
Hypoxic State of Cells and Immunosenescence: A Focus on the Role of the HIF Signaling Pathway

 , ,
, ,  ,
,
Abstract
:1. Introduction
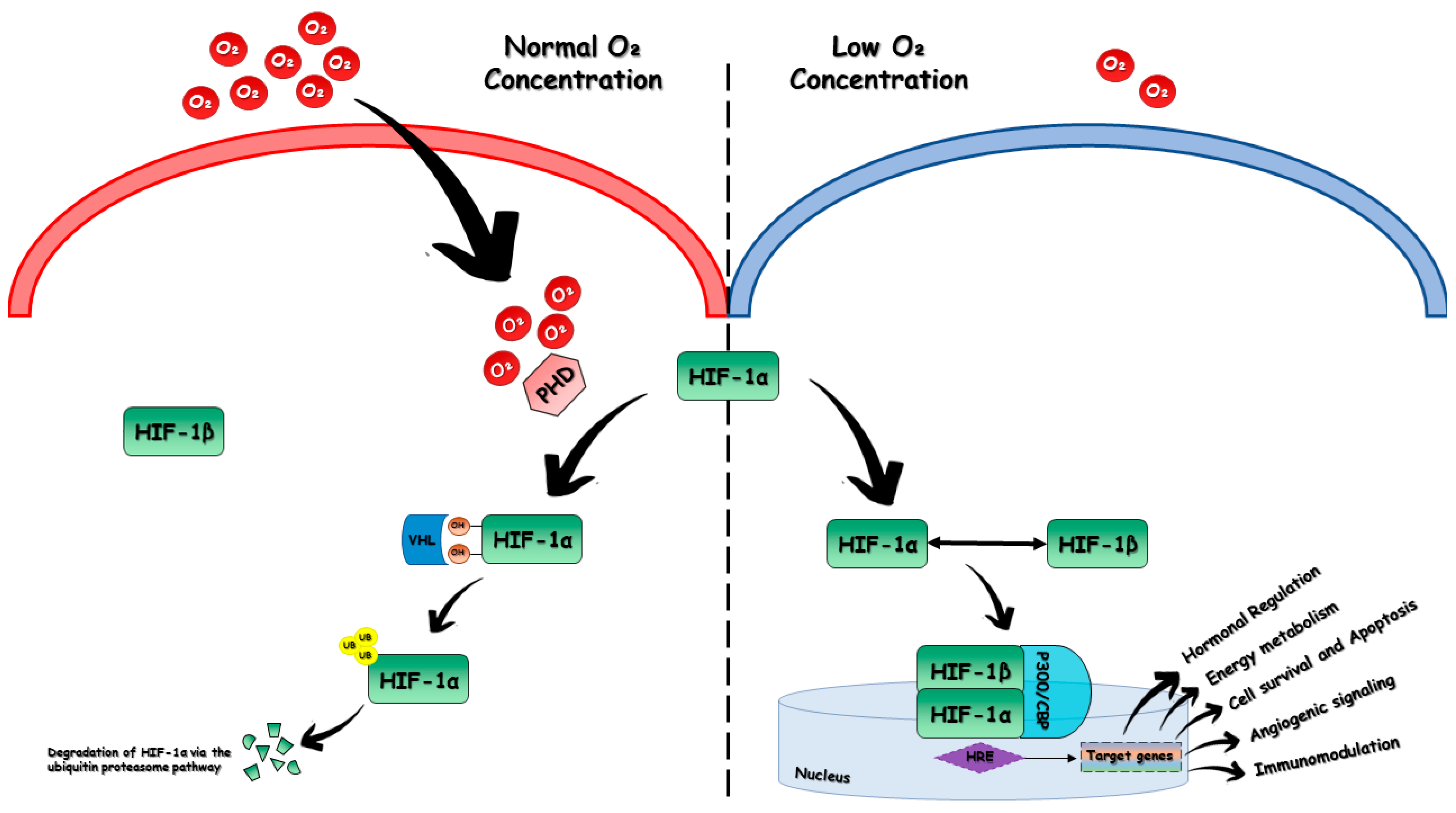
2. HIF Structure and Biology
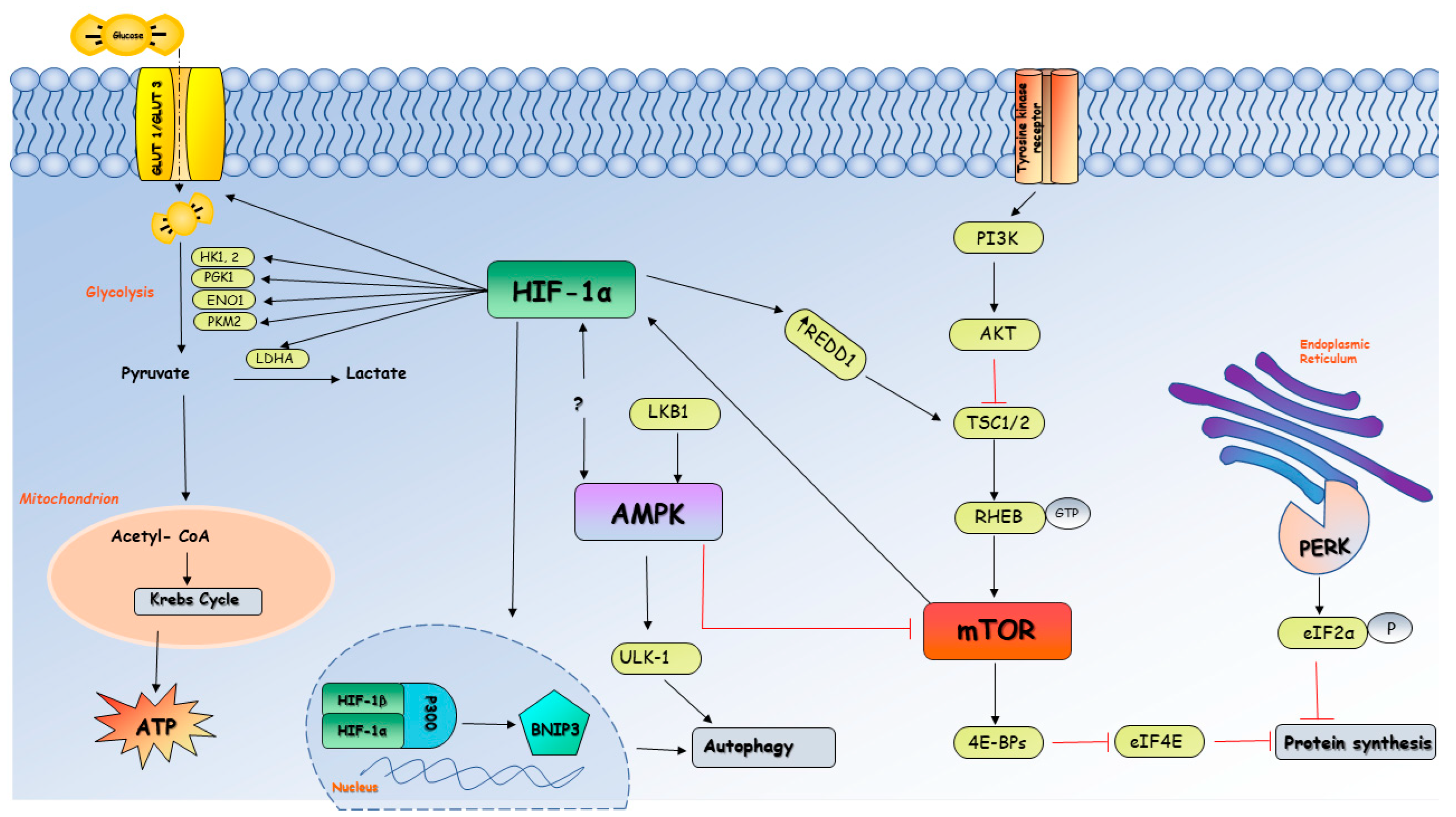
3. Reduced Oxygen Supply and Cellular Energy Status
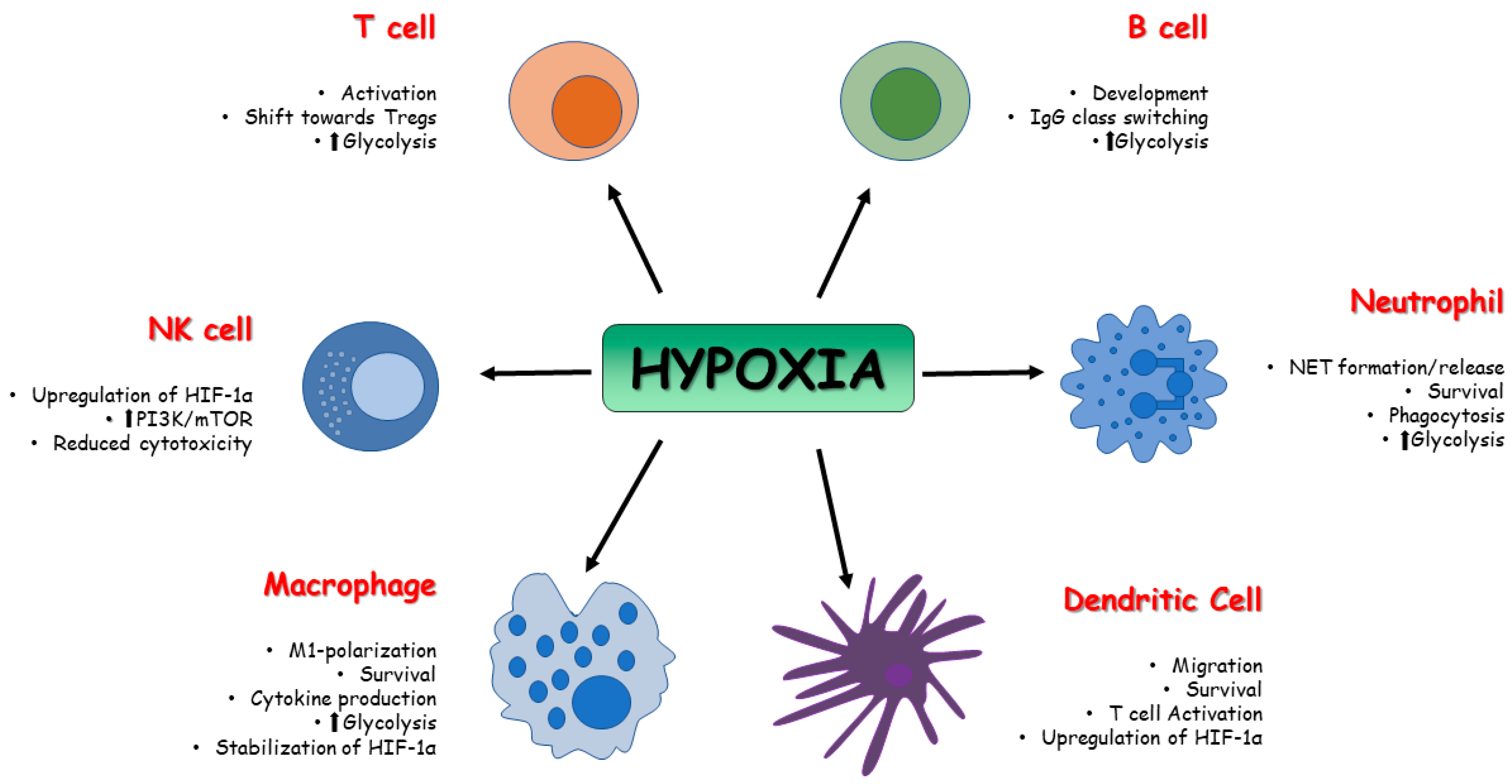
4. HIFs and Immunosenescence
5. Hypoxia, Aging, and Biomarkers
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| AMPK | AMP-activated protein kinase |
| COX | Cytochrome c oxidase |
| DC | Dendritic cells |
| DRP1 | Dynamin-related protein 1 |
| ENO1 | Enolase |
| ETC | Mitochondrial electron transport chain |
| FIH-1 | Factor-inhibiting HIF-1 |
| HIF-1α | Hypoxia-inducible factor 1 subunit α |
| HIF-3α | Hypoxia-inducible factor 3 subunit α |
| HIFs | Hypoxia-inducible factors |
| HIGD1A | Hypoxia-inducible gene domain family member 1A |
| HK1-2 | Hexokinases1-2 |
| ISCU1-2 | Iron-sulfur cluster assembly proteins 1–2 |
| LDHA | Lactate dehydrogenase A |
| LKB1 | Liver kinase B1 |
| LPIN1 | Lipin-1 |
| MFN1 | Mitofusin-1 |
| mTOR | Mammalian target of rapamycin |
| NETs | Neutrophil extracellular traps |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer cells |
| OPA1 | Optic atrophy 1 |
| PD-1 | Programmed death-1 |
| PD-L1 | Programmed death-ligand 1 |
| PERK | Protein kinase R-like ER kinase |
| PGK1 | Phosphoglycerate kinase |
| PHD | Prolyl hydroxylase proteins |
| PMNs | Polymorphonuclear neutrophils |
| ROS | Reactive oxygen species |
| SDHD | Subunit D of succinate dehydrogenase complex |
| TME | Tumor microenvironment |
| VHL | von Hippel–Lindau tumor suppressor protein |
References
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Otero-Albiol, D.; Carnero, A. Cellular senescence or stemness: Hypoxia flips the coin. J. Exp. Clin. Cancer Res. 2021, 40, 243. [Google Scholar] [CrossRef]
- Dzhalilova, D.S.; Makarova, O.V. The Role of Hypoxia-Inducible Factor in the Mechanisms of Aging. Biochemistry 2022, 87, 995–1014. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.P.; Tohari, S.; Ho, A.; Brenner, S.; Venkatesh, B. Characterization of a hypoxia-response element in the Epo locus of the pufferfish, Takifugurubripes. Mar. Genom. 2010, 3, 63–70. [Google Scholar] [CrossRef]
- Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Protein kinases and the hypoxia-inducible factor-1, two switches in angiogenesis. Curr. Pharm. Des. 2003, 9, 531–541. [Google Scholar] [CrossRef]
- Button, E.L.; Bersten, D.C.; Whitelaw, M.L. HIF has Biff—Crosstalk between HIF1a and the family of bHLH/PAS proteins. Exp. Cell Res. 2017, 356, 141–145. [Google Scholar] [CrossRef]
- Graham, A.M.; McCracken, K.G. Convergent evolution on the hypoxia-inducible factor (HIF) pathway genes EGLN1 and EPAS1 in high-altitude ducks. Heredity 2019, 122, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersten, D.C.; Sullivan, A.E.; Peet, D.J.; Whitelaw, M.L. bHLH-PAS proteins in cancer. Nat. Rev. Cancer 2013, 13, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Kunej, T. Integrative Map of HIF1A Regulatory Elements and Variations. Genes 2021, 12, 1526. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Huang, J.; Li, Z.; Gong, Y.; Zou, B.; Liu, X.; Ding, L.; Li, P.; Zhu, Z.; et al. HIF-2α upregulation mediated by hypoxia promotes NAFLD-HCC progression by activating lipid synthesis via the PI3K-AKT-mTOR pathway. Aging 2019, 11, 10839–10860. [Google Scholar] [CrossRef]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [Green Version]
- Tolonen, J.P.; Heikkilä, M.; Malinen, M.; Lee, H.M.; Palvimo, J.J.; Wei, G.H.; Myllyharju, J. A long hypoxia-inducible factor 3 isoform 2 is a transcription activator that regulates erythropoietin. Cell Mol. Life Sci. 2020, 77, 3627–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.H.; Yang, H.; Ivan, M.; Gertler, F.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of an HIF-1alpha -pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhong, Z.F.; Wang, S.P.; Vong, C.T.; Yu, B.; Wang, Y.T. HIF-1: Structure, biology and natural modulators. Chin. J. Nat. Med. 2021, 19, 521–527. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Sim, J.; Cowburn, A.S.; Palazon, A.; Madhu, B.; Tyrakis, P.A.; Macías, D.; Bargiela, D.M.; Pietsch, S.; Gralla, M.; Evans, C.E.; et al. The Factor Inhibiting HIF Asparaginyl Hydroxylase Regulates Oxidative Metabolism and Accelerates Metabolic Adaptation to Hypoxia. Cell Metab. 2018, 27, 898–913.e7. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2022, 13, 50. [Google Scholar] [CrossRef]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 15–22. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. AMPK-mTOR Signaling and Cellular Adaptations in Hypoxia. Int. J. Mol. Sci. 2021, 22, 9765. [Google Scholar] [CrossRef]
- Cooper, C.E. The steady-state kinetics of cytochrome c oxidation by cytochrome oxidase. Biochim. Biophys. Acta 1990, 1017, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Puisségur, M.P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.Z.; Liu, J.; Wang, G.Q.; Pan, H.; Huang, T.Z.; Liu, R.; Zhang, Y. HIG1 domain family member 1A is a crucial regulator of disorders associated with hypoxia. Mitochondrion 2023, 69, 171–182. [Google Scholar] [CrossRef]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Meng, Q.; Sun, Q.; Xu, Z.X.; Zhou, H.; Wang, Y. LKB1 deficiency-induced metabolic reprogramming in tumorigenesis and non-neoplastic diseases. Mol. Metab. 2021, 44, 101131. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective. Oxid. Med. Cell. Longev. 2019, 2019, 8730816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zid, B.M.; Rogers, A.N.; Katewa, S.D.; Vargas, M.A.; Kolipinski, M.C.; Lu, T.A.; Benzer, S.; Kapahi, P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 2009, 139, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Beucken, T.; Magagnin, M.G.; Jutten, B.; Seigneuric, R.; Lambin, P.; Koritzinsky, M.; Wouters, B.G. Translational control is a major contributor to hypoxia induced gene expression. Radiother. Oncol. 2011, 99, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Pierrevelcin, M.; Fuchs, Q.; Lhermitte, B.; Messé, M.; Guérin, E.; Weingertner, N.; Martin, S.; Lelong-Rebel, I.; Nazon, C.; Dontenwill, M.; et al. Focus on Hypoxia-Related Pathways in Pediatric Osteosarcomas and Their Druggability. Cells 2020, 9, 1998. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Quan, C.; He, Y.L.; Cao, Y.; Chen, Y.; Wang, Y.F.; Wu, L.Y. Autophagy regulated by the HIF/REDD1/mTORC1 signaling is progressively increased during erythroid differentiation under hypoxia. Front. Cell Dev. Biol. 2022, 10, 896893. [Google Scholar] [CrossRef]
- Stallone, G.; Infante, B.; Prisciandaro, C.; Grandaliano, G. mTOR and Aging: An Old Fashioned Dress. Int. J. Mol. Sci. 2019, 20, 2774. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Xu, L.; Zhang, X.; Shi, C.; Qiao, S.; Ma, Z.; Yuan, J. Snapshot: Implications for mTOR in Aging-related Ischemia/Reperfusion Injury. Aging Dis. 2019, 10, 116–133. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yu, Y.; Luo, X.; Wang, J.; Lan, X.; Liu, P.; Feng, Y.; Jian, W. Myocardial Ischemia-Reperfusion Injury: Therapeutics from a Mitochondria-Centric Perspective. Cardiology. 2021, 146, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Shiva, N.; Sharma, N.; Kulkarni, Y.A.; Mulay, S.R.; Gaikwad, A.B. Renal ischemia/reperfusion injury: An insight on in vitro and in vivo models. Life Sci. 2020, 256, 117860. [Google Scholar] [CrossRef]
- Chen-Yoshikawa, T.F. Ischemia-Reperfusion Injury in Lung Transplantation. Cells 2021, 10, 1333. [Google Scholar] [CrossRef]
- Hausburg, M.A.; Banton, K.L.; Roman, P.E.; Salgado, F.; Baek, P.; Waxman, M.J.; Tanner, A.; Yoder, J.; Bar-Or, D. Effects of propofol on ischemia-reperfusion and traumatic brain injury. J. Crit. Care 2020, 56, 281–287. [Google Scholar] [CrossRef]
- Saidi, R.F.; Kenari, S.K. Liver ischemia/reperfusion injury: An overview. J. Investig. Surg. 2014, 27, 366–379. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Yiang, G.-T.; Liao, W.-T.; Tsai, A.P.Y.; Cheng, Y.-L.; Cheng, P.-W.; Li, C.-Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Tasset, I.; Arias, E.; Pampliega, O.; Wong, E.; Martinez-Vicente, M.; Cuervo, A.M. Autophagy and the hallmarks of aging. Ageing Res. Rev. 2021, 72, 101468. [Google Scholar] [CrossRef]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef]
- Howell, N.J.; Tennant, D.A. The role of HIFs in ischemia-reperfusion injury. Hypoxia 2014, 2, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.A.; Jung, S.Y.; Yang, K.J.; Im, D.S.; Jeong, K.H.; Park, C.W.; Hwang, H.S. Cilastatin Preconditioning Attenuates Renal Ischemia-Reperfusion Injury via Hypoxia Inducible Factor-1α Activation. Int. J. Mol. Sci. 2020, 21, 3583. [Google Scholar] [CrossRef]
- Bernhardt, W.M.; Ca, V.; Kany, S.; Ju, J.S.; Weidemann, A.; Warnecke, C.; Arend, M.; Klaus, S.; Gu, V.; Amann, K.; et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol. 2006, 17, 1970–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Zhong, H.; Bosch-Marce, M.; Fox-Talbot, K.; Wang, L.; Wei, C.; Trush, M.A.; Semenza, G.L. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1 alpha. Cardiovasc. Res. 2008, 77, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Wu, X.; Dai, Y.; Teng, J.; Fang, Y.; Hu, J.; Zou, J.; Liang, M.; Ding, X. MicroRNA-21 Is Required for Local and Remote Ischemic Preconditioning in Multiple Organ Protection Against Sepsis. Crit. Care Med. 2017, 45, e703–e710. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF-1α in myocardial ischemia-reperfusion injury (Review). Mol. Med. Rep. 2021, 23, 352. [Google Scholar] [CrossRef]
- Lu, N.; Li, X.; Tan, R.; An, J.; Cai, Z.; Hu, X.; Wang, F.; Wang, H.; Lu, C.; Lu, H. HIF-1α/Beclin1-Mediated Autophagy Is Involved in Neuroprotection Induced by Hypoxic Preconditioning. J. Mol. Neurosci. 2018, 66, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Hepper, H.J.; Sieber, C.; Walger, P.; Bahrmann, P.; Singler, K. Infections in the elderly. Crit. Care Clin. 2013, 29, 757–774, Erratum in Crit. Care Clin. 2014, 30, xiii. [Google Scholar] [CrossRef]
- Chong, Y.; Ikematsu, H.; Yamaji, K.; Nishimura, M.; Kashiwagi, S.; Hayashi, J. Age-related accumulation of Ig V(H) gene somatic mutations in peripheral B cells from aged humans. Clin. Exp. Immunol. 2003, 133, 59–66. [Google Scholar] [CrossRef]
- Mahbub, S.; Brubaker, A.L.; Kovacs, E.J. Aging of the Innate Immune System: An Update. Curr. Immunol. Rev. 2011, 7, 104–115. [Google Scholar] [CrossRef]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Mortezaee, K.; Majidpoor, J.; Kharazinejad, E. The impact of hypoxia on tumor-mediated bypassing anti-PD-(L)1 therapy. Biomed. Pharmacother. 2023, 162, 114646. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657, Erratum in Cell 2003, 113, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinska, E.; Stockmann, C. Hypoxia, Metabolism and Immune Cell Function. Biomedicines 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, F.R.; Lewis, A.; Elks, P.M. If it’s not one thing, HIF’s another: Immunoregulation by hypoxia inducible factors in disease. FEBS J. 2020, 287, 3907–3916. [Google Scholar] [CrossRef]
- Sadiku, P.; Walmsley, S.R. Hypoxia and the regulation of myeloid cell metabolic imprinting: Consequences for the inflammatory response. EMBO Rep. 2019, 20, e47388. [Google Scholar] [CrossRef]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Watts, E.R.; Walmsley, S.R. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol. Med. 2019, 25, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.L.; Kelly, B.; Logan, A.; Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [Green Version]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.; Du, J.; Liu, F.; Liu, Y.; Liu, S.; Xie, L.; Wang, D.Y.; Ba, L. Activation of the mTOR/HIF-1α/VEGF axis promotes M1 macrophage polarization in non-eosinophilic chronic rhinosinusitis with nasal polyps. Allergy 2022, 77, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Lewis, C.E. Hypoxia regulates macrophage functions in inflammation. J. Immunol. 2005, 175, 6257–6263. [Google Scholar] [CrossRef] [Green Version]
- Fangradt, M.; Hahne, M.; Gaber, T.; Strehl, C.; Rauch, R.; Hoff, P.; Löhning, M.; Burmester, G.R.; Buttgereit, F. Human monocytes and macrophages differ in their mechanisms of adaptation to hypoxia. Arthritis Res. Ther. 2012, 14, R181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef] [Green Version]
- Naldini, A.; Morena, E.; Pucci, A.; Miglietta, D.; Riboldi, E.; Sozzani, S.; Carraro, F. Hypoxia affects dendritic cell survival: Role of the hypoxia-inducible factor-1α and lipopolysaccharide. J. Cell. Physiol. 2012, 227, 587–595. [Google Scholar] [CrossRef]
- Snyder, J.P.; Amiel, E. Regulation of Dendritic Cell Immune Function and Metabolism by Cellular Nutrient Sensor Mammalian Target of Rapamycin (mTOR). Front. Immunol. 2019, 9, 3145. [Google Scholar] [CrossRef] [Green Version]
- Köhler, T.; Reizis, B.; Johnson, R.S.; Weighardt, H.; Förster, I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur. J. Immunol. 2012, 42, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Block, H.; Rossaint, J.; Zarbock, A. The Fatal Circle of NETs and NET-Associated DAMPs Contributing to Organ Dysfunction. Cells 2022, 11, 1919. [Google Scholar] [CrossRef] [PubMed]
- Willson, J.A.; Arienti, S.; Sadiku, P.; Reyes, L.; Coelho, P.; Morrison, T.; Rinaldi, G.; Dockrell, D.H.; Whyte, M.K.B.; Walmsley, S.R. Neutrophil HIF-1α stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood 2022, 139, 281–286. [Google Scholar] [CrossRef] [PubMed]
- McInturff, A.M.; Cody, M.J.; Elliott, E.A.; Glenn, J.W.; Rowley, J.W.; Rondina, M.T.; Yost, C.C. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood 2012, 120, 3118–3125. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Sun, S.; Chen, Y.; Tian, R.; Chen, E.; Tan, R.; Wang, X.; Liu, Z.; Liu, J.; Qu, H. Immune effects of PI3K/Akt/HIF-1α-regulated glycolysis in polymorphonuclear neutrophils during sepsis. Crit. Care 2022, 26, 29. [Google Scholar] [CrossRef]
- Tang, Y.Y.; Wang, D.C.; Wang, Y.Q.; Huang, A.F.; Xu, W.D. Emerging role of hypoxia-inducible factor-1α in inflammatory autoimmune diseases: A comprehensive review. Front. Immunol. 2023, 13, 1073971. [Google Scholar] [CrossRef]
- Thompson, A.A.; Elks, P.M.; Marriott, H.M.; Eamsamarng, S.; Higgins, K.R.; Lewis, A.; Williams, L.; Parmar, S.; Shaw, G.; McGrath, E.E.; et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood 2014, 123, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.F. Hypoxia and the Tumor Microenvironment. Technol. Cancer Res. Treat. 2021, 20, 15330338211036304. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Fink, T.; Ebbesen, P.; Koppelhus, U.; Zachar, V. Natural killer cell-mediated basal and interferon-enhanced cytotoxicity against liver cancer cells is significantly impaired under in vivo oxygen conditions. Scand. J. Immunol. 2003, 58, 607–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, H.; Potempa, M.; Gotthardt, D.; Lanier, L.L. Cutting Edge: IL-2-Induced Expression of the Amino Acid Transporters SLC1A5 and CD98 Is a Prerequisite for NKG2D-Mediated Activation of Human NK Cells. J. Immunol. 2017, 199, 1967–1972. [Google Scholar] [CrossRef] [Green Version]
- Cluff, E.; Magdaleno, C.C.; Fernandez, E.; House, T.; Swaminathan, S.; Varadaraj, A.; Rajasekaran, N. Hypoxia-inducible factor-1 alpha expression is induced by IL-2 via the PI3K/mTOR pathway in hypoxic NK cells and supports effector functions in NKL cells and ex vivo expanded NK cells. Cancer Immunol. Immunother. 2022, 71, 1989–2005. [Google Scholar] [CrossRef]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef]
- Zheng, X.; Qian, Y.; Fu, B.; Jiao, D.; Jiang, Y.; Chen, P.; Shen, Y.; Zhang, H.; Sun, R.; Tian, Z.; et al. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat. Immunol. 2019, 20, 1656–1667. [Google Scholar] [CrossRef]
- Kasler, H.; Verdin, E. How inflammaging diminishes adaptive immunity. Nat. Aging 2021, 1, 24–25. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 2021, 22, 687–698. [Google Scholar] [CrossRef]
- Gaber, T.; Chen, Y.; Krauß, P.L.; Buttgereit, F. Metabolism of T Lymphocytes in Health and Disease. Int. Rev. Cell Mol. Biol. 2019, 342, 95–148. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Bruzzese, L.; Fromonot, J.; By, Y.; Durand-Gorde, J.M.; Condo, J.; Kipson, N.; Guieu, R.; Fenouillet, E.; Ruf, J. NF-κB enhances hypoxia-driven T-cell immunosuppression via upregulation of adenosine A(2A) receptors. Cell Signal. 2014, 26, 1060–1067. [Google Scholar] [CrossRef]
- Chen, Y.; Gaber, T. Hypoxia/HIF Modulates Immune Responses. Biomedicines 2021, 9, 260. [Google Scholar] [CrossRef]
- Li, L.; Feng, C.; Qin, J.; Li, D.; Liu, M.; Han, S.; Zheng, B. Regulation of humoral immune response by HIF-1α-dependent metabolic reprogramming of the germinal center reaction. Cell Immunol. 2021, 367, 104409. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Rudemiller, N.P.; Crowley, S.D. The role of chemokines in hypertension and consequent target organ damage. Pharmacol. Res. 2017, 119, 404–411. [Google Scholar] [CrossRef]
- Imanishi, M.; Tomita, S.; Ishizawa, K.; Kihira, Y.; Ueno, M.; Izawa-Ishizawa, Y.; Ikeda, Y.; Yamano, N.; Tsuchiya, K.; Tamaki, T. Smooth muscle cell-specific Hif-1α deficiency suppresses angiotensin II-induced vascular remodelling in mice. Cardiovasc. Res. 2014, 102, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; Wei, M.; Jiao, S.; Song, Y.; Wang, X.; Xie, G.; Taranto, J.; Liu, Y.; Duan, Y.; Yu, B.; et al. Hypoxia inducible factor 1α in vascular smooth muscle cells promotes angiotensin II-induced vascular remodeling via activation of CCL7-mediated macrophage recruitment. Cell Death Dis. 2019, 10, 544. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Damiani, E.; Brugè, F.; Cirilli, I.; Marcheggiani, F.; Olivieri, F.; Armeni, T.; Cianfruglia, L.; Giuliani, A.; Orlando, P.; Tiano, L. Modulation of Oxidative Status by Normoxia and Hypoxia on Cultures of Human Dermal Fibroblasts: How Does It Affect Cell Aging? Oxid. Med. Cell Longev. 2018, 2018, 5469159. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Gonzalez-Freire, M.; Fabbri, E.; Simonsick, E.; Tanaka, T.; Moore, Z.; Salimi, S.; Sierra, F.; de Cabo, R. Measuring biological aging in humans: A quest. Aging Cell 2020, 19, e13080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moaddel, R.; Ubaida-Mohien, C.; Tanaka, T.; Lyashkov, A.; Basisty, N.; Schilling, B.; Semba, R.D.; Franceschi, C.; Gorospe, M.; Ferrucci, L. Proteomics in aging research: A roadmap to clinical, translational research. Aging Cell 2021, 20, e13325. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Biomarker | Function | In Hypoxic Cells |
|---|---|---|
| β-galactosidase | Lysosomal hydrolase | Reduced expression |
| p16 | Tumor suppressor gene | Reduced expression |
| CCL7 (chemokine C-C motif ligand 7) | Chemokine involved in monocyte recruitment | Increased expression |
| IGF1 (insulin-like growth factor-1) | Growth factor | Increased expression |
| AGEs (advanced glycation end- products) | Molecules involved in vascular complications | Increased expression |
| RAGEs (advanced glycation end-product receptors) | Molecules involved in vascular complications | Increased expression |
| FOXO (Forkhead box O3) | Transcription factors | Increased expression |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troise, D.; Infante, B.; Mercuri, S.; Netti, G.S.; Ranieri, E.; Gesualdo, L.; Stallone, G.; Pontrelli, P. Hypoxic State of Cells and Immunosenescence: A Focus on the Role of the HIF Signaling Pathway. Biomedicines 2023, 11, 2163. https://doi.org/10.3390/biomedicines11082163
Troise D, Infante B, Mercuri S, Netti GS, Ranieri E, Gesualdo L, Stallone G, Pontrelli P. Hypoxic State of Cells and Immunosenescence: A Focus on the Role of the HIF Signaling Pathway. Biomedicines. 2023; 11(8):2163. https://doi.org/10.3390/biomedicines11082163
Chicago/Turabian StyleTroise, Dario, Barbara Infante, Silvia Mercuri, Giuseppe Stefano Netti, Elena Ranieri, Loreto Gesualdo, Giovanni Stallone, and Paola Pontrelli. 2023. "Hypoxic State of Cells and Immunosenescence: A Focus on the Role of the HIF Signaling Pathway" Biomedicines 11, no. 8: 2163. https://doi.org/10.3390/biomedicines11082163