 ± 0.37 | 1.28 ± 0.35 | 0.98 ± 0.14 a | 0.99 ± 0.22 a,b | 1.51 ± 0.19 c,d |
| ApoB (mmol/L) | 1.50 ± 0.19 | 0.90 ± 0.16 * | 1.64 ± 0.19 | 1.11 ± 0.22 * | 0.95 ± 0.18 a,c |
| CRP (mg/L) | 2.79 ± 2.45 | 1.72 ± 1.64 * | 2.73 ± 2.51 | 1.56 ± 1.30 * | 1.02 ± 0.63 a,c |
| Leptin (µg/L) | 36.3 ± 30.3 | 43.9 ± 32.3 | 28.6 ± 33.7 | 29.5 ± 30,0 | 33.6 ± 29.7 |
| Adiponectin (µg/L) | 10.9 ± 2.5 | 11.1 ± 2.7 | 10.2 ± 2.1 | 10.3 ± 1.7 | 10.2 ± 2.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puig, N.; Miñambres, I.; Benítez, S.; Gil, P.; Grau-Agramunt, M.; Rivas-Urbina, A.; Pérez, A.; Sánchez-Quesada, J.L. Familial Combined Hyperlipidemia (FCH) Patients with High Triglyceride Levels Present with Worse Lipoprotein Function Than FCH Patients with Isolated Hypercholesterolemia. Biomedicines 2020, 8, 6. https://doi.org/10.3390/biomedicines8010006
Puig N, Miñambres I, Benítez S, Gil P, Grau-Agramunt M, Rivas-Urbina A, Pérez A, Sánchez-Quesada JL. Familial Combined Hyperlipidemia (FCH) Patients with High Triglyceride Levels Present with Worse Lipoprotein Function Than FCH Patients with Isolated Hypercholesterolemia. Biomedicines. 2020; 8(1):6. https://doi.org/10.3390/biomedicines8010006
Chicago/Turabian StylePuig, Núria, Inka Miñambres, Sonia Benítez, Pedro Gil, Margarida Grau-Agramunt, Andrea Rivas-Urbina, Antonio Pérez, and José Luis Sánchez-Quesada. 2020. "Familial Combined Hyperlipidemia (FCH) Patients with High Triglyceride Levels Present with Worse Lipoprotein Function Than FCH Patients with Isolated Hypercholesterolemia" Biomedicines 8, no. 1: 6. https://doi.org/10.3390/biomedicines8010006