
Role of Endothelium in Cardiovascular Sequelae of Long COVID
 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
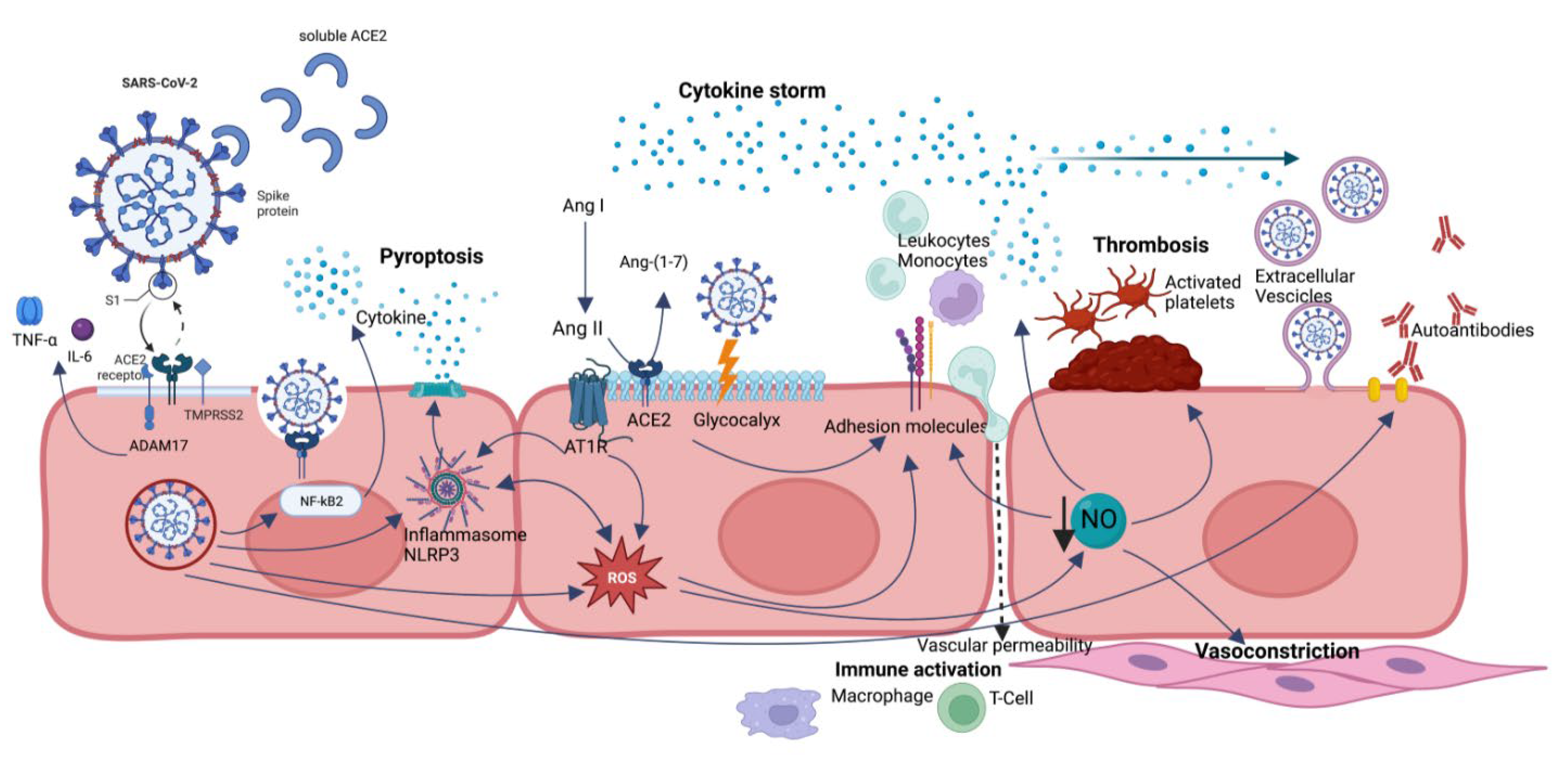
2. Endothelium and SARS-CoV-2 Infection
3. Long COVID
3.1. Epidemiology and Risk Factors
3.2. General and Endothelial Pathogenetic Mechanisms
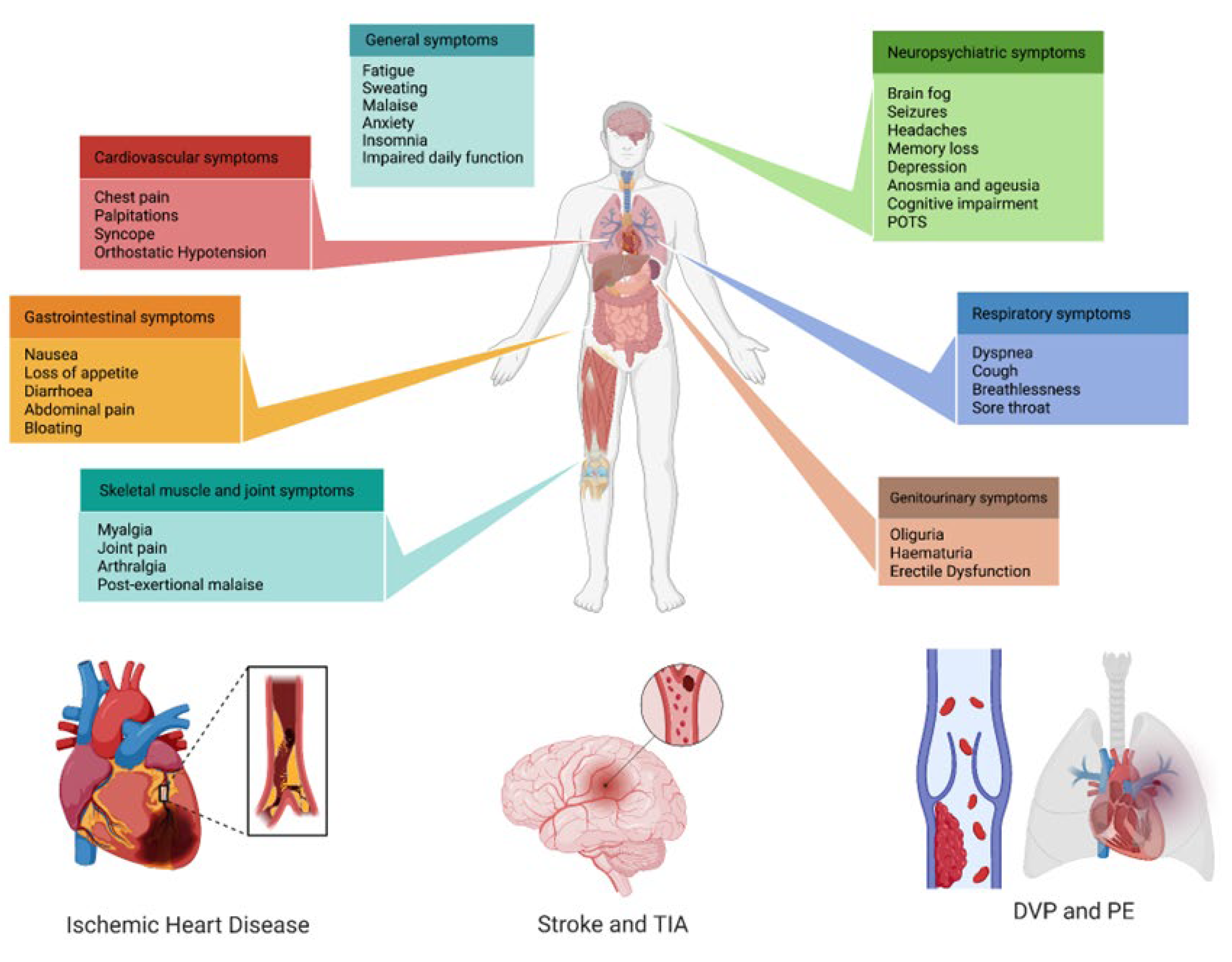
3.3. Clinical Manifestations of Long COVID
4. Cardiovascular Involvement in Long COVID
4.1. Venous Thromboembolism
4.2. Cardio- and Cerebrovascular Diseases, Myopericarditis, and Dysrhythmias
5. Clinical Insights and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D′Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Guillaume, N.; Jacob, V.; Mentaverri, R.; Kamel, S.; Boullier, A.; Slama, M. The Endothelium and COVID-19: An Increasingly Clear Link Brief Title: Endotheliopathy in COVID-19. Int. J. Mol. Sci. 2022, 23, 6196. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Zimmer, S.; Steinmetz, M.; Asdonk, T.; Motz, I.; Coch, C.; Hartmann, E.; Barchet, W.; Wassmann, S.; Hartmann, G.; Nickenig, G. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ. Res. 2011, 108, 1358–1366. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Evans, P.C.; Rainger, G.E.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial dysfunction in COVID-19: A position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, 116, 2177–2184. [Google Scholar] [CrossRef]
- COVID-19 Rapid Guideline: Managing the Long-Term Effects of COVID-19; National Institute for Health and Care Excellence (NICE): London, UK, 2020.
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef]
- Blagova, O.; Varionchik, N.; Zaidenov, V.; Savina, P.; Sarkisova, N. Anti-heart antibodies levels and their correlation with clinical symptoms and outcomes in patients with confirmed or suspected diagnosis COVID-19. Eur. J. Immunol. 2021, 51, 893–902. [Google Scholar] [CrossRef]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral myocarditis—Diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 2015, 12, 670–680. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Gladka, M.M.; Maack, C. The endothelium as Achilles′ heel in COVID-19 patients. Cardiovasc. Res. 2020, 116, e195–e197. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stolzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Hattori, Y.; Hattori, K.; Machida, T.; Matsuda, N. Vascular endotheliitis associated with infections: Its pathogenetic role and therapeutic implication. Biochem. Pharmacol. 2022, 197, 114909. [Google Scholar] [CrossRef]
- Xu, S.W.; Ilyas, I.; Weng, J.P. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Y.; Luo, W.; Huang, L.; Xiao, J.; Li, F.; Qin, S.; Song, X.; Wu, Y.; Zeng, Q.; et al. A comprehensive investigation of the mRNA and protein level of ACE2, the putative receptor of SARS-CoV-2, in human tissues and blood cells. Int. J. Med. Sci. 2020, 17, 1522–1531. [Google Scholar] [CrossRef]
- Ye, M.; Wysocki, J.; Naaz, P.; Salabat, M.R.; LaPointe, M.S.; Batlle, D. Increased ACE 2 and decreased ACE protein in renal tubules from diabetic mice: A renoprotective combination? Hypertension 2004, 43, 1120–1125. [Google Scholar] [CrossRef] [Green Version]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell. Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, D.; Palmeira, J.D.F.; Argañaraz, G.A.; Argañaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 7, 576745. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell. Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [Green Version]
- De Queiroz, T.M.; Lakkappa, N.; Lazartigues, E. ADAM17-Mediated Shedding of Inflammatory Cytokines in Hypertension. Front. Pharmacol. 2020, 29, 1154. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Giustino, G.; Pinney, S.P.; Lala, A.; Reddy, V.Y.; Johnston-Cox, H.A.; Mechanick, J.I.; Halperin, J.L.; Fuster, V. Coronavirus and Cardiovascular Disease, Myocardial Injury, and Arrhythmia: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 76, 2011–2023. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Zhao, N.; Di, B.; Xu, L.L. The NLRP3 inflammasome and COVID-19: Activation, pathogenesis and therapeutic strategies. Cytokine Growth Factor Rev. 2021, 61, 2–15. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 2022, 606, 585–593. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Bujko, K.; Ciechanowicz, A.; Sielatycka, K.; Cymer, M.; Marlicz, W.; Kucia, M. SARS-CoV-2 Entry Receptor ACE2 Is Expressed on Very Small CD45- Precursors of Hematopoietic and Endothelial Cells and in Response to Virus Spike Protein Activates the Nlrp3 Inflammasome. Stem Cell Rev. Rep. 2021, 17, 266–277. [Google Scholar] [CrossRef]
- Meng, Y.; Pan, M.; Zheng, B.; Chen, Y.; Li, W.; Yang, Q.; Zheng, Z.; Sun, N.; Zhang, Y.; Li, X. Autophagy Attenuates Angiotensin II-Induced Pulmonary Fibrosis by Inhibiting Redox Imbalance-Mediated NOD-Like Receptor Family Pyrin Domain Containing 3 Inflammasome Activation. Antioxid. Redox Signal. 2019, 30, 520–541. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664, Erratum in Nat. Commun. 2021, 12, 5306. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.D.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell. Death Discov. 2021, 7, 43, Erratum in Cell. Death Discov. 2021, 7, 116. [Google Scholar] [CrossRef]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target Ther. 2021, 6, 255, Erratum in Signal Transduct Target Ther. 2021, 6, 326. [Google Scholar] [CrossRef]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Ghasemian Sorbeni, F.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422, Erratum in Lancet Respir. Med. 2020, 8, E26. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Fan, J.G. Characteristics and Mechanism of Liver Injury in 2019 Coronavirus Disease. J. Clin. Transl. Hepatol. 2020, 8, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Kaya, G.; Kaya, A.; Saurat, J.H. Clinical and Histopathological Features and Potential Pathological Mechanisms of Skin Lesions in COVID-19: Review of the Literature. Dermatopathology 2020, 7, 3–16. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506, Erratum in Lancet 2020, 395, 496. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Ruhl, L.; Pink, I.; Kühne, J.F.; Beushausen, K.; Keil, J.; Christoph, S.; Sauer, A.; Boblitz, L.; Schmidt, J.; David, S.; et al. Endothelial dysfunction contributes to severe COVID-19 in combination with dysregulated lymphocyte responses and cytokine networks. Signal Transduct. Target Ther. 2021, 6, 418. [Google Scholar] [CrossRef]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Kubes, P.; Kanwar, S.; Niu, X.; Gaboury, J.P. Nitric oxide synthesis inhibition induces leukocyte adhesion via superoxide and mast cells. FASEB J. 1993, 7, 1293–1299. [Google Scholar] [CrossRef]
- Kurose, I.; Wolf, R.; Grisham, M.B.; Aw, T.Y.; Specian, R.D.; Granger, D.N. Microvascular responses to inhibition of nitric oxide production. Role of active oxidants. Circ. Res. 1995, 76, 30–39. [Google Scholar] [CrossRef]
- Tsihlis, N.D.; Oustwani, C.S.; Vavra, A.K.; Jiang, Q.; Keefer, L.K.; Kibbe, M.R. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell Biochem. Biophys. 2011, 60, 89–97. [Google Scholar] [CrossRef]
- Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef] [Green Version]
- Kvietys, P.R.; Granger, D.N. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic. Biol. Med. 2012, 52, 556–592. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Park, S. Role of vascular smooth muscle cell in the inflammation of atherosclerosis. BMB Rep. 2014, 47, 1–7, Erratum in BMB Rep. 2016, 49, 34. [Google Scholar] [CrossRef]
- Incalza, M.A.; D′Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Staiculescu, M.C.; Foote, C.; Meininger, G.A.; Martinez-Lemus, L.A. The role of reactive oxygen species in microvascular remodeling. Int. J. Mol. Sci. 2014, 15, 23792–23835. [Google Scholar] [CrossRef] [Green Version]
- Akerström, S.; Mousavi-Jazi, M.; Klingström, J.; Leijon, M.; Lundkvist, A.; Mirazimi, A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1966–1969. [Google Scholar] [CrossRef] [Green Version]
- Green, S.J. Covid-19 accelerates endothelial dysfunction and nitric oxide deficiency. Microbes Infect. 2020, 22, 149–150. [Google Scholar] [CrossRef]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in Covid-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef] [PubMed]
- Goonewardena, S.N.; Grushko, O.G.; Wells, J.; Herty, L.; Rosenson, R.S.; Haus, J.M.; Hummel, S.L. Immune-Mediated Glycocalyx Remodeling in Hospitalized COVID-19 Patients. Cardiovasc. Drugs Ther. 2023, 37, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [Green Version]
- Aldecoa, C.; Llau, J.V.; Nuvials, X.; Artigas, A. Role of albumin in the preservation of endothelial glycocalyx integrity and the microcirculation: A review. Ann. Intensive Care 2020, 10, 85. [Google Scholar] [CrossRef]
- Dogné, S.; Flamion, B. Endothelial Glycocalyx Impairment in Disease: Focus on Hyaluronan Shedding. Am. J. Pathol. 2020, 190, 768–780. [Google Scholar] [CrossRef]
- Lipowsky, H.H. Role of the Glycocalyx as a Barrier to Leukocyte-Endothelium Adhesion. Adv. Exp. Med. Biol. 2018, 1097, 51–68. [Google Scholar] [CrossRef]
- Delgadillo, L.F.; Marsh, G.A.; Waugh, R.E. Endothelial Glycocalyx Layer Properties and Its Ability to Limit Leukocyte Adhesion. Biophys. J. 2020, 118, 1564–1575. [Google Scholar] [CrossRef]
- Jin, J.; Fang, F.; Gao, W.; Chen, H.; Wen, J.; Wen, X.; Chen, J. The Structure and Function of the Glycocalyx and Its Connection With Blood-Brain Barrier. Front. Cell. Neurosci. 2021, 15, 739699. [Google Scholar] [CrossRef]
- De Agostini, A.I.; Watkins, S.C.; Slayter, H.S.; Youssoufian, H.; Rosenberg, R.D. Localization of anticoagulantly active heparan sulfate proteoglycans in vascular endothelium: Antithrombin binding on cultured endothelial cells and perfused rat aorta. J. Cell. Biol. 1990, 111, 1293–1304. [Google Scholar] [CrossRef]
- Zou, Z.; Li, L.; Schäfer, N.; Huang, Q.; Maegele, M.; Gu, Z. Endothelial glycocalyx in traumatic brain injury associated coagulopathy: Potential mechanisms and impact. J. Neuroinflammation 2021, 18, 134. [Google Scholar] [CrossRef]
- Zha, D.; Fu, M.; Qian, Y. Vascular Endothelial Glycocalyx Damage and Potential Targeted Therapy in COVID-19. Cells 2022, 11, 1972. [Google Scholar] [CrossRef]
- Karampoor, S.; Zahednasab, H.; Farahmand, M.; Mirzaei, R.; Zamani, F.; Tabibzadeh, A.; Bouzari, B.; Ajdarkosh, H.; Nikkhah, M.; Hashemi, M.R.; et al. A possible pathogenic role of Syndecan-1 in the pathogenesis of coronavirus disease 2019 (COVID-19). Int. Immunopharmacol. 2021, 97, 107684. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Chen, Y.; Ma, J.; Yang, Y.; Aodeng, S.; Cui, Q.; Wen, K.; Xiao, M.; Xie, J.; et al. Syndecan-1, an indicator of endothelial glycocalyx degradation, predicts outcome of patients admitted to an ICU with COVID-19. Mol. Med. 2021, 27, 151. [Google Scholar] [CrossRef]
- Suzuki, K.; Okada, H.; Tomita, H.; Sumi, K.; Kakino, Y.; Yasuda, R.; Kitagawa, Y.; Fukuta, T.; Miyake, T.; Yoshida, S.; et al. Possible involvement of Syndecan-1 in the state of COVID-19 related to endothelial injury. Thromb. J. 2021, 19, 5. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef] [Green Version]
- Kinaneh, S.; Khamaysi, I.; Karram, T.; Hamoud, S. Heparanase as a potential player in SARS-CoV-2 infection and induced coagulopathy. Biosci. Rep. 2021, 41, BSR20210290. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Pavlidis, G.; Katsimbri, P.; Lambadiari, V.; Parissis, J.; Andreadou, I.; Tsoumani, M.; Boumpas, D.; Kouretas, D.; Iliodromitis, E. Tocilizumab improves oxidative stress and endothelial glycocalyx: A mechanism that may explain the effects of biological treatment on COVID-19. Food Chem. Toxicol. 2020, 145, 111694. [Google Scholar] [CrossRef]
- Wang, C.; Yu, C.; Jing, H.; Wu, X.; Novakovic, V.A.; Xie, R.; Shi, J. Long COVID: The Nature of Thrombotic Sequelae Determines the Necessity of Early Anticoagulation. Front. Cell. Infect. Microbiol. 2022, 12, 861703. [Google Scholar] [CrossRef]
- Waqas, M.Y.; Javid, M.A.; Nazir, M.M.; Niaz, N.; Nisar, M.F.; Manzoor, Z.; Bhatti, S.; Hameed, S.; Khaliq, M. Extracellular vesicles and exosome: Insight from physiological regulatory perspectives. J. Physiol. Biochem. 2022, 78, 573–580. [Google Scholar] [CrossRef]
- Yamamoto, S.; Niida, S.; Azuma, E.; Yanagibashi, T.; Muramatsu, M.; Huang, T.T.; Sagara, H.; Higaki, S.; Ikutani, M.; Nagai, Y.; et al. Inflammation-induced endothelial cell-derived extracellular vesicles modulate the cellular status of pericytes. Sci. Rep. 2015, 5, 8505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guervilly, C.; Bonifay, A.; Burtey, S.; Sabatier, F.; Cauchois, R.; Abdili, E.; Arnaud, L.; Lano, G.; Pietri, L.; Robert, T.; et al. Dissemination of extreme levels of extracellular vesicles: Tissue factor activity in patients with severe COVID-19. Blood Adv. 2021, 5, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Mezine, F.; Guerin, C.L.; Philippe, A.; Gendron, N.; Soret, L.; Sanchez, O.; Mirault, T.; Diehl, J.; Chocron, R.; Boulanger, C.; et al. Increased Circulating CD62E+ Endothelial Extracellular Vesicles Predict Severity and in- Hospital Mortality of COVID-19 Patients. Stem Cell Rev. Rep. 2023, 19, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Carfì, A.; Bernabei, R.; Landi, F. Persistent Symptoms in Patients After Acute COVID-19. J. Am. Med. Assoc. 2020, 324, 603–605. [Google Scholar] [CrossRef] [PubMed]
- WHO—World Health Organization. Post COVID-19 Condition (Long COVID). Available online: https://www.who.int/europe/news-room/fact-sheets/item/post-covid-19-condition (accessed on 7 December 2022).
- WHO—World Health Organization. Weekly Epidemiological Update on COVID-19. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19 (accessed on 25 May 2023).
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Evans, R.A.; McAuley, H.; Harrison, E.M.; Shikotra, A.; Singapuri, A.; Sereno, M.; Elneima, O.; Docherty, A.; Lone, N.; Leavy, O.; et al. Physical, cognitive, and mental health impacts of COVID-19 after hospitalisation (PHOSP-COVID): A UK multicentre, prospective cohort study. Lancet Respir. Med. 2021, 9, 1275–1287, Erratum in Lancet Respir. Med. 2022, 10, e9. [Google Scholar] [CrossRef]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.; Klaser, K.; Antonelli, M.; Canas, L.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631, Erratum in Nat. Med. 2021, 27, 1116. [Google Scholar] [CrossRef]
- Raman, B.; Bluemke, D.A.; Lüscher, T.F.; Neubauer, S. Long COVID: Post-acute sequelae of COVID-19 with a cardiovascular focus. Eur. Heart J. 2022, 43, 1157–1172. [Google Scholar] [CrossRef]
- Cervia, C.; Zurbuchen, Y.; Taeschler, P.; Ballouz, T.; Menges, D.; Hasler, S.; Adamo, S.; Raeber, M.; Bächli, E.; Rudiger, A.; et al. Immunoglobulin signature predicts risk of post-acute COVID-19 syndrome. Nat. Commun. 2022, 13, 446. [Google Scholar] [CrossRef]
- Augustin, M.; Schommers, P.; Stecher, M.; Dewald, F.; Gieselmann, L.; Gruell, H.; Horn, C.; Vanshylla, K.; Cristanziano, V.; Osebold, L.; et al. Post-COVID syndrome in non-hospitalised patients with COVID-19: A longitudinal prospective cohort study. Lancet Reg. Health Eur. 2021, 6, 100122. [Google Scholar] [CrossRef]
- Gyöngyösi, M.; Alcaide, P.; Asselbergs, F.W.; Brundel, B.J.J.M.; Camici, G.G.; Martins, P.D.C.; Ferdinandy, P.; Fontana, M.; Girao, H.; Gnecchi, M.; et al. Long COVID and the cardiovascular system—Elucidating causes and cellular mechanisms in order to develop targeted diagnostic and therapeutic strategies: A joint Scientific Statement of the ESC Working Groups on Cellular Biology of the Heart and Myocardial and Pericardial Diseases. Cardiovasc. Res. 2022, 199, 336–356. [Google Scholar] [CrossRef]
- Pinato, D.J.; Tabernero, J.; Bower, M.; Scotti, L.; Patel, M.; Colomba, E.; Dolly, S.; Loizidou, A.; Chester, J.; Mukherjee, U.; et al. OnCovid study group. Prevalence and impact of COVID-19 sequelae on treatment and survival of patients with cancer who recovered from SARS-CoV-2 infection: Evidence from the OnCovid retrospective, multicentre registry study. Lancet Oncol. 2021, 22, 1669–1680. [Google Scholar] [CrossRef]
- Daugherty, S.E.; Guo, Y.; Heath, K.; Dasmariñas, M.C.; Jubilo, K.G.; Samranvedhya, J.; Lipsitch, M.; Cohen, K. Risk of clinical sequelae after the acute phase of SARS-CoV-2 infection: Retrospective cohort study. BMJ 2021, 19, 373. [Google Scholar] [CrossRef]
- Magnusson, K.; Kristoffersen, D.T.; Dell′Isola, A.; Kiadaliri, A.; Turkiewicz, A.; Runhaar, J.; Bierma-Zeinstra, S.; Englund, M.; Magnus, P.; Kinge, J.; et al. Post-covid medical complaints following infection with SARS-CoV-2 Omicron vs Delta variants. Nat. Commun. 2022, 13, 7363. [Google Scholar] [CrossRef]
- Ayoubkhani, D.; Bosworth, M.L.; King, S.; Pouwels, K.B.; Glickman, M.; Nafilyan, V.; Zaccardi, F.; Khunti, K.; Alwan, N.; Walker, A.; et al. Risk of Long COVID in People Infected With Severe Acute Respiratory Syndrome Coronavirus 2 After 2 Doses of a Coronavirus Disease 2019 Vaccine: Community-Based, Matched Cohort Study. Open Forum Infect. Dis. 2022, 9, ofac464. [Google Scholar] [CrossRef]
- Han, E.; Gyöngyösi, M. Long COVID—Eine neue Herausforderung in der Medizin: Fokus auf Schwangerschaft und Stillzeit. J. Gynäkol. Endokrinol. 2023, 33, 7–12. [Google Scholar] [CrossRef]
- Gao, P.; Liu, J.; Liu, M. Effect of COVID-19 vaccines on reducing the risk of long COVID in the real world: A systematic review and meta-analysis. Int. J. Environ. Res. Public. Health 2022, 19, 12422. [Google Scholar] [CrossRef]
- Santos, C.A.D.; Fonseca Filho, G.G.; Alves, M.M.; Macedo, E.Y.L.; Pontes, M.G.A.; Paula, A.P.; Barreto, C.T.R.; Zeneide, F.N.; Nery, A.F.; Freitas, R.A.O.; et al. Maternal and Neonatal Outcomes Associated with Mild COVID-19 Infection in an Obstetric Cohort in Brazil. Am. J. Trop. Med. Hyg. 2022, 107, 1060. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146, Erratum in Nat. Rev. Microbiol 2023, 21, 408. [Google Scholar] [CrossRef]
- Klein, J.; Wood, J.; Jaycox, J.; Lu, P.; Dhodapkar, R.M.; Gehlhausen, J.R.; Tabachnikova, A.; Tabacof, L.; Malik, A.; Kamath, K.; et al. Distinguishing features of Long COVID identified through immune profiling. medRxiv 2022. [Google Scholar] [CrossRef]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef] [PubMed]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.; Burrell, L.; Kent, S.; Dore, G.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Lu, S.; Tang, A.F.; Durstenfeld, M.S.; Ho, H.E.; Goldberg, S.A.; Forman, C.; Munter, S.; Hoh, R.; Tai, V.; et al. Markers of Immune Activation and Inflammation in Individuals With Postacute Sequelae of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Infect. Dis. 2021, 224, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.; Khan, M.A.; Putrino, D.; Woodcock, A.; Kell, D.B.; Pretorius, E. Long COVID: Pathophysiological factors and abnormalities of coagulation. Trends Endocrinol Metab. 2023, 34, 321–344. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881–895. [Google Scholar] [CrossRef]
- Wang, E.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israleow, B.; Wong, P.; et al. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Halpert, G.; Schimke, L.F.; Ostrinski, Y.; Vojdani, A.; Baiocchi, G.C.; Freire, P.P.; Filgueiras, I.S.; Zyskind, I.; Lattin, M.T.; et al. Autoantibodies targeting GPCRs and RAS-related molecules associate with COVID-19 severity. Nat. Commun. 2022, 13, 1220. [Google Scholar] [CrossRef]
- Briquez, P.S.; Rouhani, S.J.; Yu, J.; Pyzer, A.R.; Trujillo, J.; Dugan, H.L.; Stamper, C.T.; Changrob, S.; Sperling, A.I.; Wilson, P.C.; et al. Severe COVID-19 induces autoantibodies against angiotensin II that correlate with blood pressure dysregulation and disease severity. Sci. Adv. 2022, 8, 3777. [Google Scholar] [CrossRef]
- Liotti, F.M.; Menchinelli, G.; Marchetti, S.; Posteraro, B.; Landi, F.; Sanguinetti, M.; Cattani, P. Assessment of SARS-CoV-2 RNA Test Results Among Patients Who Recovered From COVID-19 With Prior Negative Results. JAMA Intern. Med. 2021, 181, 702. [Google Scholar] [CrossRef]
- Eymieux, S.; Uzbekov, R.; Rouillé, Y.; Blanchard, E.; Hourioux, C.; Dubuisson, J.; Belouzard, S.; Roingeard, P. Secretory Vesicles Are the Principal Means of SARS-CoV-2 Egress. Cells 2021, 10, 2047. [Google Scholar] [CrossRef]
- Rosell, A.; Havervall, S.; von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated With Severity and Mortality—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 878–882. [Google Scholar] [CrossRef]
- Ahamed, J.; Laurence, J. Long COVID endotheliopathy: Hypothesized mechanisms and potential therapeutic approaches. J. Clin. Investig. 2022, 132, 161167. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Caruso, D.; Guido, G.; Zerunian, M.; Polidori, T.; Lucertini, E.; Pucciarelli, F.; Polici, M.; Rucci, C.; Bracci, B.; Nicolai, M.; et al. Post-Acute Sequelae of COVID-19 Pneumonia: Six-month Chest CT Follow-up. Radiology 2021, 301, 396–405. [Google Scholar] [CrossRef]
- Cueto-Robledo, G.; Porres-Aguilar, M.; Puebla-Aldama, D.; Barragán-Martínez, M.D.P.; Jurado-Hernández, M.Y.; García-César, M.; Torres Rojas, M.B.T.; García-Treminio, C.; Roldan-Valadez, E. Severe Pulmonary Hypertension: An Important Sequel After Severe Post-Acute COVID-19 Pneumonia. Curr. Probl. Cardiol. 2022, 47, 101004. [Google Scholar] [CrossRef]
- Kubánková, M.; Hohberger, B.; Hoffmanns, J.; Fürst, J.; Herrmann, M.; Guck, J.; Kräter, M. Physical phenotype of blood cells is altered in COVID-19. Biophys. J. 2021, 120, 2838–2847. [Google Scholar] [CrossRef]
- Østergaard, L. SARS CoV-2 related microvascular damage and symptoms during and after COVID-19: Consequences of capillary transit-time changes, tissue hypoxia and inflammation. Physiol. Rep. 2021, 9, e14726. [Google Scholar] [CrossRef]
- Moasefi, N.; Fouladi, M.; Norooznezhad, A.H.; Yarani, R.; Rahmani, A.; Mansouri, K. How could perfluorocarbon affect cytokine storm and angiogenesis in coronavirus disease 2019 (COVID-19): Role of hypoxia-inducible factor 1α. Inflamm. Res. 2021, 70, 749–752. [Google Scholar] [CrossRef]
- Von Meijenfeldt, F.A.; Havervall, S.; Adelmeijer, J.; Lundström, A.; Magnusson, M.; Mackman, N.; Thalin, C.; Lisman, T. Sustained prothrombotic changes in COVID-19 patients 4 months after hospital discharge. Blood Adv. 2021, 5, 756–759. [Google Scholar] [CrossRef]
- Ratchford, S.M.; Stickford, J.L.; Province, V.M.; Stute, N.; Augenreich, M.A.; Koontz, L.K.; Landry, K.B.; Stickford, A. Vascular alterations among young adults with SARS-CoV-2. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Renteria, H.; Travieso, A.; Sagir, A.; Martínez-Gómez, E.; Carrascosa-Granada, A.; Toya, T.; Núñez-Gil, I.; Estrada, V.; Lermani, A.; Escaned, J. In-vivo evidence of systemic endothelial vascular dysfunction in COVID-19. Int. J. Cardiol. 2021, 345, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Santoro, L.; Falsetti, L.; Zaccone, V.; Nesci, A.; Tosato, M.; Giupponi, B.; Savastano, M.C.; Moroncini, G.; Gasbarrini, A.; Landi, F.; et al. Impaired Endothelial Function in Convalescent Phase of COVID-19: A 3 Month Follow Up Observational Prospective Study. J. Clin. Med. 2022, 11, 1774. [Google Scholar] [CrossRef] [PubMed]
- Chioh, F.W.; Fong, S.W.; Young, B.E.; Wu, K.X.; Siau, A.; Krishnan, S.; Chan, Y.H.; Carissimo, G.; Teo, L.l.; Gao, F.; et al. Convalescent COVID-19 patients are susceptible to endothelial dysfunction due to persistent immune activation. eLife 2021, 10, e64909. [Google Scholar] [CrossRef]
- Bermejo-Martin, J.F.; Almansa, R.; Torres, A.; González-Rivera, M.; Kelvin, D.J. COVID-19 as a cardiovascular disease: The potential role of chronic endothelial dysfunction. Cardiovasc. Res. 2020, 116, 132–133. [Google Scholar] [CrossRef]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, 1091, Erratum in BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [Green Version]
- Santoliquido, A.; Porfidia, A.; Nesci, A.; De Matteis, G.; Marrone, G.; Porceddu, E.; Cammà, G.; Giarretta, I.; Fantoni, M.; Landi, F.; et al. Incidence of deep vein thrombosis among non-ICU patients hospitalized for COVID-19 despite pharmacological thromboprophylaxis. J. Thromb. Haemost. 2020, 18, 2358–2363. [Google Scholar] [CrossRef]
- Llitjos, J.; Leclerc, M.; Chochois, C.; Monsallier, J.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef]
- Kamal, M.; Abo Omirah, M.; Hussein, A.; Saeed, H. Assessment and characterisation of post-COVID-19 manifestations. Int. J. Clin. Pract. 2021, 75, e13746. [Google Scholar] [CrossRef]
- Kotecha, T.; Knight, D.S.; Razvi, Y.; Kumar, K.; Vimalesvaran, K.; Thornton, G.; Patel, R.; Chacko, L.; Brown, J.T.; Coyle, C.; et al. Patterns of myocardial injury in recovered troponin-positive COVID-19 patients assessed by cardiovascular magnetic resonance. Eur. Heart J. 2021, 42, 1866–1878. [Google Scholar] [CrossRef]
- Nasserie, T.; Hittle, M.; Goodman, S.N. Assessment of the Frequency and Variety of Persistent Symptoms Among Patients With COVID-19. JAMA Netw. Open. 2021, 4, 2111417. [Google Scholar] [CrossRef]
- Monje, M.; Iwasaki, A. The neurobiology of long COVID. Neuron 2022, 110, 3484–3496. [Google Scholar] [CrossRef]
- Raj, S.R.; Arnold, A.C.; Barboi, A.; Claydon, V.E.; Limberg, J.K.; Lucci, V.E.M.; Numan, M.; Peltier, A.; Snapper, H.; Vernino, S. Long-COVID postural tachycardia syndrome: An American Autonomic Society statement. Clin. Auton. Res. 2021, 31, 365–368. [Google Scholar] [CrossRef]
- Lai, C.C.; Hsu, C.K.; Yen, M.Y.; Lee, P.I.; Ko, W.C.; Hsueh, P.R. Long COVID: An inevitable sequela of SARS-CoV-2 infection. J. Microbiol. Immunol. Infect. 2023, 56, 1–9. [Google Scholar] [CrossRef]
- Yang, T.; Yan, M.Z.; Li, X.; Lau, E.H.Y. Sequelae of COVID-19 among previously hospitalized patients up to 1 year after discharge: A systematic review and meta-analysis. Infection 2022, 50, 1067–1109. [Google Scholar] [CrossRef]
- Leo, F.; Wormanns, D.; Grohé, C. COVID-19 aus Sicht der Pneumologie—Langzeitfolgen und Implikationen für die pneumologische Nachsorge. [COVID-19: A Pneumological Point of View—Long-Term Sequelae of COVID-19—Implications For Follow-up In Respiratory Medicine]. Dtsch. Med. Wochenschr. 2020, 145, 1086–1092. [Google Scholar] [CrossRef]
- Guler, S.A.; Ebner, L.; Aubry-Beigelman, C.; Bridevaux, P.O.; Brutsche, M.; Clarenbach, C.; Garzoni, C.; Geiser, T.K.; Lenoir, A.; Mancinetti, M.; et al. Pulmonary function and radiological features 4 months after COVID-19: First results from the national prospective observational Swiss COVID-19 lung study. Eur. Respir. J. 2021, 57, 2003690. [Google Scholar] [CrossRef]
- Cortés-Telles, A.; López-Romero, S.; Figueroa-Hurtado, E.; Pou-Aguilar, Y.N.; Wong, A.W.; Milne, K.M.; Ryerson, C.J.; Guenette, J.A. Pulmonary function and functional capacity in COVID-19 survivors with persistent dyspnoea. Respir. Physiol. Neurobiol. 2021, 288, 103644. [Google Scholar] [CrossRef]
- Mo, X.; Jian, W.; Su, Z.; Chen, M.; Peng, H.; Peng, P.; Lei, C.; Chen, R.; Zhong, N.; Li, S. Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur. Respir. J. 2020, 55, 2001217. [Google Scholar] [CrossRef]
- Qin, W.; Chen, S.; Zhang, Y.; Dong, F.; Zhang, Z.; Hu, B.; Zhu, Z.; Li, F.; Wang, X.; Wang, Y.; et al. Diffusion capacity abnormalities for carbon monoxide in patients with COVID-19 at 3-month follow-up. Eur. Respir. J. 2021, 58, 2003677. [Google Scholar] [CrossRef]
- Zaccone, V.; Falsetti, L.; Nitti, C.; Gentili, T.; Marchetti, A.; Pier Santelli, M.N.; Sampaolesi, M.; Riccomi, F.; Raponi, A.; Salvi, A. The Prognostic Role of Procalcitonin in Critically Ill Patients Admitted in a Medical Stepdown Unit: A Retrospective Cohort Study. Sci. Rep. 2020, 10, 4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, J.L.; Yang, W.; Ito, K.; Matte, T.D.; Shaman, J.; Kinney, P.L. Seasonal Influenza Infections and Cardiovascular Disease Mortality. JAMA Cardiol. 2016, 1, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, T.Y.; Redwood, S.; Prendergast, B.; Chen, M. Coronaviruses and the cardiovascular system: Acute and long-term implications. Eur. Heart J. 2020, 41, 1798–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef]
- Mahmud, E.; Dauerman, H.L.; Welt, F.G.P.; Messenger, J.C.; Rao, S.V.; Grines, C.; Mattu, A.; Kirtane, A.J.; Jauhar, R.; Meraj, P.; et al. Management of acute myocardial infarction during the COVID-19 pandemic. Catheter. Cardiovasc. Interv. 2020, 96, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.W.; Belin de Chantemèle, E.J.; Weintraub, N.L. Perivascular Adipocytes in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2220–2227. [Google Scholar] [CrossRef]
- Loebel, M.; Grabowski, P.; Heidecke, H.; Bauer, S.; Hanitsch, L.G.; Wittke, K.; Maisel, C.; Reinke, P.; Volk, H.; Fluge, Ø.; et al. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav. Immun. 2016, 52, 32–39. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Von Meijenfeldt, F.A.; Havervall, S.; Adelmeijer, J.; Lundström, A.; Rudberg, A.S.; Magnusson, M.; Mackman, N.; Thalin, C.; Lisman, T. Prothrombotic changes in patients with COVID-19 are associated with disease severity and mortality. Res. Pract. Thromb. Haemost. 2020, 5, 132–141. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, A.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef]
- Laurence, J.; Nuovo, G.; Racine-Brzostek, S.E.; Seshadri, M.; Elhadad, S.; Crowson, A.N.; Mulvey, J.J.; Harp, J.; Ahamed, J.; Magro, C. Premortem Skin Biopsy Assessing Microthrombi, Interferon Type I Antiviral and Regulatory Proteins, and Complement Deposition Correlates with Coronavirus Disease 2019 Clinical Stage. Am. J Pathol. 2022, 192, 1282–1294. [Google Scholar] [CrossRef]
- Wójcik, K.; Bazan-Socha, S.; Celejewska-Wójcik, N.; Górka, K.; Lichołai, S.; Polok, K.; Stachura, T.; Zaręba, L.; Dziedzic, R.; Gradzikiewicz, A.; et al. Decreased protein C activity, lower ADAMTS13 antigen and free protein S levels accompanied by unchanged thrombin generation potential in hospitalized COVID-19 patients. Thromb Res. 2023, 223, 80–86. [Google Scholar] [CrossRef]
- Townsend, L.; Fogarty, H.; Dyer, A.; Martin-Loeches, I.; Bannan, C.; Nadarajan, P.; Bergin, C.; O′Farrelly, C.; Conlon, N.; Bourke, N.M.; et al. Prolonged elevation of D-dimer levels in convalescent COVID-19 patients is independent of the acute phase response. J. Thromb. Haemost. 2021, 19, 1064–1070. [Google Scholar] [CrossRef]
- Giannis, D.; Allen, S.L.; Tsang, J.; Flint, S.; Pinhasov, T.; Williams, S.; Tan, G.; Thakur, R.; Leung, C.; Snyder, M.; et al. Postdischarge thromboembolic outcomes and mortality of hospitalized patients with COVID-19: The CORE-19 registry. Blood 2021, 137, 2838–2847. [Google Scholar] [CrossRef]
- Engelen, M.M.; Vandenbriele, C.; Balthazar, T.; Claeys, E.; Gunst, J.; Guler, I.; Jacquemin, M.; Janssens, S.; Lorent, N.; Liesenborghs, L.; et al. Venous Thromboembolism in Patients Discharged after COVID-19 Hospitalization. Semin. Thromb. Hemost. 2021, 47, 362–371. [Google Scholar] [CrossRef]
- Patell, R.; Bogue, T.; Koshy, A.; Bindal, P.; Merrill, M.; Aird, W.C.; Bauer, K.A.; Zwicker, J.I. Postdischarge thrombosis and hemorrhage in patients with COVID-19. Blood 2020, 136, 1342–1346. [Google Scholar] [CrossRef]
- Taccone, F.S.; Gevenois, P.A.; Peluso, L.; Pletchette, Z.; Lheureux, O.; Brasseur, A.; Garufi, A.; Talamonti, M.; Motte, S.; Nobile, L.; et al. Higher Intensity Thromboprophylaxis Regimens and Pulmonary Embolism in Critically Ill Coronavirus Disease 2019 Patients. Crit. Care Med. 2020, 48, e1087–e1090. [Google Scholar] [CrossRef]
- Arslan, Y.; Yilmaz, G.; Dogan, D.; Hasirci, M.; Cetindogan, H.; Ocal, N.; Savasci, U.; Fidan, G.; Tasci, C. The effectiveness of early anticoagulant treatment in Covid-19 patients. Phlebology 2021, 36, 384–391. [Google Scholar] [CrossRef]
- Rentsch, C.T.; Beckman, J.A.; Tomlinson, L.; Gellad, W.F.; Alcorn, C.; Kidwai-Khan, F.; Skanderson, M.; Brittani, E.; King, J.T., Jr.; Ho, Y.L.; et al. Early initiation of prophylactic anticoagulation for prevention of coronavirus disease 2019 mortality in patients admitted to hospital in the United States: Cohort study. BMJ 2021, 372, n311. [Google Scholar] [CrossRef]
- Ramacciotti, E.; Barile Agati, L.; Calderaro, D.; Aguiar, V.C.R.; Spyropoulos, A.C.; de Oliveira, C.C.C.; Dos Santos, J.L.; Volpiani, G.G.; Sobreira, M.L.; Joviliano, E.E.; et al. Rivaroxaban versus no anticoagulation for post-discharge thromboprophylaxis after hospitalisation for COVID-19 (MICHELLE): An open-label, multicentre, randomised, controlled trial. Lancet 2022, 399, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Moores, L.K.; Tritschler, T.; Brosnahan, S.; Carrier, M.; Collen, J.F.; Doerschug, K.; Holley, A.B.; Jimenez, D.; Le Gal, G.; Rali, P.; et al. Prevention, Diagnosis, and Treatment of VTE in Patients With Coronavirus Disease 2019. Chest 2020, 158, 1143–1163. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A.; Tseng, E.K.; Nieuwlaat, R.; Angchaisuksiri, P.; Blair, C.; Dane, K.; Davila, J.; DeSancho, M.T.; Diuguid, D.; Griffin, D.O.; et al. American Society of Hematology living guidelines on the use of anticoagulation for thromboprophylaxis in patients with COVID-19: July 2021 update on postdischarge thromboprophylaxis. Blood Adv. 2022, 6, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Der Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Spyropoulos, A.C.; Levy, J.H.; Ageno, W.; Connors, J.M.; Hunt, B.J.; Iba, T.; Levi, M.; Samama, C.M.; Thachil, J.; Giannis, D.; et al. Scientific and Standardization Committee communication: Clinical guidance on the diagnosis, prevention, and treatment of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1859–1865. [Google Scholar] [CrossRef]
- Lee, K.W.; Yusof Khan, A.H.K.; Ching, S.M.; Chia, P.K.; Loh, W.C.; Abdul Rashid, A.M.; Baharin, J.; Inche Mat, L.N.; Wan Sulaiman, W.A.; Devaraj, N.K.; et al. Stroke and Novel Coronavirus Infection in Humans: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 579070. [Google Scholar] [CrossRef]
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long covid—Mechanisms, risk factors, and management. BMJ 2021, 374, n1648, Erratum in BMJ. 2021, 374, n1944. [Google Scholar] [CrossRef]
- Lewek, J.; Jatczak-Pawlik, I.; Maciejewski, M.; Jankowski, P.; Banach, M. COVID-19 and cardiovascular complications—The preliminary results of the LATE-COVID study. Arch. Med. Sci. 2021, 17, 818–822. [Google Scholar] [CrossRef]
- Venturelli, S.; Benatti, S.V.; Casati, M.; Binda, F.; Zuglian, G.; Imeri, G.; Conti, C.; Biffi, A.M.; Spada, M.S.; Bondi, E.; et al. Surviving COVID-19 in Bergamo province: A post-acute outpatient re-evaluation. Epidemiol. Infect. 2021, 149, e32. [Google Scholar] [CrossRef]
- Sonnweber, T.; Boehm, A.; Sahanic, S.; Pizzini, A.; Aichner, M.; Sonnweber, B.; Kurz, K.; Koppelstätter, S.; Haschka, D.; Petzer, V.; et al. Persisting alterations of iron homeostasis in COVID-19 are associated with non-resolving lung pathologies and poor patients′ performance: A prospective observational cohort study. Respir. Res. 2020, 21, 276. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoro, L.; Zaccone, V.; Falsetti, L.; Ruggieri, V.; Danese, M.; Miro, C.; Di Giorgio, A.; Nesci, A.; D’Alessandro, A.; Moroncini, G.; et al. Role of Endothelium in Cardiovascular Sequelae of Long COVID. Biomedicines 2023, 11, 2239. https://doi.org/10.3390/biomedicines11082239
Santoro L, Zaccone V, Falsetti L, Ruggieri V, Danese M, Miro C, Di Giorgio A, Nesci A, D’Alessandro A, Moroncini G, et al. Role of Endothelium in Cardiovascular Sequelae of Long COVID. Biomedicines. 2023; 11(8):2239. https://doi.org/10.3390/biomedicines11082239
Chicago/Turabian StyleSantoro, Luca, Vincenzo Zaccone, Lorenzo Falsetti, Vittorio Ruggieri, Martina Danese, Chiara Miro, Angela Di Giorgio, Antonio Nesci, Alessia D’Alessandro, Gianluca Moroncini, and et al. 2023. "Role of Endothelium in Cardiovascular Sequelae of Long COVID" Biomedicines 11, no. 8: 2239. https://doi.org/10.3390/biomedicines11082239