

GSDME in Endothelial Cells: Inducing Vascular Inflammation and Atherosclerosis via Mitochondrial Damage and STING Pathway Activation
 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Models and Procedures
2.2. Hematoxylin and Eosin (HE) Staining
2.3. Immunofluorescence
2.4. Cell Culture
2.5. Small Interfering RNA (siRNA) Transfection
2.6. Monocyte–Endothelial Cell Adhesion Assay
2.7. Western Blot Analysis
2.8. Statistical Analysis
3. Results
3.1. GSDME Promoted Vascular Inflammation and Atherosclerosis in ApoE−/− Mice
3.2. GSDME Mediated Ox-LDL-Induced Inflammation in HUVECs
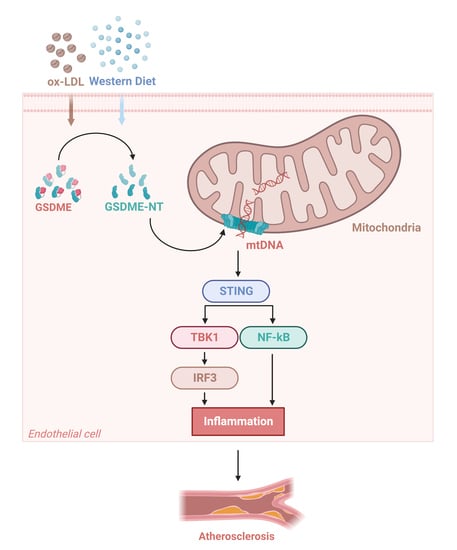
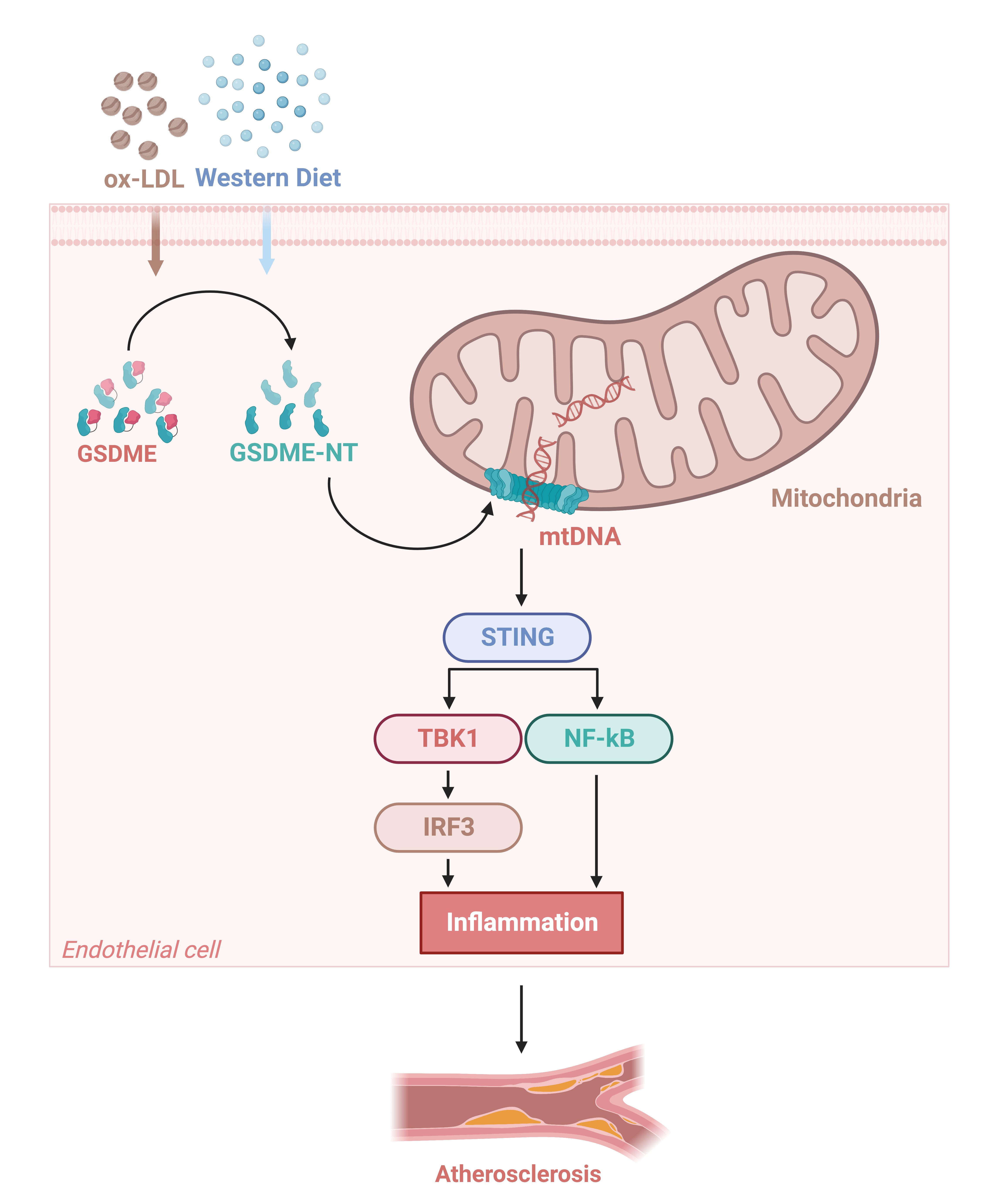
3.3. GSDME Damaged Mitochondria in Endothelial Cells and Activated STING Pathway
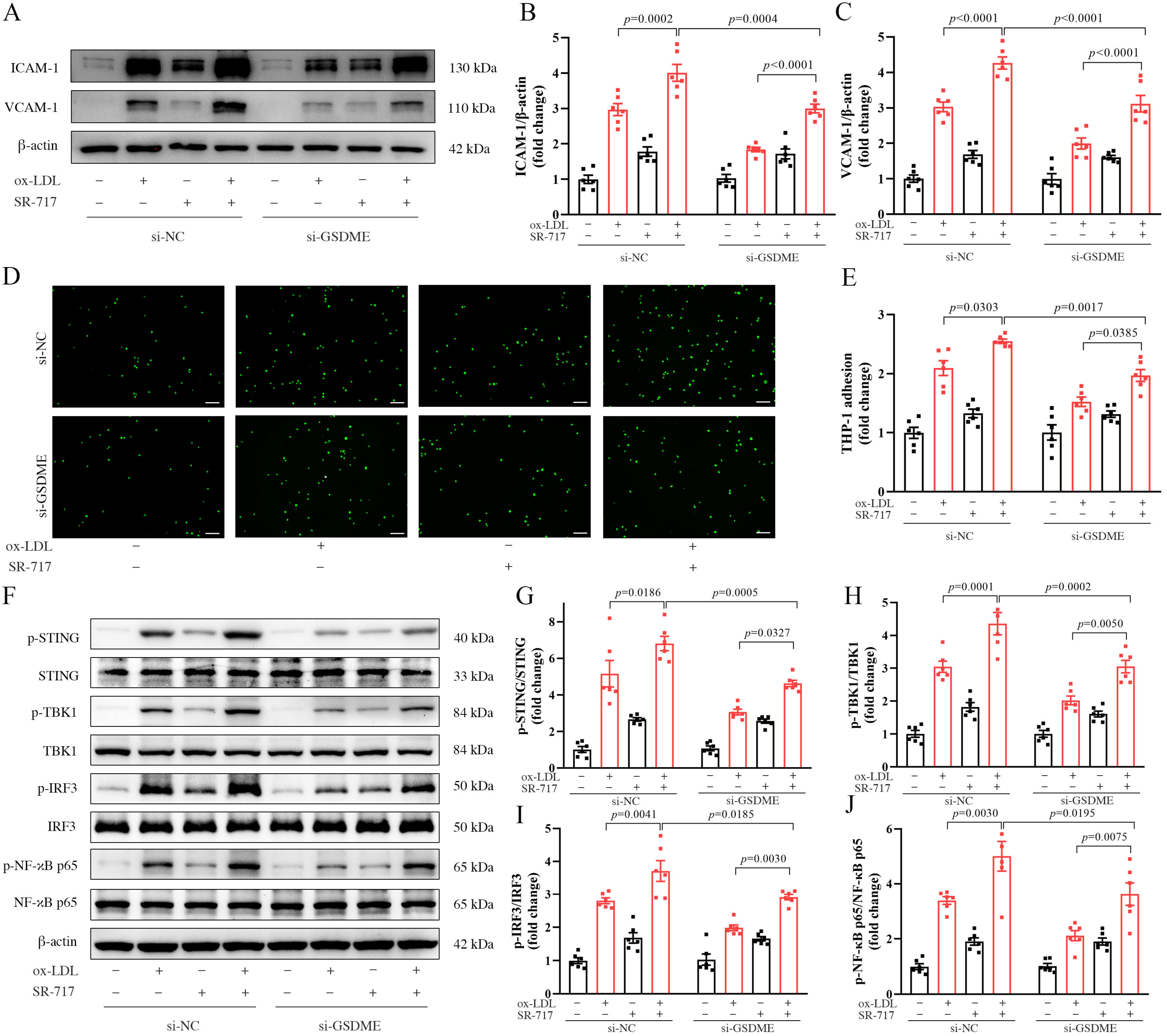
3.4. Activation of the STING Pathway Altered the Protective Effects of GSDME Deficiency on Atherogenesis and Endothelial Inflammation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yu, P.; Jiang, H.; Yang, X.; Zhao, J.; Zou, Y.; Ge, J. The Essential Role of Pin1 via NF-κB Signaling in Vascular Inflammation and Atherosclerosis in ApoE−/− Mice. Int. J. Mol. Sci. 2017, 18, 644. [Google Scholar] [CrossRef]
- Su, E.; Yu, P.; Zhang, B.; Zhang, A.; Xie, S.; Zhang, C.; Li, S.; Zou, Y.; Liu, M.; Jiang, H.; et al. Endothelial Intracellular ANG (Angiogenin) Protects Against Atherosclerosis by Decreasing Endoplasmic Reticulum Stress. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 305–325. [Google Scholar] [CrossRef]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Liu, X.; Lieberman, J. Knocking ‘em Dead: Pore-Forming Proteins in Immune Defense. Annu. Rev. Immunol. 2020, 38, 455–485. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116, Correction in Nature 2016, 540, 150. [Google Scholar] [CrossRef]
- Qian, Z.; Zhao, Y.; Wan, C.; Deng, Y.; Zhuang, Y.; Xu, Y.; Zhu, Y.; Lu, S.; Bao, Z. Pyroptosis in the Initiation and Progression of Atherosclerosis. Front. Pharmacol. 2021, 12, 652963. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Zhang, Y.; Xu, Y.; Yang, W.Y.; Jiang, X.; Sha, X.; Cheng, X.; Wang, J.; Qin, X.; Yu, J.; et al. Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ. Res. 2016, 118, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Opoku, E.; Traughber, C.A.; Zhang, D.; Iacano, A.J.; Khan, M.; Han, J.; Smith, J.D.; Gulshan, K. Gasdermin D Mediates Inflammation-Induced Defects in Reverse Cholesterol Transport and Promotes Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 715211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, B.; Li, D.; Wang, G.; Han, X.; Sun, X. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem. Biophys. Res. Commun. 2018, 495, 1418–1425. [Google Scholar] [CrossRef]
- Yao, F.; Jin, Z.; Zheng, Z.; Lv, X.; Ren, L.; Yang, J.; Chen, D.; Wang, B.; Yang, W.; Chen, L.; et al. HDAC11 promotes both NLRP3/caspase-1/GSDMD and caspase-3/GSDME pathways causing pyroptosis via ERG in vascular endothelial cells. Cell Death Discov. 2022, 8, 112. [Google Scholar] [CrossRef]
- Ueda, K.; Sakai, C.; Ishida, T.; Morita, K.; Kobayashi, Y.; Horikoshi, Y.; Baba, A.; Okazaki, Y.; Yoshizumi, M.; Tashiro, S.; et al. Cigarette smoke induces mitochondrial DNA damage and activates cGAS-STING pathway: Application to a biomarker for atherosclerosis. Clin. Sci. 2023, 137, 163–180, Correction in Clin. Sci. 2023, 137, 353. [Google Scholar] [CrossRef]
- Khwaja, B.; Thankam, F.G.; Agrawal, D.K. Mitochondrial DAMPs and altered mitochondrial dynamics in OxLDL burden in atherosclerosis. Mol. Cell Biochem. 2021, 476, 1915–1928. [Google Scholar] [CrossRef]
- de Torre-Minguela, C.; Gómez, A.I.; Couillin, I.; Pelegrín, P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J. 2021, 35, e21757. [Google Scholar] [CrossRef]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486. [Google Scholar] [CrossRef]
- Guo, Y.; Gu, R.; Gan, D.; Hu, F.; Li, G.; Xu, G. Mitochondrial DNA drives noncanonical inflammation activation via cGAS-STING signaling pathway in retinal microvascular endothelial cells. Cell Commun. Signal 2020, 18, 172. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- An, C.; Sun, F.; Liu, C.; Huang, S.; Xu, T.; Zhang, C.; Ge, S. IQGAP1 promotes mitochondrial damage and activation of the mtDNA sensor cGAS-STING pathway to induce endothelial cell pyroptosis leading to atherosclerosis. Int. Immunopharmacol. 2023, 123, 110795. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cheng, Z.; Huang, B.; Luo, S.; Guo, Y. Palmitic acid promotes endothelial-to-mesenchymal transition via activation of the cytosolic DNA-sensing cGAS-STING pathway. Arch. Biochem. Biophys. 2022, 727, 109321. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.N.; Yu, C.; Vartabedian, V.F.; Jia, Y.; Kumar, M.; Gamo, A.M.; Vernier, W.; Ali, S.H.; Kissai, M.; Lazar, D.C.; et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 2020, 369, 993–999. [Google Scholar] [CrossRef]
- Wen, S.; Wang, Z.H.; Zhang, C.X.; Yang, Y.; Fan, Q.L. Caspase-3 Promotes Diabetic Kidney Disease Through Gasdermin E-Mediated Progression to Secondary Necrosis During Apoptosis. Diabetes Metab. Syndr. Obes. 2020, 13, 313–323. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, R.; Huang, L.; Xu, Y.; Su, M.; Chen, J.; Geng, L.; Xu, W.; Gong, S. CD147 Aggravated Inflammatory Bowel Disease by Triggering NF-κB-Mediated Pyroptosis. Biomed. Res. Int. 2020, 2020, 5341247. [Google Scholar] [CrossRef]
- Liu, J.; Zhong, Y.; Liu, H.; Yang, H.; Lu, P.; Shi, Y.; Wang, X.; Zheng, W.; Yu, X.; Xu, Y.; et al. Oncostatin M sensitizes keratinocytes to UVB-induced inflammation via GSDME-mediated pyroptosis. J. Dermatol. Sci. 2021, 104, 95–103. [Google Scholar] [CrossRef]
- Yin, Y.; Li, X.; Sha, X.; Xi, H.; Li, Y.-F.; Shao, Y.; Mai, J.; Virtue, A.; Lopez-Pastrana, J.; Meng, S.; et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 804–816. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; Yang, H.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef]
- Mao, C.; Li, D.; Zhou, E.; Zhang, J.; Wang, C.; Xue, C. Nicotine exacerbates atherosclerosis through a macrophage-mediated endothelial injury pathway. Aging 2021, 13, 7627–7643. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lan, B.; Zheng, T.; Yang, L.; Zhang, X.; Cheng, L.; Tuerhongjiang, G.; Yuan, Z.; Wu, Y. GSDME-mediated pyroptosis promotes the progression and associated inflammation of atherosclerosis. Nat. Commun. 2023, 14, 929. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; He, X.; Wu, L.-M.; Zhang, R.-Y.; Li, L.-M.; Wu, C.-M.; Lu, Y.-B.; Hu, B.; Shi, C.; Lu, Z.-F.; et al. MLKL Aggravates Ox-LDL-Induced Cell Pyroptosis via Activation of NLRP3 Inflammasome in Human Umbilical Vein Endothelial Cells. Inflammation 2020, 43, 2222–2231. [Google Scholar] [CrossRef]
- Temizoz, B.; Kuroda, E.; Ohata, K.; Jounai, N.; Ozasa, K.; Kobiyama, K.; Aoshi, T.; Ishii, K.J. TLR9 and STING agonists synergistically induce innate and adaptive type-II IFN. Eur. J. Immunol. 2015, 45, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef] [PubMed]
- Luchner, M.; Reinke, S.; Milicic, A. TLR Agonists as Vaccine Adjuvants Targeting Cancer and Infectious Diseases. Pharmaceutics 2021, 13, 142. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; VanPortfliet, J.J.; Chen, Y.F.; Bryant, J.D.; Li, Y.; Fails, D.; Torres-Odio, S.; Ragan, K.B.; Deng, J.; Mohan, A.; et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell 2023, 186, 3013–3032. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, S.; Su, E.; Song, X.; Xue, J.; Yu, P.; Zhang, B.; Liu, M.; Jiang, H. GSDME in Endothelial Cells: Inducing Vascular Inflammation and Atherosclerosis via Mitochondrial Damage and STING Pathway Activation. Biomedicines 2023, 11, 2579. https://doi.org/10.3390/biomedicines11092579
Xie S, Su E, Song X, Xue J, Yu P, Zhang B, Liu M, Jiang H. GSDME in Endothelial Cells: Inducing Vascular Inflammation and Atherosclerosis via Mitochondrial Damage and STING Pathway Activation. Biomedicines. 2023; 11(9):2579. https://doi.org/10.3390/biomedicines11092579
Chicago/Turabian StyleXie, Shiyao, Enyong Su, Xiaoyue Song, Junqiang Xue, Peng Yu, Baoli Zhang, Ming Liu, and Hong Jiang. 2023. "GSDME in Endothelial Cells: Inducing Vascular Inflammation and Atherosclerosis via Mitochondrial Damage and STING Pathway Activation" Biomedicines 11, no. 9: 2579. https://doi.org/10.3390/biomedicines11092579