The Role and Function of HDL in Patients with Chronic Kidney Disease and the Risk of Cardiovascular Disease


{kind=link}
Abstract
:1. Introduction
2. HDL Structure, Role, and Function
2.1. Mature HDL Formation
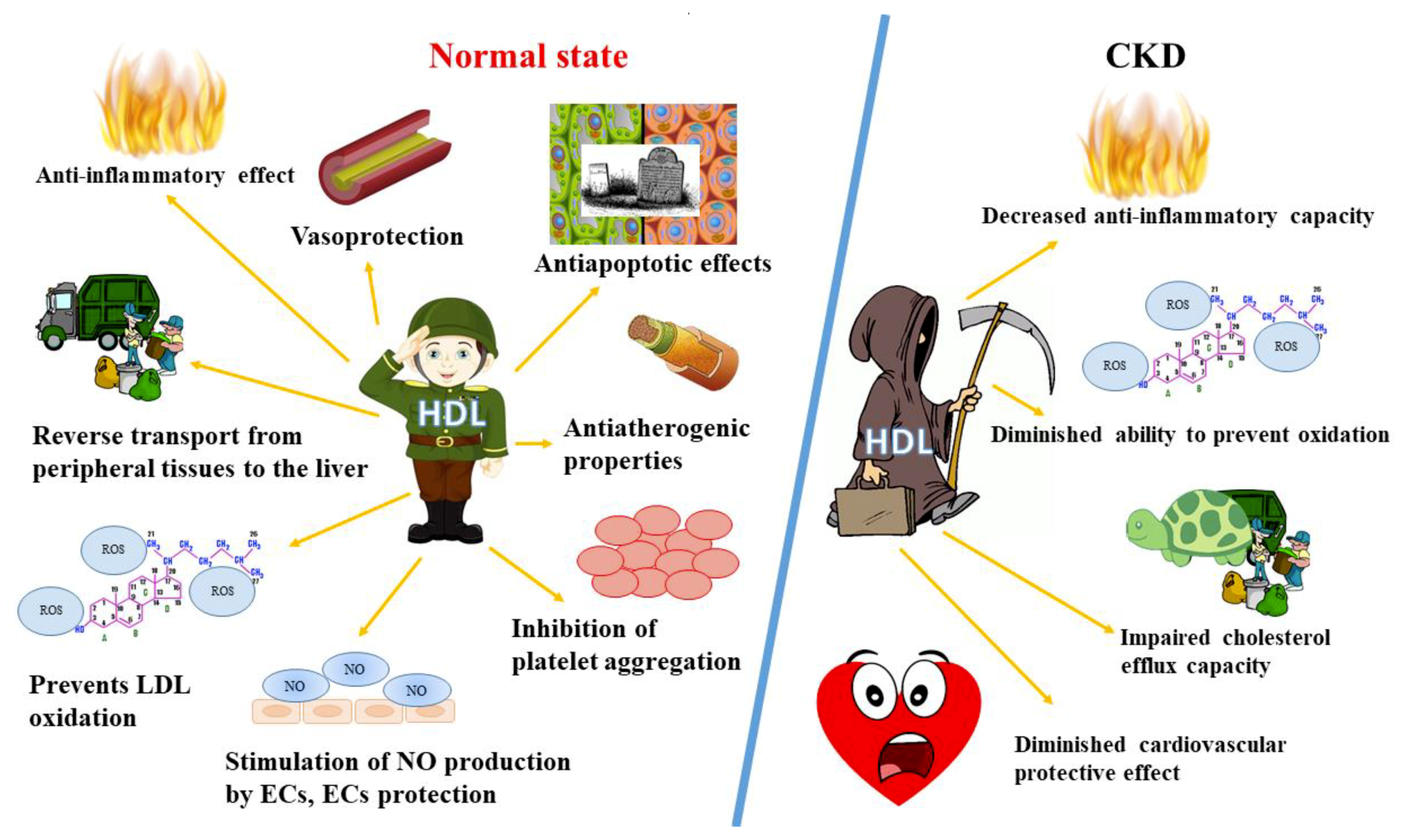
2.2. HDL Role and Functions
3. HDL Disturbances
3.1. Chronic Kidney Disease
3.2. Nephrotic Syndrome
3.3. Diabetic Nephropathy
3.4. Kidney Transplantation
3.5. HDL and the Progression of Chronic Kidney Disease
3.6. Postmenopausal Women
4. Cardiovascular Risk Related to Modifications of HDL Particles in CKD Patients
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Takemoto, Y.; Toshihide, N. Economic issues of chronic kidney disease. In CKD-Associated Complications: Progress in the Last Half Century; Nakanishi, T., Kuragano, T., Eds.; Karger Medical and Scientific Publishers: Basel, Switzerland, 2019. [Google Scholar]
- Siemens, T.A.; Riella, M.C.; Moraes, T.P.; Riella, C.V. APOL1 risk variants and kidney disease: What we know so far. J. Bras. Nefrol. 2018, 40, 388–402. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Chronic Kidney Disease in the United States. 2019. Available online: https://www.cdc.gov/kidneydisease/publications-resources/2019-national-facts.html (accessed on 20 September 2019).
- Silva, C.J.C.; de Alencar, R.P.; Tenório, L.M.; da Silva, N.L.; de Barros, A.F.; Marinho, C.R.M.; de Souza Pinto, R. Cardiovascular Risk in Patients with Chronic Kidney Disease is Associated with Decrease of HDL Levels. Int. J. Clin. Cardiol. 2018, 5, 115. [Google Scholar] [CrossRef]
- Subbiah, A.K.; Chhabra, Y.K.; Mahajan, S. Cardiovascular disease in patients with chronic kidney disease: A neglected subgroup. Heart Asia 2016, 8, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharabas, I.; Siddiqi, N. Cardiovascular disease risk profiles comparison among dialysis patients. Saudi J. Kidney Dis. Transplant. 2016, 27, 692–700. [Google Scholar]
- Dounousi, E.; Duni, A.; Marinaki, S.; Boletis, J.N. Framing and managing cardiovascular risk in chronic kidney disease: From native to transplanted kidney. Contin. Cardiol. Educ. 2017, 3, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Carrero, J.J.; Axelsson, J.; Lindholm, B.; Heimbürger, O.; Massy, Z. Emerging biomarkers for evaluating cardiovascular risk inthe chronic kidney disease patient: How do new pieces fit into the uremic puzzle? Clin. J. Am. Soc. Nephrol. 2008, 3, 505–2521. [Google Scholar] [CrossRef] [Green Version]
- Sarnak, M.J.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U.; et al. Conference Participants. J. Am. Coll. Cardiol. 2019, 74, 1823–1838. [Google Scholar] [CrossRef]
- Jardine, A.G.; Gaston, R.S.; Fellstrom, B.C.; Holdaas, H. Prevention of cardiovascular disease in adult recipients of kidney transplants. Lancet 2011, 378, 1419–1427. [Google Scholar] [CrossRef]
- Bulbul, M.C.; Dagel, T.; Afsar, B.; Ulusu, N.N.; Kuwabara, M.; Covic, A.; Kanbay, M. Disorders of Lipid Metabolism in Chronic Kidney Disease. Blood Purif. 2018, 46, 44–152. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Causes of dysregulation of lipid metabolism in chronic renal failure. Semin. Dial. 2009, 22, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and antithrombotic actions of HDL. Circ. Res. 2006, 98, 1352–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermúdez-López, M.; Arroyo, D.; Betriu, À.; Masana, L.; Fernández, E.; Valdivielso, J.M. New perspectives on CKD-induced dyslipidemia. Expert Opin. Ther. Targets 2017, 21, 967–976. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; MDText.com, Inc.: South Dartmouth, MA, USA, 2019. [Google Scholar]
- Domínguez Coello, S.; Cabrera De León, A.; Bosa Ojeda, F.; Pérez Méndez, L.I.; Díaz González, L.; Aguirre-Jaime, A.J. High density lipoprotein cholesterol increases with living altitude. Int. J. Epidemiol. 2000, 29, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, J.C.; Crook, D.; Godsland, I.F. Influence of age and menopause on serum lipids and lipoproteins in healthy women. Atherosclerosis 1993, 98, 83–90. [Google Scholar] [CrossRef]
- Karathanasis, S.K.; Freeman, L.A.; Gordon, S.M.; Remaley, A.T. The Changing Face of HDL and the Best Way to Measure it. Clin. Chem. 2017, 63, 196–210. [Google Scholar] [CrossRef] [Green Version]
- Ossoli, A.; Simonelli, S.; Varrenti, M.; Morici, N.; Oliva, F.; Stucchi, M.; Gomaraschi, M.; Strazzella, A.; Arnaboldi, L.; Thomas, M.; et al. Recombinant LCAT (Lecithin:Cholesterol Acyltransferase) Rescues Defective HDL (High-Density Lipoprotein)-Mediated Endothelial Protection in Acute Coronary Syndrome. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 915–924. [Google Scholar] [CrossRef]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Diallo, D.; Delbosc, S.; Genève, C.; Zappella, N.; Yong-Sang, J.; Patche, J.; Harrois, A.; Hamada, S.; Denamur, E.; et al. High-density lipoprotein (HDL) particle size and concentration changes in septic shock patients. Ann. Intensive Care 2019, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Eren, E.; Yilmaz, N.; Aydin, O. High Density Lipoprotein and it’s Dysfunction. Open Clin. Biochem. J. 2012, 6, 78–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, P.P.; Barter, P.J.; Rosenson, R.S.; Boden, W.E.; Chapman, M.J.; Cuchel, M.; D’Agostino, R.B.; Davidson, M.H.; Davidson, W.S.; Heinecke, J.W.; et al. High-density lipoproteins: A consensus statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 484–525. [Google Scholar] [CrossRef] [PubMed]
- Rached, F.H.; Chapman, M.J.; Kontush, A. HDL particle subpopulations: Focus on biological function. BioFactors 2015, 41, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Nessler, K.; Windak, A.; Grzybczak, R.; Nessler, M.B.; Siniarski, A.; Gajos, G. High-density lipoprotein (HDL) cholesterol – more complicated than we think? Ann. Agric. Environ. Med. 2018, 25, 517–526. [Google Scholar] [CrossRef]
- Riwanto, M.; Rohrer, L.; Roschitzki, B.; Besler, C.; Mocharla, P.; Mueller, M.; Perisa, D.; Heinrich, K.; Altwegg, L.; von Eckardstein, A.; et al. Altered activation of endothelial anti- and pro-apoptotic pathways by high-density lipoprotein from patients with coronary artery disease: Role of HDL-proteome remodeling. Circulation 2013, 127, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Rader, D.J. Molecular regulation of HDL metabolism and function: Implications for novel therapies. J. Clin. Investig. 2006, 116, 3090–3100. [Google Scholar] [CrossRef] [Green Version]
- Didichenko, S.A.; Navdaev, A.V.; Cukier, A.M.; Gille, A.; Schuetz, P.; Spycher, M.O.; Thérond, P.; Chapman, M.J.; Kontush, A.; Wright, S.D. Enhanced HDL Functionality in Small HDL Species Produced Upon Remodeling of HDL by Reconstituted HDL, CSL112: Effects on Cholesterol Efflux, Anti-Inflammatory and Antioxidative Activity. Circ. Res. 2016, 119, 751–763. [Google Scholar] [CrossRef]
- Garner, B.; Waldeck, A.R.; Witting, P.K.; Rye, K.A.; Stocker, R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J. Biol. Chem. 1998, 273, 6088–6095. [Google Scholar] [CrossRef] [Green Version]
- Linton, M.F.; Tao, H.; Linton, E.F.; Yancey, P.G. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Linton MF, Tao H, Linton EF, Yancey PG. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Trends Endocrinol. Metab. 2017, 28, 461–472. [Google Scholar] [CrossRef]
- Shih, D.M.; Xia, Y.R.; Wang, X.P.; Miller, E.; Castellani, L.W.; Subbanagounder, G.; Cheroutre, H.; Faull, K.F.; Berliner, J.A.; Witztum, J.L.; et al. Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J. Biol. Chem. 2000, 275, 17527–17535. [Google Scholar] [CrossRef] [Green Version]
- Tward, A.; Xia, Y.R.; Wang, X.P.; Shi, Y.S.; Park, C.; Castellani, L.W.; Lusis, A.J.; Shih, D.M. Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation 2002, 106, 484–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riwanto, M.; Rohrer, L.; von Eckardstein, A.; Landmesser, U. Dysfunctional HDL: From structure-function-relationships to biomarkers. Handb. Exp. Pharmacol. 2015, 224, 337–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017, 19, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.; Lordan, R.; Zabetakis, I. Inflammation, not Cholesterol, Is a Cause of Chronic Disease. Nutrients 2018, 10, 604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.; Giordano, S.; Datta, G. Role of HDL-Associated Proteins and Lipids in the Regulation of Inflammation, Advances in Lipoprotein Research, Turgay Isbir. IntechOpen. 2017. Available online: https://www.intechopen.com/books/advances-in-lipoprotein-research/role-of-hdl-associated-proteins-and-lipids-in-the-regulation-of-inflammation/ (accessed on 10 August 2019).
- Rye, K.A.; Barter, P.J. Antiinflammatory actions of HDL: A new insight. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1890–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tall, A.R.; Yvan-Charvet, L.; Terasaka, N.; Pagler, T.; Wang, N. HDL, ABC transporters and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008, 7, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uittenbogaard, A.; Shaul, P.W.; Yuhanna, I.S.; Blair, A.; Smart, E.J. High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J. Biol. Chem. 2000, 275, 11278–11283. [Google Scholar] [CrossRef] [Green Version]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D.M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Investig. 2011, 121, 2693–2708. [Google Scholar] [CrossRef] [Green Version]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein E-A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K.A.; Chapman, M.J.; Kontush, A. HDL3-mediated inactivation of LDL-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein A-I and HDL particle surface lipid rigidity: Relevance to inflammation and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2169–2175. [Google Scholar] [CrossRef] [Green Version]
- Sacre, S.M.; Stannard, A.K.; Owen, J.S. Apolipoprotein E (apoE) isoforms differentially induce nitric oxide production in endothelial cells. FEBS Lett. 2003, 540, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Ophir, G.; Amariglio, N.; Jacob-Hirsch, J.; Elkon, R.; Rechavi, G.; Michaelson, D.M. Apolipoprotein E4 enhances brain inflammation by modulation of the NF-kappaB signaling cascade. Neurobiol. Dis. 2005, 20, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Recalde, D.; Ostos, M.A.; Badell, E.; Garcia-Otin, A.L.; Pidoux, J.; Castro, G.; Zakin, M.M.; Scott-Algara, D. Human apolipoprotein A-IV reduces secretion of proinflammatory cytokines and atherosclerotic effects of a chronic infection mimicked by lipopolysaccharide. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 756–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vowinkel, T.; Mori, M.; Krieglstein, C.F.; Russell, J.; Saijo, F.; Bharwani, S.; Turnage, R.H.; Davidson, W.S.; Tso, P.; Granger, D.N.; et al. Apolipoprotein A-IV inhibits experimental colitis. J. Clin. Investig. 2004, 114, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perségol, L.; Darabi, M.; Dauteuille, C.; Lhomme, M.; Chantepie, S.; Rye, K.A.; Therond, P.; Chapman, M.J.; Salvayre, R.; Nègre-Salvayre, A.; et al. Small dense HDLs display potent vasorelaxing activity; reflecting their elevated content of sphingosine-1-phosphate. J. Lipid Res. 2018, 59, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Bisoendial, R.J.; Hovingh, G.K.; Levels, J.H.; Lerch, P.G.; Andresen, I.; Hayden, M.R.; Kastelein, J.J.; Stroes, E.S. Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation 2003, 107, 2944–2948. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Yin, H.; Liu, P.; Zhang, H.; She, M. Essential role of HDL on endothelial progenitor cell proliferation with PI3K/Akt/cyclin D1 as the signal pathway. Exp. Biol. Med. 2010, 235, 1082–1092. [Google Scholar] [CrossRef]
- Kratzer, A.; Giral, H.; Landmesser, U. High-density lipoproteins as modulators of endothelial cell functions: Alterations in patients with coronary artery disease. Cardiovasc. Res. 2014, 103, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Robbesyn, F.; Garcia, V.; Augé, N.; Vieira, O.; Frisach, M.-F.; Salvayre, R.; Nègre-Salvayre, A. HDL counterbalance the proinflammatory effect of oxidized LDL by inhibiting intracellular reactive oxygen species rise; proteasome activation; and subsequent NF-κB activation in smooth muscle cells. FASEB J. 2003, 17, 743–745. [Google Scholar] [CrossRef] [Green Version]
- Mackness, B.; Hine, D.; Liu, Y.; Mastorikou, M.; Mackness, M. Paraoxonase-1 inhibits oxidised LDL-induced MCP-1 production by endothelial cells. Biochem. Biophys. Res. Commun. 2004, 318, 680–683. [Google Scholar] [CrossRef]
- Kajani, S.; Curley, S.; McGillicuddy, F.C. Unravelling HDL-Looking beyond the Cholesterol Surface to the Quality Within. IJMS 2018, 19, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Stoep, M.; Korporaal, S.J.A.; Van Eck, M. High-density lipoprotein as a modulator of platelet and coagulation responses. Cardiovasc. Res. 2014, 103, 362–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabczyk, M.; Hondo, L.; Krzek, M.; Undas, A. High-density cholesterol and apolipoprotein AI as modifiers of plasma fibrin clot properties in apparently healthy individuals. Blood Coagul. Fibrinolysis 2013, 24, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Kaba, N.K.; Francis, C.W.; Moss, J.; Zareba, W.; Oakes, D.; Knox, K.L.; Fernandez, I.D.; Rainwater, D.L. Effects of lipids and lipid-lowering therapy on hemostatic factors in patients with myocardial infarction. J. Thromb. Haemost. 2004, 2, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Asselbergs, F.W.; Williams, S.M.; Hebert, P.R.; Coffey, C.S.; Hillege, H.L.; Navis, G.; Vaughan, D.E.; van Gilst, W.H.; Moore, J.H. Gender-specific correlations of plasminogen activator inhibitor-1 and tissue plasminogen activator levels with cardiovascular disease-related traits. J. Thromb. Haemost. 2007, 5, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Banfi, C.; Pirillo, A.; Tremoli, E.; Hamsten, A.; Catapano, A.L.; Eriksson, P. Oxidised-HDL3 induces the expression of PAI-1 in human endothelial cells. Role of p38MAPK activation and mRNA stabilization. Br. J. Haematol. 2004, 127, 97–104. [Google Scholar] [CrossRef]
- Vaziri, N.D. Lipotoxicity and impaired high-density lipoprotein-mediated reverse cholesterol transport in chronic kidney disease. J. Ren. Nutr. 2010, 20, S35–S43. [Google Scholar] [CrossRef]
- Femlak, M.; Gluba-Brzózka, A.; Ciałkowska-Rysz, A.; Rysz, J. The role and function of HDL in patients with diabetes mellitus and the related cardiovascular risk. Lipids Health Dis. 2017, 16, 207. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.; Speer, T.; Colin, S.; Charakida, M.; Zewinger, S.; Staels, B.; Chinetti-Gbaguidi, G.; Hettrich, I.; Rohrer, L.; O’Neill, F.; et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J. Am. Soc. Nephrol. 2014, 25, 2658–2668. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, G.; Bacchetti, T.; Negre-Salvayre, A.; Salvayre, R.; Dousset, N.; Curatola, G. Structural modifications of HDL and functional consequences. Atherosclerosis 2006, 184, 1–7. [Google Scholar] [CrossRef]
- Bardagjy, A.S.; Steinberg, F.M. Relationship between HDL Functional Characteristics and Cardiovascular Health and Potential Impact of Dietary Patterns: A Narrative Review. Nutrients 2019, 11, 1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiesa, S.T.; Charakida, M. High-Density Lipoprotein Function and Dysfunction in Health and Disease. Cardiovasc. Drug Ther. 2019, 33, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, H.; Hirano, T.; Ueda, M.; Kojima, S.; Mashiba, S.; Hayase, Y.; Michihata, T.; Shibata, T. High-Density Lipoprotein Subfractions and Their Oxidized Subfraction Particles in Patients with Chronic Kidney Disease. J. Atheroscler. Thromb. 2016, 23, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolle, M.; Huang, T.; Schuchardt, M.; Jankowski, V.; Prufer, N.; Jankowski, J.; Tietge, U.J.; Zidek, W.; van der Giet, M. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc. Res. 2012, 94, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Ćwiklińska, A.; Cackowska, M.; Wieczorek, E.; Król, E.; Kowalski, R.; Kuchta, A.; Kortas-Stempak, B.; Gliwińska, A.; Dąbkowski, K.; Zielińska, J.; et al. Progression of Chronic Kidney Disease Affects HDL Impact on Lipoprotein Lipase (LPL)-Mediated VLDL Lipolysis Efficiency. Kidney Blood Press. Res. 2018, 43, 970–978. [Google Scholar] [CrossRef]
- Wieczorek, E.; Cwiklinska, A.; Cackowska, M.; Kuchta, A.; Krol, E.; Kortas-Stempak, B.; Gliwinska, A.; Dabkowski, K.; Jankowski, M. The impact of HDL concentration on efficiency of lipoprotein lipase (LPL)-mediated VLDL lipolysis in patients with chronic kidney disease. Atherosclerosis 2017, 263, e210. [Google Scholar] [CrossRef]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein 63 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Tang, C.; Heinecke, J.W.; Oram, J.F. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J. Lipid Res. 2010, 51, 1849–1858. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D. HDL abnormalities in nephrotic syndrome and chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D. Dyslipidemia of chronic renal failure: The nature, mechanisms, and potential consequences. Am. J. Physiol. Renal. Physiol. 2006, 290, F262–F272. [Google Scholar] [CrossRef]
- Muls, E.; Rosseneu, M.; Daneels, R.; Schurgers, M.; Boelaert, J. Lipoprotein distribution and composition in the human nephrotic syndrome. Atherosclerosis 1985, 54, 225–237. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, M.; He, D.; Zhao, X.; Zhang, W.; Wei, L.; Huang, E.; Ji, L.; Zhang, M.; Willard, B.; et al. HDL in diabetic nephropathy has less effect in endothelial repairing than diabetes without complications. Lipids Health Dis. 2016, 15, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.; Ray, U.; Yu, R.; Hudspeth, A.; Smillie, M.; Jordan, N.; Bartle, J. Kidney Function as a Determinant of HDL and Triglyceride Concentrations in the Australian Population. J. Clin. Med. 2016, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopecky, C.; Haidinger, M.; Birner-Grünberger, R.; Darnhofer, B.; Kaltenecker, C.C.; Marsche, G.; Holzer, M.; Weichhart, T.; Antlanger, M.; Kovarik, J.J.; et al. Restoration of Renal Function Does Not Correct Impairment of Uremic HDL Properties. JASN 2015, 26, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.S.; Tan, L.; Long, J.L.; Davidson, W.S. Proteomic diversity of high density lipoproteins: Our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 2013, 54, 2575–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pasquale, C.G.; Arnolda, L.F.; Doyle, I.R.; Aylward, P.E.; Chew, D.P.; Bersten, A.D. Plasma surfactant protein-B: A novel biomarker in chronic heart failure. Circulation 2004, 110, 1091–1096. [Google Scholar] [CrossRef] [Green Version]
- Otocka-Kmiecik, A.; Mikhailidis, D.P.; Nicholls, S.J.; Davidson, M.; Rysz, J.; Banach, M. Dysfunctional HDL: A novel important diagnostic and therapeutic target in cardiovascular disease? Prog. Lipid Res. 2012, 51, 314–324. [Google Scholar] [CrossRef]
- Khera, A.V.; Rader, D.J. Future therapeutic directions in reverse cholesterol transport. Curr. Atheroscler. Rep. 2010, 12, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Kawachi, K.; Kataoka, H.; Manabe, S.; Mochizuki, T.; Nitta, K. Low HDL cholesterol as a predictor of chronic kidney disease progression: A cross-classification approach and matched cohort analysis. Heart Vessels 2019, 34, 1440–1455. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Thériault, S.; Walsh, M.; Paré, G. HDL Cholesterol, LDL Cholesterol, and Triglycerides as Risk Factors for CKD: A Mendelian Randomization Study. Am. J. Kidney Dis. 2018, 71, 166–172. [Google Scholar] [CrossRef]
- Bairey Merz, C.N.; Ramineni, T.; Leong, D. Sex-specific risk factors for cardiovascular disease in women-making cardiovascular disease real. Curr. Opin. Cardiol. 2018, 33, 500–505. [Google Scholar] [CrossRef] [PubMed]
- de Kat, A.C.; Dam, V.; Onland-Moret, N.C.; Eijkemans, M.J.C.; Broekmans, F.J.M.; Van Der Schouw, Y.T. Unraveling the associations of age and menopause with cardiovascular risk factors in a large population-based study. BMC Med. 2017, 15, 2. [Google Scholar]
- Wang, Q.; Ferreira, D.L.S.; Nelson, S.M.; Sattar, N.; Ala-Korpela, M.; Lawlor, D.A. Metabolic characterization of menopause: Cross-sectional and longitudinal evidence. BMC Med. 2018, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Shah, C. Oommen ER study of cardiovascular risk factors in pre and postmenopausal women. IJPBS 2012, 3, 560–570. [Google Scholar]
- Deepthi, S.; Naidu, J.; Narayan, A.R. Relationship between estrogen and lipid profile status in postmenopausal women. IJABPT 2012, 3, 230–234. [Google Scholar]
- Pardhe, B.D.; Ghimire, S.; Shakya, J.; Pathak, S.; Shakya, S.; Bhetwal, A.; Khanal, P.R.; Parajuli, N.P. Elevated Cardiovascular Risks among Postmenopausal Women: A Community Based Case Control Study from Nepal. Biochem. Res. Int. 2017, 2017, 1–5. [Google Scholar] [CrossRef]
- Varu, D.M.S.; Vegad, D.A.M.; Jani, D.H.A.; Savalia, D.C.V.; Joshi, D.V.S. A comparative study of serum lipid profile between premenopausal and postmenopausal women. Natl. J. Integr. Res. Med. 2012, 3, 43–45. [Google Scholar]
- Shende, S.S.; Iyer, C.; Mahajan, V.V.; Kute, P.; Sonare, A.; Gandhi, I. Effect of duration on lipid profile status in post-menopausal women. Health 2014, 2, 90–94. [Google Scholar]
- Xu, W.; Li, C.; Qian, G.; Huang, Y.; Zhao, L. Association of metabolic syndrome with chronic kidney disease in premenopausal and postmenopausal women. Nan Fang Yi Ke Da Xue Xue Bao 2019, 39, 861–866. [Google Scholar] [CrossRef]
- Lew, J.; Sanghavi, M.; Ayers, C.R.; McGuire, D.K.; Omland, T.; Atzler, R.; Gore, M.O.; Neeland, I.; Berry, J.D.; Khera, A.; et al. Sex-based differences in cardiometabolic biomarkers. Circulation 2017, 135, 544–555. [Google Scholar] [CrossRef]
- Igweh, J.C.; Nwagha, I.U.; Okaro, J.M. The effects of menopause on the serum lipid profile of normal females of South East Nigeria. Niger. J. Physiol. Sci. 2005, 20, 48–53. [Google Scholar] [PubMed]
- Derby, C.A.; Crawford, S.L.; Pasternak, R.C.; Sowers, M.; Sternfeld, B.; Matthews, K.A. Lipid changes during the menopause transition in relation to age and weight: The Study of Women’s Health Across the Nation. Am. J. Epidemiol. 2009, 169, 1352–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, L.J.; Bugiardini, R.; Merz, C.N. Women and ischemic heart disease: Evolving knowledge. J. Am. Coll. Cardiol. 2009, 54, 1561–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Khoudary, S.R.; Wang, L.; Brooks, M.M.; Thurston, R.C.; Derby, C.A.; Matthews, K.A. Increase HDL-C level over the menopausal transition is associated with greater atherosclerotic progression. J. Clin. Lipidol. 2016, 10, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Zago, V.; Sanguinetti, S.; Brites, F.; Berg, G.; Verona, J.; Basilio, F.; Wikinski, R.; Schreier, L. Impaired high density lipoprotein antioxidant activity in healthy postmenopausal women. Atherosclerosis 2004, 177, 203–210. [Google Scholar] [CrossRef]
- Bots, M.L.; Elwood, P.C.; Nikitin, Y.; Salonen, J.T.; Freire de Concalves, A.; Inzitari, D.; Sivenius, J.; Benetou, V.; Tuomilehto, J.; Koudstaal, P.J.; et al. Total and HDL cholesterol and risk of stroke. EUROSTROKE: A collaborative study among research centres in Europe. J. Epidemiol. Community Health 2002, 56, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Temgoua, M.N.; Danwang, C.; Agbor, V.N.; Noubiap, J.J. Prevalence, incidence and associated mortality of cardiovascular disease in patients with chronic kidney disease in low- and middle-income countries: A protocol for a systematic review and meta-analysis. BMJ Open 2017, 7, e016412. [Google Scholar] [CrossRef]
- House, A.A.; Wanner, C.; Sarnak, M.J.; Piña, I.L.; McIntyre, C.W.; Komenda, P.; Kasiske, B.L.; Deswal, A.; deFilippi, C.R.; Cleland, J.G.F.; et al. Heart failure in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019, 95, 1304–1317. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular disease in dialysis patients. Nephrol. Dial. Transplant. 2018, 33 (Suppl. 3), iii28–iii34. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology; mechanisms; and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Modi, Z.J.; Lu, Y.; Ji, N.; Kapke, A.; Selewski, D.T.; Dietrich, X.; Abbott, K.; Nallamothu, B.K.; Schaubel, U.E.; Saran, R.; et al. Risk of Cardiovascular Disease and Mortality in Young Adults With End-stage Renal Disease: An Analysis of the US Renal Data System. JAMA Cardiol. 2019, 4, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanner, C.; Krane, V.; März, W.; Olschewski, M.; Mann, J.F.E.; Ruf, G.; Ritz, E. for the German Diabetes and Dialysis Study. Investigators Atorvastatin in Patients with Type 2 Diabetes Mellitus Undergoing Hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Fellström, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.-W.; Chevaile, A.; Cobbe, S.M.; Grönhagen-Riska, C.; et al. for the AURORA Study Group. Rosuvastatin and Cardiovascular Events in Patients Undergoing Hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baigent, C.; Landray, M.J.; Reith, C.; Emberson, J.; Wheeler, D.C.; Tomson, C.; Wanner, C.; Krane, V.; Cass, A.; Craig, J.; et al. SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): A randomised placebo-controlled trial. Lancet 2011, 377, 2181–2192. [Google Scholar] [CrossRef] [Green Version]
- Asselbergs, F.W.; Diercks, G.F.; Hillege, H.L.; van Boven, A.J.; Janssen, W.M.; Voors, A.A.; de Zeeuw, D.; de Jong, P.E.; van Veldhuisen, D.J.; van Gilst, W.H.; et al. Effects of fosinopril and pravastatin on cardiovascular events in subjects with microalbuminuria. Circulation 2004, 110, 2809–2816. [Google Scholar] [CrossRef] [Green Version]
- Luscher, T.F.; Landmesser, U.; von Eckardstein, A.; Fogelman, A.M. High-density lipoprotein: Vascular protective effects; dysfunction; and potential as therapeutic target. Circ. Res. 2014, 114, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.; Philip, P. CANHEART: Is HDL cholesterol a cardiovascular specific risk factor? Glob. Cardiol. Sci. Pract. 2016, 2016, e201634. [Google Scholar] [CrossRef] [Green Version]
- Shoji, T.; Masakane, I.; Watanabe, Y.; Iseki, K.; Tsubakihara, Y.; Committee of Renal Data Registry; Japanese Society for Dialysis Therapy. Elevated non-high-density lipoprotein cholesterol (non-HDL-C) predicts atherosclerotic cardiovascular events in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2011, 6, 1112–1120. [Google Scholar] [CrossRef] [Green Version]
- Zewinger, S.; Speer, T.; Kleber, M.E.; Scharnagl, H.; Woitas, R.; Lepper, P.M.; Pfahler, K.; Seiler, S.; Heine, G.H.; März, W.; et al. HDL cholesterol is not associated with lower mortality in patients with kidney dysfunction. J. Am. Soc. Nephrol. 2014, 25, 1073–1082. [Google Scholar] [CrossRef] [Green Version]
- Silbernagel, G.; Genser, B.; Drechsler, C.; Scharnagl, H.; Grammer, T.B.; Stojakovic, T.; Krane, V.; Ritz, E.; Wanner, C.; März, W. HDL cholesterol; apolipoproteins; and cardiovascular risk in hemodialysis patients. J. Am. Soc. Nephrol. 2015, 26, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, F. HDL in CKD-The Devil Is in the Detail. J. Am. Soc. Nephrol. 2018, 29, 1356–1371. [Google Scholar] [CrossRef] [PubMed]
- Moradi, H.; Streja, E.; Kashyap, M.L.; Vaziri, N.D.; Fonarow, G.C.; Kalantar-Zadeh, K. Elevated high-density lipoprotein cholesterol and cardiovascular mortality in maintenance hemodialysis patients. Nephrol. Dial. Transplant. 2014, 29, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, B.C.; Kronenberg, F.; Beddhu, S.; Cheung, A.K. Lipoprotein metabolism and lipid management in chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1246–1261. [Google Scholar] [CrossRef]
- Moradi, H.; Vaziri, N.D.; Kashyap, M.L.; Said, H.M.; Kalantar-Zadeh, K. Role of HDL dysfunction in end-stage renal disease: A double-edged sword. J. Ren. Nutr. 2013, 23, 203–206. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, R.D.; McAllister, C.J.; Kovesdy, C.P.; Derose, S.F.; Kopple, J.D.; Kalantar-Zadeh, K. Association between serum lipids and survival in hemodialysis patients and impact of race. J. Am. Soc. Nephrol. 2007, 18, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia alters HDL composition and function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Kaseda, R.; Jabs, K.; Hunley, T.E.; Jones, D.; Bian, A.; Allen, R.M.; Vickers, K.C.; Yancey, P.G.; Linton, M.F.; Fazio, S.; et al. Dysfunctional high-density lipoproteins in children with chronic kidney disease. Metabolism 2015, 64, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Nakanishi, S.; Yoneda, M.; Awaya, T.; Yamane, K.; Hirano, T.; Kohno, N. Associations between small dense LDL, HDL subfractions (HDL2; HDL3) and risk of atherosclerosis in Japanese-Americans. J. Atheroscler. Thromb. 2012, 19, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Asztalos, B.F.; Collins, D.; Cupples, L.A.; Demissie, S.; Horvath, K.V.; Bloomfield, H.E.; Robins, S.J.; Schaefer, E.J. Value of high-density lipoprotein (HDL) subpopulations in predicting recurrent cardiovascular events in the Veterans Affairs HDL Intervention Trial. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2185–2191. [Google Scholar] [CrossRef]
- Kuchta, A.; Ćwiklińska, A.; Czaplińska, M.; Wieczorek, E.; Kortas-Stempak, B.; Gliwińska, A.; Dąbkowski, K.; Sałaga-Zaleska, K.; Mickiewicz, A.; Dębska-Ślizień, A.; et al. Plasma Levels of Preβ1-HDL Are Significantly Elevated in Non-Dialyzed Patients with Advanced Stages of Chronic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchta, A.; Strzelecki, A.; Ćwiklińska, A.; Gruchała, M.; Zdrojewski, Z.; Kortas-Stempak, B.; Wieczorek, E.; Gliwińska, A.; Dąbkowski, K.; Jankowski, M. HDL subpopulations containing apoA-I without apoA-II (LpA-I) in patients with angiographically proven coronary artery disease. J. Cardiol. 2017, 3, 523–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, X.-M.; Niu, D.-M.; Wu, J.; Yuan, Y.-L.; Song, J.-X.; Wang, J.-J. Elevated levels of preβ1-high-density lipoprotein are associated with cholesterol ester transfer protein; the presence and severity of coronary artery disease. Lipids Health Dis. 2017, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florens, N.; Calzada, C.; Lyasko, E.; Juillard, L.; Soulage, C.O. Modified Lipids and Lipoproteins in Chronic Kidney Disease: A New Class of Uremic Toxins. Toxins 2016, 8, 376. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, L.; Simonelli, S.; Conca, P.; Busnach, G.; Cabibbe, M.; Gesualdo, L.; Gigante, M.; Penco, S.; Veglia, F.; Franceschini, G. Acquired lecithin:cholesterol acyltransferase deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J. Intern. Med. 2015, 5, 552–561. [Google Scholar] [CrossRef]
- Wang, K.; Zelnick, L.R.; Hoofnagle, A.N.; Vaisar, T.; Henderson, C.M.; Imrey, P.B.; Robinson-Cohen, C.; de Boer, I.H.; Shiu, Y.T.; Himmelfarb, J.; et al. Alteration of HDL Protein Composition with Hemodialysis Initiation. Clin. J. Am. Soc. Nephrol. 2018, 13, 1225–1233. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Yancey, P.G.; Ikizler, T.A.; Jerome, W.G.; Kaseda, R.; Cox, B.; Bian, A.; Shintani, A.; Fogo, A.B.; Linton, M.F.; et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J. Am. Coll. Cardiol. 2012, 60, 2372–2379. [Google Scholar] [CrossRef] [Green Version]
- Untersteller, K.; Meissl, S.; Trieb, M.; Emrich, I.E.; Zawada, A.M.; Holzer, M.; Knuplez, E.; Fliser, D.; Heine, G.H.; Marsche, G. HDL functionality and cardiovascular outcome among nondialysis chronic kidney disease patients. J. Lipid Res. 2018, 59, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Birner-Gruenberger, R.; Schittmayer, M.; Holzer, M.; Marsche, G. Understanding high-density lipoprotein function in disease: Recent advances in proteomics unravel the complexity of its composition and biology. Prog. Lipid Res. 2014, 56, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Marsche, G.; Saemann, M.D.; Heinemann, A.; Holzer, M. Inflammation alters HDL composition and function: Implications for HDL-raising therapies. Pharmacol. Ther. 2013, 137, 341–351. [Google Scholar] [CrossRef]
- Holzer, M.; Schilcher, G.; Curcic, S.; Trieb, M.; Ljubojevic, S.; Stojakovic, T.; Scharnagl, H.; Kopecky, C.M.; Rosenkranz, A.R.; Heinemann, A.; et al. Dialysis modalities and HDL composition and function. J. Am. Soc. Nephrol. 2015, 26, 2267–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, B.; de Boer, I.; Tang, C.; Mayer, P.S.; Zelnick, L.; Afkarian, M.; Heinecke, J.W.; Himmelfarb, J. A cluster of proteins implicated in kidney disease is increased in high-density lipoprotein isolated from hemodialysis subjects. J. Proteome Res. 2015, 14, 2792–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Bick, A.G.; Flannick, J.; Friedman, D.J.; Genovese, G.; Parfenov, M.G.; Depalma, S.R.; Gupta, N.; Gabriel, S.B.; Taylor, H.A., Jr.; et al. Increased burden of cardiovascular disease in carriers of APOL1 genetic variants. Circ. Res. 2014, 114, 845–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, W.S.; Silva, R.A.; Chantepie, S.; Lagor, W.R.; Chapman, M.J.; Kontush, A. Proteomic analysis of defined hdl subpopulations reveals particle-specific protein clusters: Relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 870–876. [Google Scholar] [CrossRef]
- Peralta, C.A.; Katz, R.; DeBoer, I.; Ix, J.; Sarnak, M.; Kramer, H.; Siscovick, D.; Shea, S.; Szklo, M.; Shlipak, M. Racial and ethnic differences in kidney function decline among persons without chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.-H.; Ge, M.; Merscher, S.; Rosenberg, A.Z.; Desante, M.; Roshanravan, H.; Okamoto, K.; Shin, M.K.; Hoek, M.; Fornoni, A.; et al. APOL1 renal risk variants promote cholesterol accumulation in tissues and cultured macrophages from APOL1 transgenic mice. PLoS ONE 2019, 14, e0211559. [Google Scholar] [CrossRef]
- Freedman, B.I.; Langefeld, C.D.; Murea, M.; Ma, L.; Otvos, J.D.; Turner, J.; Antinozzi, P.A.; Divers, J.; Hicks, P.J.; Bowden, N.W.; et al. Apolipoprotein L1 nephropathy risk variants associate with HDL subfraction concentration in African Americans. Nephrology, dialysis, transplantation: Official publication of the European Dialysis and Transplant Association—European Renal Association. Nephrol. Dial. Transplant. 2011, 26, 3805–3810. [Google Scholar] [CrossRef]
- Kollerits, B.; Krane, V.; Drechsler, C.; Lamina, C.; März, W.; Ritz, E.; Wanner, C.; Kronenberg, F. German Diabetes and Dialysis Study Investigators. Apolipoprotein A-IV concentrations and clinical outcomes in haemodialysis patients with type 2 diabetes mellitus-a post hoc analysis of the 4D Study. J. Intern. Med. 2012, 272, 592–600. [Google Scholar] [CrossRef]
- Kopecky, C.; Genser, B.; Drechsler, C.; Krane, V.; Kaltenecker, C.C.; Hengstschläger, M.; März, W.; Wanner, C.; Säemann, M.D.; Weichhart, T. Quantification of HDL proteins; cardiac events; and mortality in patients with type 2 diabetes on hemodialysis. Clin. J. Am. Soc. Nephrol. 2015, 10, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Willeit, P.; Freitag, D.F.; Laukkanen, J.A.; Chowdhury, S.; Gobin, R.; Mayr, M.; Di Angelantonio, E.; Chowdhury, R. Asymmetric dimethylarginine and cardiovascular risk: Systematic review and meta-analysis of 22 prospective studies. J. Am. Heart Assoc. 2015, 4, e001833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, S.; Sonntag, S.R.; Lieb, W.; Maas, R. Asymmetric and symmetric dimethylarginine as risk markers for total mortality and cardiovascular outcomes: A systematic review and meta-analysis of prospective studies. PLoS ONE 2016, 11, e0165811. [Google Scholar] [CrossRef] [PubMed]
- Descamps-Latscha, B.; Witko-Sarsat, V.; Nguyen-Khoa, T.; Nguyen, A.T.; Gausson, V.; Mothu, N.; London, G.M.; Jungers, P. Advanced oxidation protein products as risk factors for atherosclerotic cardiovascular events in nondiabetic predialysis patients. Am. J. Kidney Dis. 2005, 45, 39–47. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Kopple, J.D.; Kamranpour, N.; Fogelman, A.M.; Navab, M. HDL-inflammatory index correlates with poor outcome in hemodialysis patients. Kidney Int. 2007, 72, 1149–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keidar, S.; Bogner, I.; Gamliel-Lazarovich, A.; Leiba, R.; Fuhrman, B.; Kouperberg, E. High plasma high-density lipoprotein levels, very low cardiovascular risk profile, and subclinical carotid atherosclerosis in postmenopausal women. J. Clin. Lipidol. 2009, 3, 345–350. [Google Scholar] [CrossRef]
- Muka, T.; Oliver-Williams, C.; Kunutsor, S.; Laven, J.S.E.; Fauser, B.C.J.M.; Chowdhury, R.; Kavousi, M.; Franco, O.H. Association of age at onset of menopause and time since onset of menopause with cardiovascular outcomes, intermediate vascular traits, and all-cause mortality: A systematic review and meta-analysis. JAMA Cardiol. 2016, 1, 767–776. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rysz, J.; Gluba-Brzózka, A.; Rysz-Górzyńska, M.; Franczyk, B. The Role and Function of HDL in Patients with Chronic Kidney Disease and the Risk of Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 601. https://doi.org/10.3390/ijms21020601
Rysz J, Gluba-Brzózka A, Rysz-Górzyńska M, Franczyk B. The Role and Function of HDL in Patients with Chronic Kidney Disease and the Risk of Cardiovascular Disease. International Journal of Molecular Sciences. 2020; 21(2):601. https://doi.org/10.3390/ijms21020601
Chicago/Turabian StyleRysz, Jacek, Anna Gluba-Brzózka, Magdalena Rysz-Górzyńska, and Beata Franczyk. 2020. "The Role and Function of HDL in Patients with Chronic Kidney Disease and the Risk of Cardiovascular Disease" International Journal of Molecular Sciences 21, no. 2: 601. https://doi.org/10.3390/ijms21020601