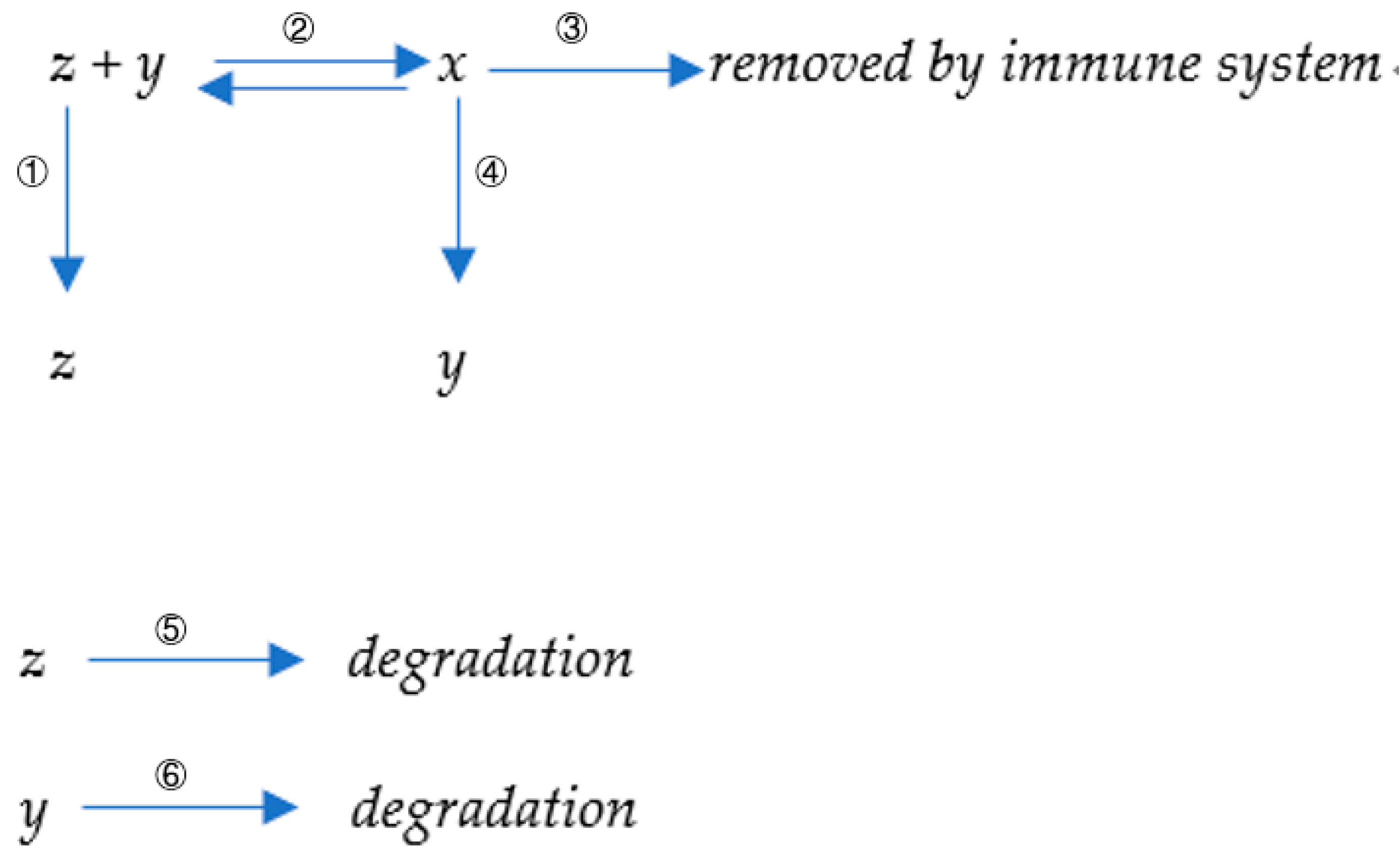
3.1. Possible Physical Mechanisms of Clonal Deletion
Clonal deletion of B-cells is a critical regulatory process in the immune system, aimed at eliminating potentially pathogenic B-cells to maintain immune balance and self-tolerance. This process is referred to as B-cell negative selection.
In the bone marrow, hematopoietic stem cells undergo a series of differentiation stages to generate mature B-cells. During this process, B-cells undergo scrutiny based on their reactivity to self-antigens and foreign antigens, selectively retaining cells with a normal immune function that do not pose a risk of self-damage. Specifically, clonal deletion involves two main mechanisms: self-antigen recognition and immune regulatory signaling.
Self-antigen recognition refers to the binding of antigens by the B-cell receptor (BCR) present on the surface of B-cells, leading to an immune response. In early development, B-cells in the bone marrow encounter a variety of self-antigens and employ mechanisms of self-antigen recognition to determine their interaction. If the affinity between a B-cell’s BCR and self-antigens is excessively high, resulting in hyperactive immune reactions, it may lead to autoimmune diseases. To prevent such scenarios, these overly reactive B-cells undergo negative selection. During negative selection, multiple regulatory mechanisms are initiated to eliminate B-cells with potential harm. One mechanism involves exposing these B-cells to high concentrations of self-antigens, inducing them towards apoptotic (cell death) pathways. Additionally, immune regulatory cells such as regulatory T-cells also participate by secreting regulatory cytokines to trigger B-cell apoptosis or restrict their survival and proliferation capabilities, thereby suppressing the expansion of abnormal clones.
Our model proposes a possible new mechanism for clonal deletion. The fate of B-cells primarily depends on two aspects: the binding affinity between their encoded antibodies and the corresponding antigens, and the stimulatory capacity of the resulting peptides from antigen cleavage on the corresponding helper T-cells.
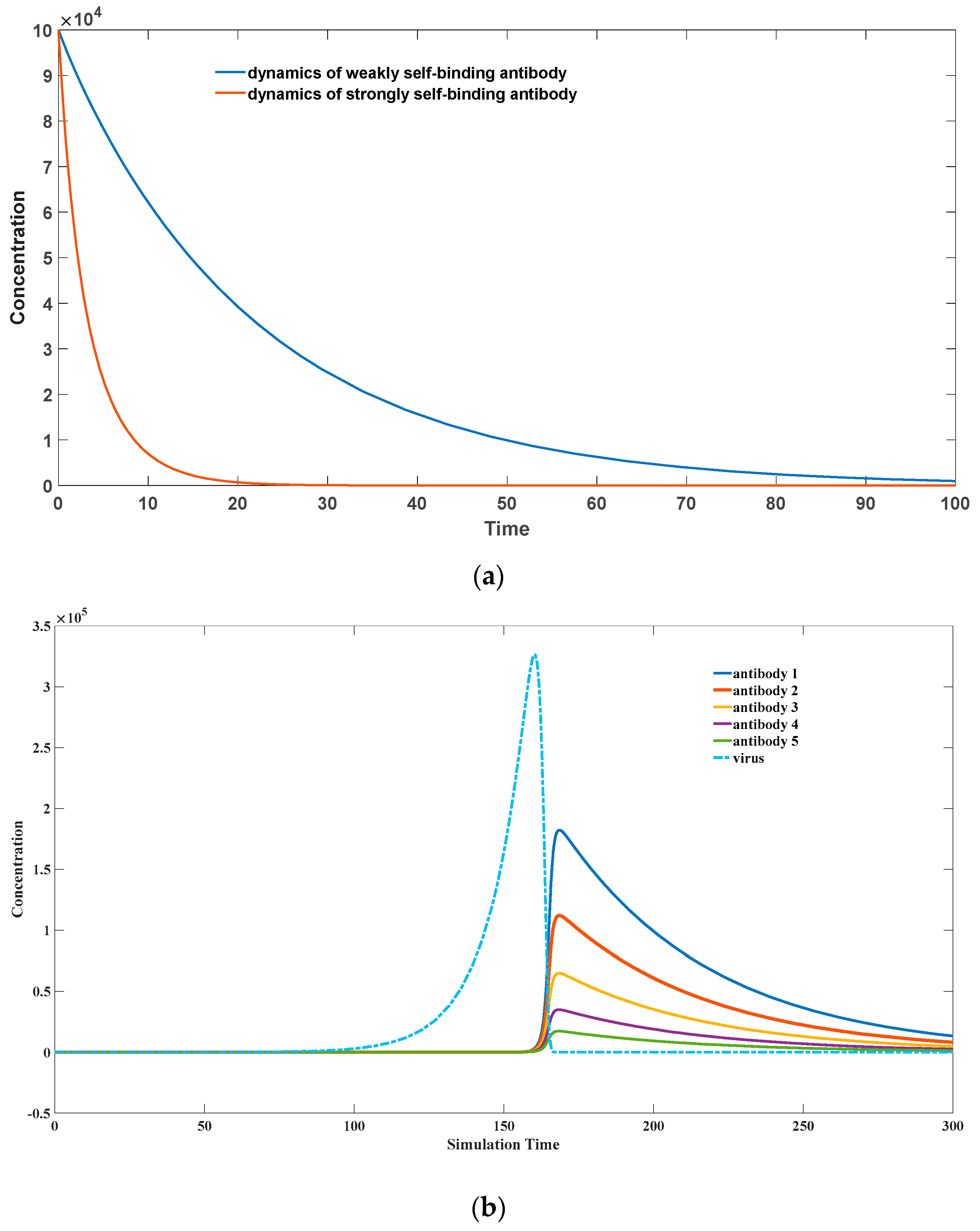
For exogenous antigens, the peptide sequences formed after antigen cleavage often possess strong stimulating effects on Th cells. This stimulation prompts T-cells to secrete a large amount of growth factors, which in turn promotes the division and proliferation of the corresponding Th cells. As B-cells are spatially linked to these Th cells, the growth factors also strongly stimulate the proliferation of B-cells, leading to the amplification of the corresponding antibodies. Although antigen-bound B-cells can be easily recognized and eliminated by other immune cells such as NK cells or the complement system, the proliferative effect of Th cells surpasses the rate of elimination. Hence, the overall dynamics still exhibit a rapid expansion trend, as depicted in
Figure 3a.
Specifically, when the peptide’s stimulatory effect on Th cells is weak (k4 = 0.1) for self-antigenic substances, high-affinity antibodies (k2 = 10−8) are cleared more rapidly than low-affinity antibodies (k2 = 10−9). This is because high-affinity antibodies more readily form antigen–antibody complexes with self-antigens, leading to their further clearance by the immune system, accompanied by the elimination of the corresponding B-cells. For self-antigenic substances, their peptide sequences generated after antigen cleavage have a very weak stimulatory effect on Th cells, thus are unable to induce Th cells to secrete a large amount of growth factors for proliferation. However, due to the high binding affinity between the antibody and the antigen, the corresponding B-cells quickly cover the antigenic substances, accelerating their apoptosis. At this stage, there is also binding between these B-cells and Th cells, causing the physical co-elimination of both cell types. The overall dynamics demonstrate a rapid deletion trend of antibodies.
Distinct from pathogenic microorganisms, self-antigenic substances lack the ability to undergo rapid expansion and are not easily eliminated in a short period of time. Their concentration can remain relatively stable over an extended period. Therefore, in our simulation, we consider their concentration to be constant. Conversely, as shown in
Figure 3b, for exogenous microbial infections, the higher the binding affinity of the antibodies, the faster they proliferate. This ensures the rapid selection and proliferation of high-affinity antibodies by our immune system.
Additionally, we observe that antibodies with high self-binding activity undergo rapid deletion. Many experimental studies have reported that clonal deletion is not a complete elimination of antibodies with strong self-binding activity, especially during the early stages of B-cell development [
27,
28,
29]. Clonal deletion is thus considered to be a progressive process. Therefore, we lean towards the theory that the dominant factor driving clonal deletion are the antibody dynamics reflected in this model, rather than additional special mechanisms to handle B-cells with high self-binding activity.
The choice of parameters has a profound impact on the simulation outcomes. Typically, parameter values are determined through literature mining or experimental fitting. Regrettably, specific experimental data pertaining to the clonal deletion of B-cells in the context of our model remains elusive, thus diminishing the model’s veracity. Nevertheless, an advantage of our model is its qualitative explication of certain phenomena, wherein consistent trends emerge within a given parameter range.
Regarding
Figure 3a, the initial concentration of antigen–antibody complexes (denoted as
x(0)) is assumed to be zero. The initial concentration of antibodies (denoted as
y(0)) is approximately 10
5, while the initial concentration of endogenous antigenic material (denoted as
z(0)) is approximately 3 × 10
7. These initial parameter values draw inspiration from our prior publications [
24,
30]. Importantly, the overall trend exhibits low sensitivity to the precise selection of initial concentrations. Specifically, when the initial antibody concentration (
y(0)) resides in the order of 10
5 and the initial endogenous antigenic material concentration (
z(0)) resides in the order of 10
7, a consistent trend emerges whereby antibodies exhibiting strong affinity for self-antigens are promptly eliminated. For instance, when contemplating somatic hypermutation in somatic cells,
Figure 4 and
Figure 5 stipulate a selection of
z(0) at 2 × 10
6. Similar principles guide the selection of other parameters. Employing available data on the kinetics of antigen–antibody binding, the dissociation coefficient
assumes an extremely minute value, namely 10
−18. Correspondingly, the forward binding coefficient
is constrained to the range elucidated in
Section 2.2, spanning from 10
−5 to 10
−13, based on binding affinity considerations and the equilibrium constant
Kd. A larger value signifies a heightened binding affinity. In our scenario, a value of 10
−8 is elected to denote robustly binding antibodies, while 10
−9 signifies weakly binding antibodies in
Figure 3a. The parameter
captures the rate at which antigen–antibody complexes are cleared. Its value ought to reside between 0 and 1, with larger values indicating swifter clearance rates. Notably, this rate must significantly exceed that of natural antibody decay. Consequently, we adopt a value of 0.02 for the natural decay rate of antibodies
, while the clearance rate of antigen–antibody complexes assumes a value of 0.5. The parameter
depicts the stimulating effect of antigen–antibody complexes on antibody regeneration. Herein, a diminutive value of 0.1 is chosen as the coefficient governing self-antigen–antibody complex-induced antibody regeneration. For exogenous antigens, this stimulation coefficient should be substantially augmented.
Figure 3.
(
a) A possible scenario of clonal deletion. The parameter sets we used are:
z(0) = 3 × 10
7,
= 0,
= 10
5,
= 10
−8
for a strongly binding antibody,
= 10
−9 for a weakly binding antibody,
= 10
−18,
= 0.5,
= 0.1,
= 0.02. (
b) Selection of good binding antibodies toward pathogen infection (Adopted from our previous publication [
24]). The parameter sets we used are:
x(0) = 0,
y(0) = 1,
z(0) = 1,
w = 1,
= 10
−5,
= 10
−14,
= 1,
= 2,
= 0.02,
= 0.02, for antibody 1;
= 9 × 10
−6,
= 9 × 10
−15 for antibody 2;
= 8 × 10
−6,
= 8 × 10
−15 for antibody 3;
= 7 × 10
−6,
= 7 × 10
−15 for antibody 4;
= 6 × 10
−6,
= 6 × 10
−15 for antibody 5.
Figure 3.
(
a) A possible scenario of clonal deletion. The parameter sets we used are:
z(0) = 3 × 10
7,
= 0,
= 10
5,
= 10
−8
for a strongly binding antibody,
= 10
−9 for a weakly binding antibody,
= 10
−18,
= 0.5,
= 0.1,
= 0.02. (
b) Selection of good binding antibodies toward pathogen infection (Adopted from our previous publication [
24]). The parameter sets we used are:
x(0) = 0,
y(0) = 1,
z(0) = 1,
w = 1,
= 10
−5,
= 10
−14,
= 1,
= 2,
= 0.02,
= 0.02, for antibody 1;
= 9 × 10
−6,
= 9 × 10
−15 for antibody 2;
= 8 × 10
−6,
= 8 × 10
−15 for antibody 3;
= 7 × 10
−6,
= 7 × 10
−15 for antibody 4;
= 6 × 10
−6,
= 6 × 10
−15 for antibody 5.
3.2. Somatic Hypermutation Accelerates Self-Immune Response Elimination
Somatic hypermutation (SHM) is a pivotal process occurring in B-cells during their maturation within the germinal centers of secondary lymphoid organs, including the spleen and lymph nodes. It plays a fundamental role in generating high-affinity antibodies and promoting immune responses.
The primary outcome of somatic hypermutation is the induction of point mutations within the variable region of immunoglobulin genes encoding the antigen-binding site of antibodies. This diversifies the repertoire of antibodies generated by B-cells, enabling recognition and binding to a wide range of antigens with enhanced specificity.
Somatic hypermutation involves the activation-induced cytidine deaminase (AID) enzyme, which targets the variable region of immunoglobulin genes. AID converts cytosine residues in the DNA sequence to uracil through deamination [
31,
32]. The presence of uracil triggers DNA repair mechanisms such as base excision repair (BER) and mismatch repair (MMR), which introduce random nucleotide substitutions during the repair process. These substitutions contribute to the generation of a diverse set of antibody variants.
The introduction of somatic mutations creates a pool of B-cells with varying affinities for the antigen. Through a process known as affinity maturation, B-cells with higher-affinity antibodies are preferentially selected for survival and proliferation, while those with lower-affinity or non-functional antibodies undergo apoptosis. During affinity maturation, B-cells presenting antibodies with greater affinity for the antigen are more likely to receive survival signals from T follicular helper (Tfh) cells and follicular dendritic cells [
23]. These cells provide essential survival and proliferation signals, including cytokines and antigen presentation, respectively, ensuring the expansion and persistence of B-cell clones producing high-affinity antibodies. By iteratively undergoing rounds of somatic hypermutation, selection, and clonal expansion, the immune system progressively enhances its capacity to produce antibodies with increasing affinity for the antigen. This iterative process allows B-cells to finely tune their immune responses and generate highly specific and effective antibodies against invading pathogens.
It is important to note that somatic hypermutation is tightly regulated, as excessive or uncontrolled mutations can lead to the production of autoantibodies, contributing to the development of autoimmune diseases. Consequently, multiple mechanisms exist to prevent the generation of self-reactive antibodies, including clonal deletion, receptor editing, and regulatory T-cell control. Somatic hypermutation represents a critical mechanism in humoral immune responses. It introduces random nucleotide substitutions within immunoglobulin genes, resulting in the generation of a diverse antibody repertoire. Through selection and clonal expansion, B-cells producing high-affinity antibodies are favored, leading to a more efficient immune response [
33,
34,
35]. However, the precise regulation of somatic hypermutation is necessary to prevent deleterious autoimmune reactions.
In recent years, it has been discovered that somatic hypermutation, a type of event involving extensive genetic recombination in immune cells, occurs continuously and differs from germ-line genetic recombination. Even mature B-cells, when proliferating and generating offspring, display antibody sequences that are different from the parental generation [
36,
37,
38]. This ongoing somatic hypermutation not only increases antibody diversity to combat pathogen infections but also plays a role in rapidly eliminating self-immune reactions. Experimental evidence has shown that mice lacking somatic hypermutation exhibit strong long-term autoimmune responses when injected with autoreactive antibodies produced by B-cells. In contrast, mice with normal somatic hypermutation capabilities can swiftly suppress these intense self-immune reactions [
39]. This indicates the crucial role of somatic hypermutation in maintaining self-tolerance.
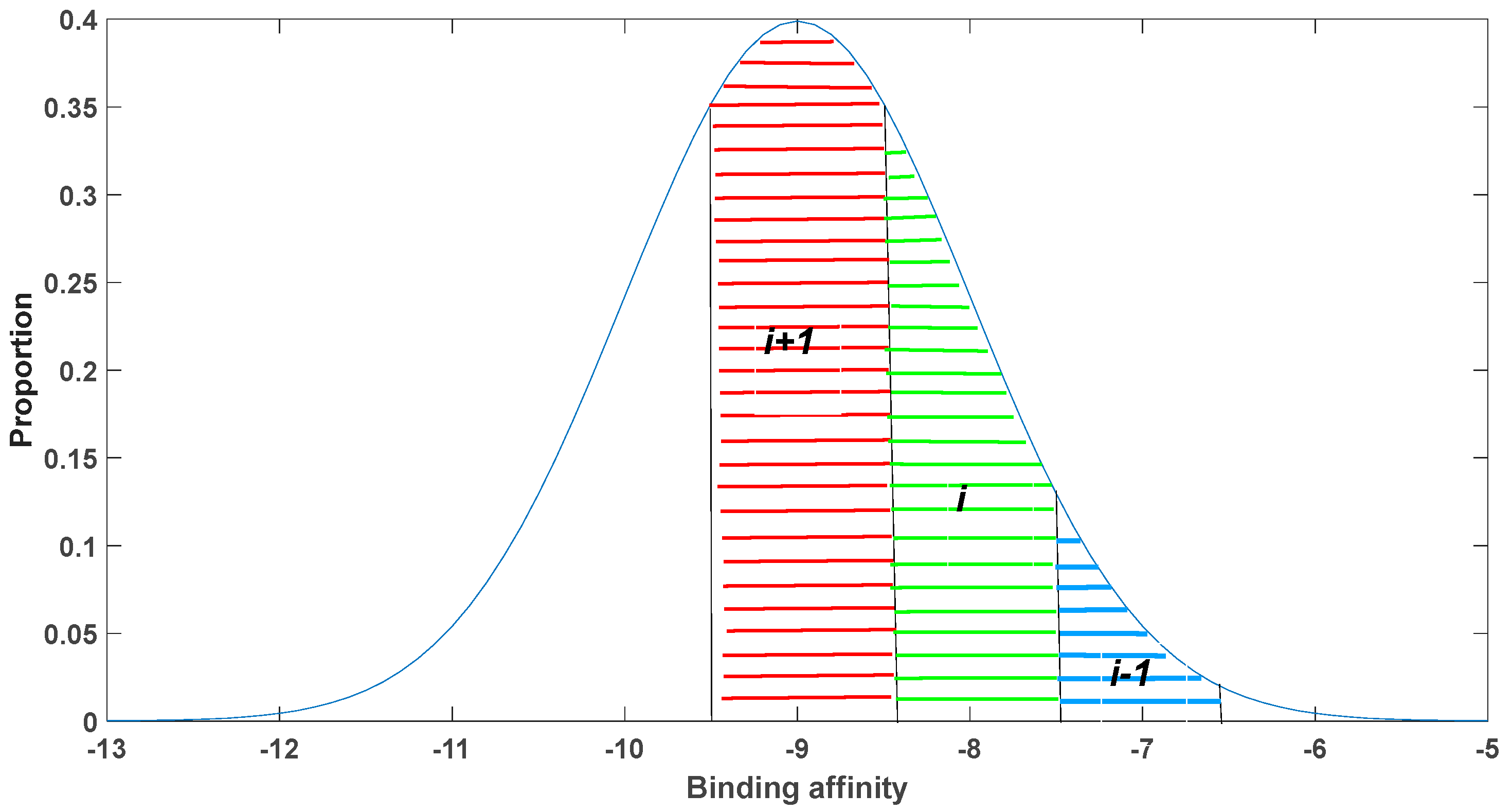
Our model confirms the positive influence of somatic hypermutation in preserving self-tolerance. To construct this model, we initially developed a model to assess the impact of mutations on binding affinity. Accurate calculation of the effects of mutations on binding affinity can be obtained by statistically analyzing the changes in binding affinity caused by numerous point mutations. Such statistics can be derived from the experimental data or estimated by computational methods based on the calculated binding energy after mutation. For simplicity, in our study, we utilized an existing database and roughly estimated that the binding coefficients between antigens and antibodies follow a normal distribution with a mean of −9 and a variance of 1. This distribution assumes that most mutations tend to move towards moderate binding affinities. When the binding affinity is stronger than the average value, the probability of mutations leading to even stronger binding gradually decreases. Conversely, when the binding affinity is weaker than the average value, the probability of mutations resulting in stronger binding increases gradually.
We investigated the reciprocal conversion and kinetic characteristics of antibodies with different binding affinities using a discrete approach. In our model, as described in
Section 2.2, we examined the dynamics of antibody populations based on their binding affinities. From
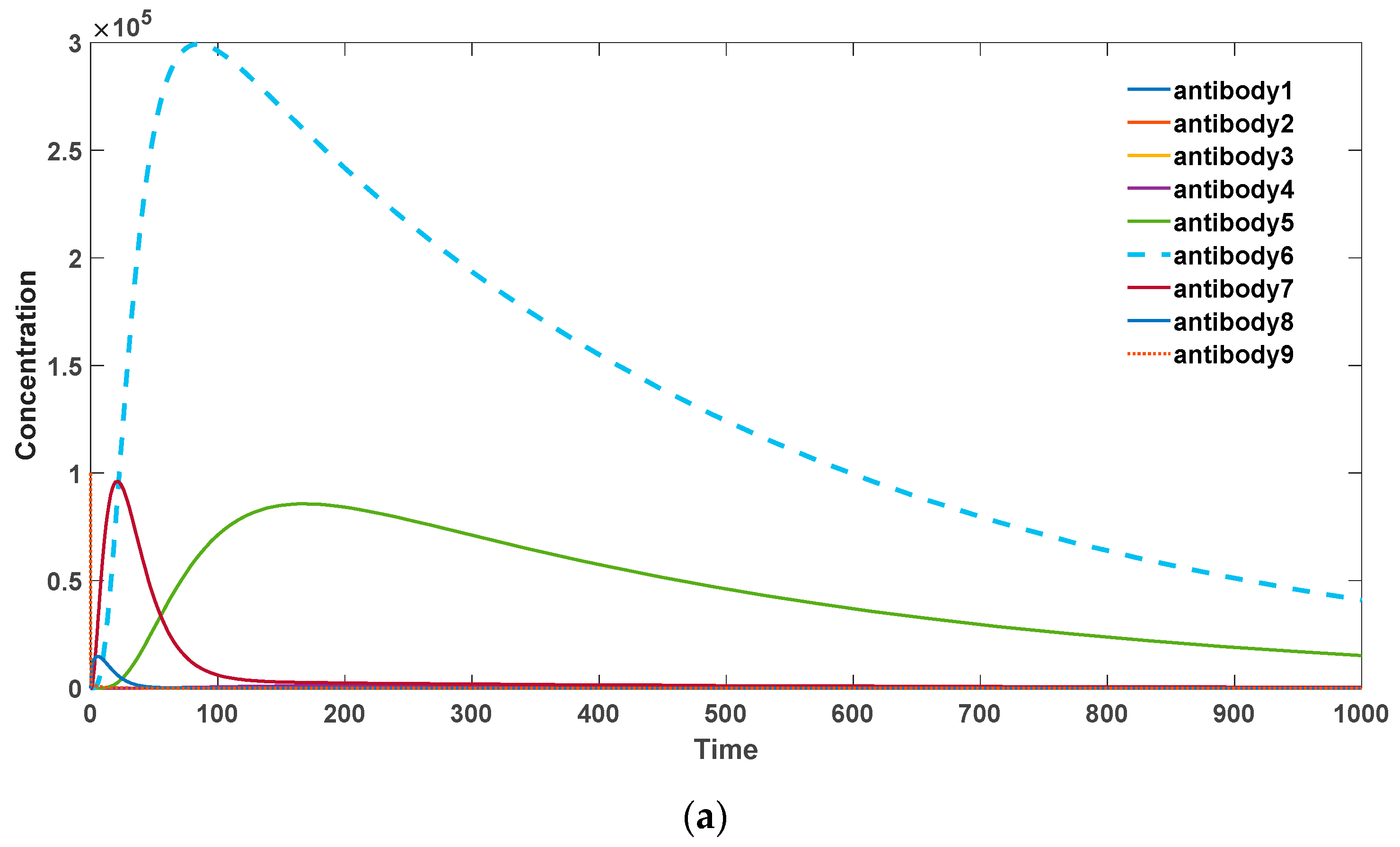
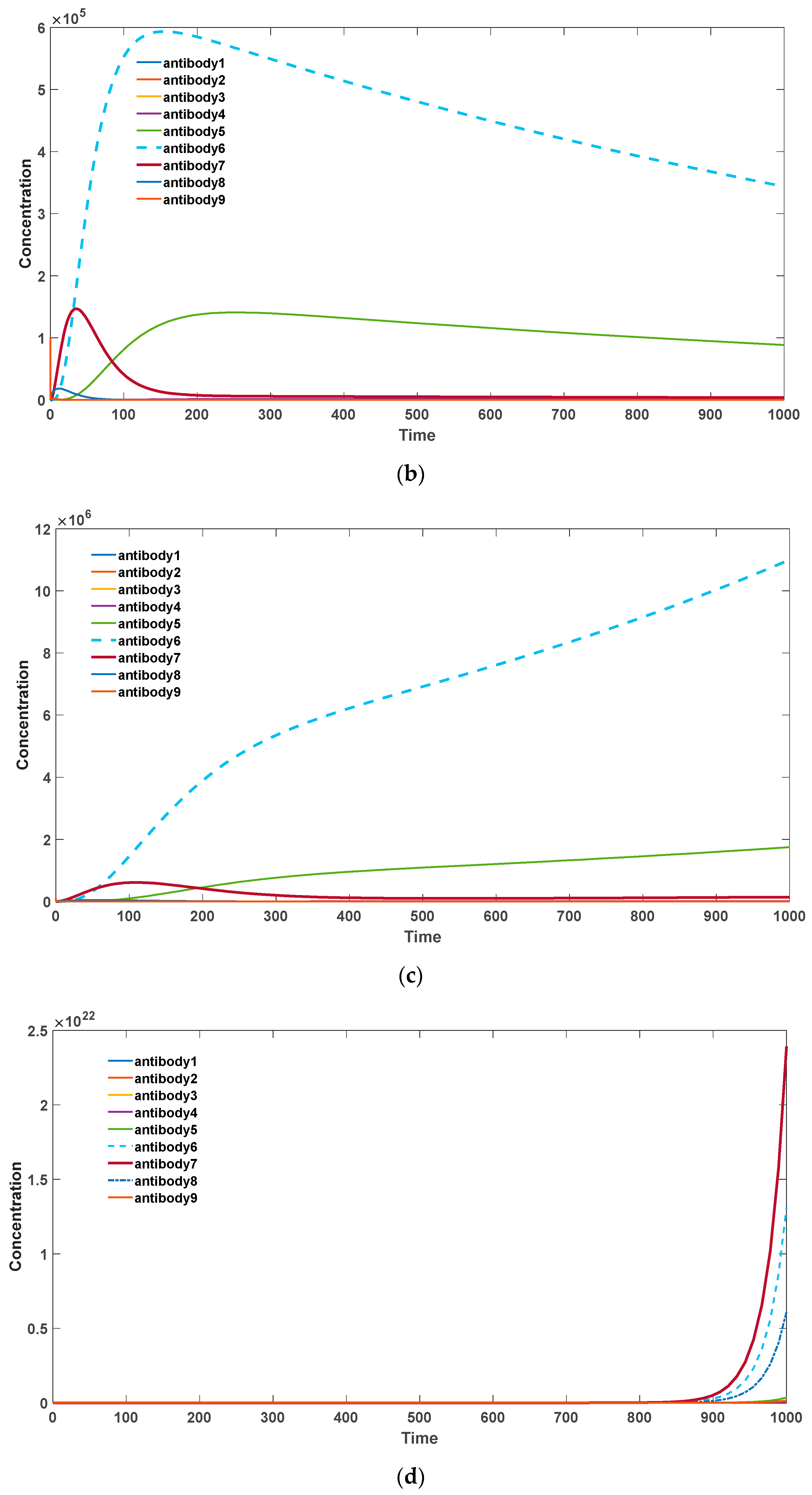
Figure 4, it can be observed that when there is a strong self-binding antibody present, somatic hypermutation accelerates the elimination of these self-binding antibodies. In this scenario, mutations tend to drive the antibodies towards lower binding affinities.
Specifically, as the probability of somatic hypermutation decreases, antibodies with higher binding activities (antibodies 6, 7, 8) undergo substantial proliferation, leading to an overall increase in the antibody levels. Consequently, this may result in the occurrence of severe autoimmune diseases. In the presence of significant somatic hypermutation, as shown in
Figure 4a, antibodies with various binding activities eventually diminish. This represents the gradual disappearance of inflammatory reactions associated with autoimmune responses, which is consistent with experimental evidence. Additionally, the total quantity of antibodies declines more rapidly compared to the absence of somatic hypermutation.
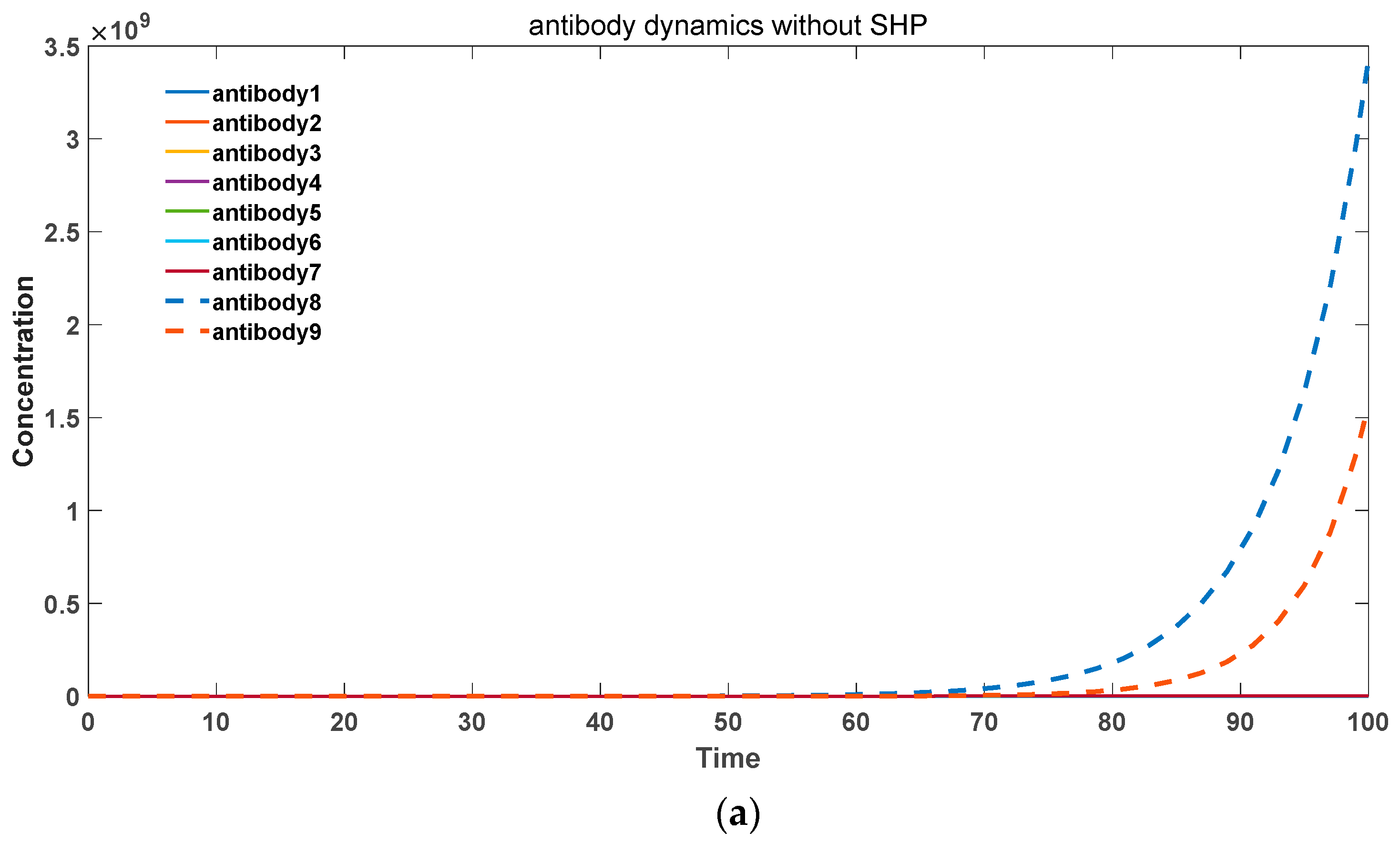
In contrast, for exogenous pathogenic microbial antigens, somatic hypermutation accelerates the generation of a large quantity of high-affinity antibodies to combat infections. This scenario is depicted in
Figure 5. Specifically, in
Figure 5a, for the low binding activity antibody 1 (
k2 = 10–13), without somatic hypermutation, it is unable to undergo amplification and generate more high-affinity antibodies. Moreover, it fails to achieve rapid proliferation and gradually becomes eliminated. In
Figure 5b, in the presence of somatic hypermutation, the low activity antibody 1 can evolve into various high-affinity antibodies and undergo rapid expansion. Therefore, somatic hypermutation plays a crucial and positive role in the immune system’s response to exogenous microbial infections.
The main cause of this distinction lies in the difference in the peptide sequences presented by antigen-presenting cells (APCs). Exogenous peptide sequences often elicit strong T-helper cell responses, whereas endogenous self-peptide sequences typically induce weaker T-helper cell responses. In our model, this distinction is mainly manifested by the different values of k4.
Figure 4.
(a) Antibody dynamics with SHM rate equal to 0.4. (b) Antibody dynamics with SHM rate equal to 0.3. (c)
Antibody dynamics with SHM rate equal to 0.2. (d) Antibody dynamics with SHM rate equal to 0.1. The parameter sets we used are: z(0) = 2 × 106, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0,
= 0, = 0, = 105,
= 10−13, = 10−12,
= 10−11,
= 10−10, = 10−9,
= 10−8, = 10−7,
= 10−6,
= 10−5,
= 10−18,
= 0.5, = 0.6, = 0.02, p
= 0.4 in (a), p = 0.3 in (b), p = 0.2 in (c), p = 0.1 in (d).
Figure 4.
(a) Antibody dynamics with SHM rate equal to 0.4. (b) Antibody dynamics with SHM rate equal to 0.3. (c)
Antibody dynamics with SHM rate equal to 0.2. (d) Antibody dynamics with SHM rate equal to 0.1. The parameter sets we used are: z(0) = 2 × 106, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0,
= 0, = 0, = 105,
= 10−13, = 10−12,
= 10−11,
= 10−10, = 10−9,
= 10−8, = 10−7,
= 10−6,
= 10−5,
= 10−18,
= 0.5, = 0.6, = 0.02, p
= 0.4 in (a), p = 0.3 in (b), p = 0.2 in (c), p = 0.1 in (d).
Figure 5.
(a) Antibody dynamics without SHM in foreign antigen environments. (b) Antibody dynamics with SHM rate equal to 0.5 in foreign antigen environments. The parameter sets we used are: z(0) = 2 × 106,
= 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 105,
= 10−13,
= 10−12,
= 10−11,
= 10−10,
= 10−9,
= 10−8,
= 10−7, = 10−6,
= 10−5,
= 10−18, = 0.5, = 1.1, =
0.02, p = 0 in (a), p = 0.5 in (b).
Figure 5.
(a) Antibody dynamics without SHM in foreign antigen environments. (b) Antibody dynamics with SHM rate equal to 0.5 in foreign antigen environments. The parameter sets we used are: z(0) = 2 × 106,
= 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 0, = 105,
= 10−13,
= 10−12,
= 10−11,
= 10−10,
= 10−9,
= 10−8,
= 10−7, = 10−6,
= 10−5,
= 10−18, = 0.5, = 1.1, =
0.02, p = 0 in (a), p = 0.5 in (b).
3.3. Theory of Immuno-Driven Aging
There are currently many theories about aging, such as the free radical theory [
40], the telomere attrition theory [
41], and programming aging theory [
42]. In recent years, through studies in omics and transcriptomics, new characteristics of aging cells have been discovered, such as high expression of cell inflammatory factors [
43,
44,
45]. Many of these chronic inflammations are mediated by the immune system. Different from the traditional immunosenescence theory [
21,
22], our proposed theory of immune-driven aging aims to explain that it may be difficult for the immune system to maintain its homeostasis, and over time, the types, composition, and proportions of antibodies will undergo significant changes. The immune system tends to produce more autoantibodies with strong self-binding affinity. As these autoantibodies accumulate, they can lead to various chronic inflammations, which in turn accelerate cellular aging, resulting in the release of a greater amount of self-antigens and more severe inflammatory reactions, ultimately leading to organismal death. Many experiments have shown that antibodies in the human body undergo continuous changes over time. For example, peptide microarray tests for the antibody-peptide binding strength in serum have shown a significant increase in antibody binding to certain specific sequences in the elderly population, while a noticeable decrease in binding strength to other peptides [
46]. Somatic hypermutation may be one of the reasons for the evolution of the antibody repertoire, as experiments have indicated that sequence changes brought about by somatic hypermutation are not random but subject to selective pressure [
36,
37,
38]. This also validates our earlier theory of antibody kinetics, namely the role of autoantigens in maintaining memory cells [
24]. Antibodies evolve towards a higher binding affinity to autoantigens. To further elucidate this theory, we used mathematical simulations to model the changing trends of antibody composition over time, taking into account the influence of somatic hypermutation. We continue to use our Model 2, and the initial composition and proportion of antibodies released into peripheral tissues is a key consideration. We assume that the initial composition of antibodies follows a normal distribution as described in our Model 2, and we simulate changes in antibody total quantity and composition proportions under external environmental pressures. We test the characteristics of antibody changes at different somatic hypermutation frequencies.
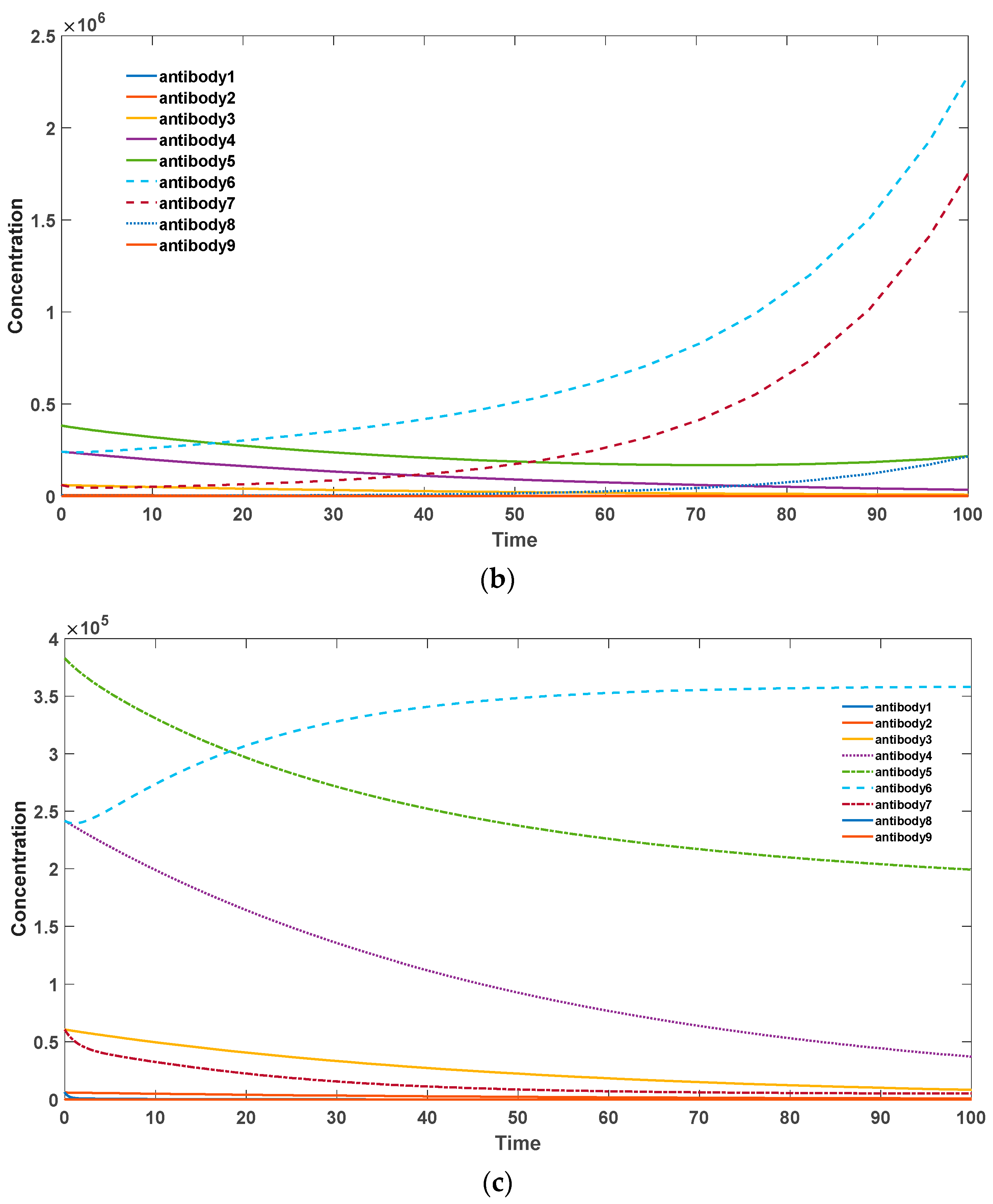
In
Figure 6 it can be observed that somatic hypermutation in somatic cells can prevent the excessive accumulation of autoantibodies. When the mechanism of somatic hypermutation is completely inhibited, antibodies with a high binding activity to self-antigens undergo rapid and abundant proliferation, leading to the onset of autoimmune diseases. This point has been well demonstrated by the knockout experiments of AID enzyme in mice, where mice lacking the AID enzyme exhibited strong autoimmune diseases at a young age [
39]. Elevating the frequency of somatic hypermutation in an appropriate range can effectively reduce the accumulation of high-affinity autoantibodies while controlling the overall accumulation of antibodies.
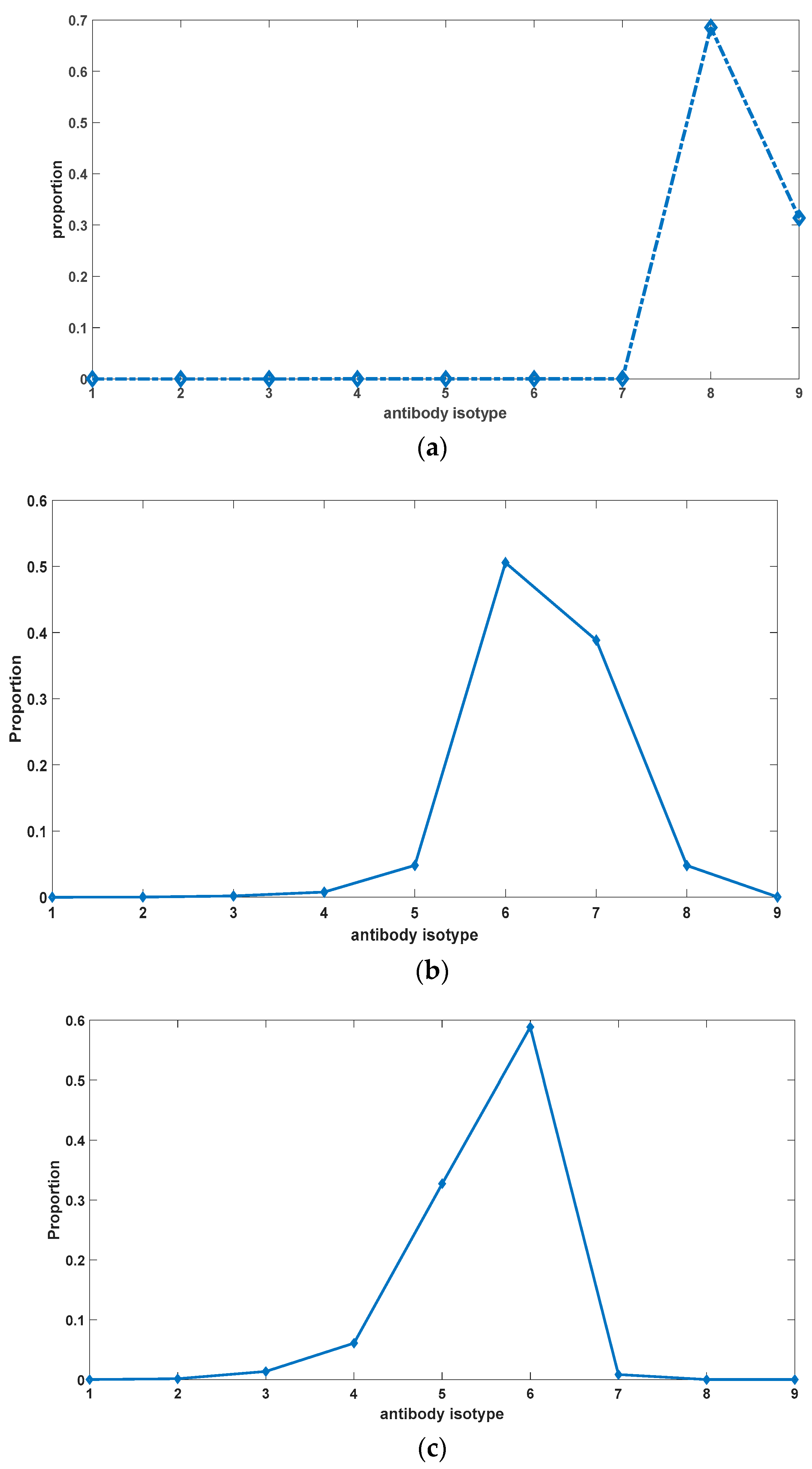
Figure 7 illustrates the alterations in the proportions of different antibody types under varying rates of somatic hypermutation in somatic cells. It is evident from
Figure 7 that somatic hypermutation serves not only to impede the excessive accumulation of autoantibodies but also exerts an influence on the overall composition of antibodies. Complete abrogation of somatic hypermutation leads to the swift evolution of autoantibodies towards heightened binding affinity. Somatic hypermutation acts as an inhibitory force against this evolutionary progression. Nevertheless, it is worth noting that the trend of antibody evolution towards enhanced self-reactivity appears refractory to reversal, as evidenced by a rightward shift in the distribution of antibody constituents relative to the initial state. Moreover, with the passage of time, the proportion of antibodies exhibiting potent binding affinity to self-antigens progressively augments (as depicted in
Figure 8), giving rise to an irreversible alteration in antibody composition. This phenomenon potentially underlies pivotal factors contributing to organismal senescence and the emergence of geriatric chronic inflammation.
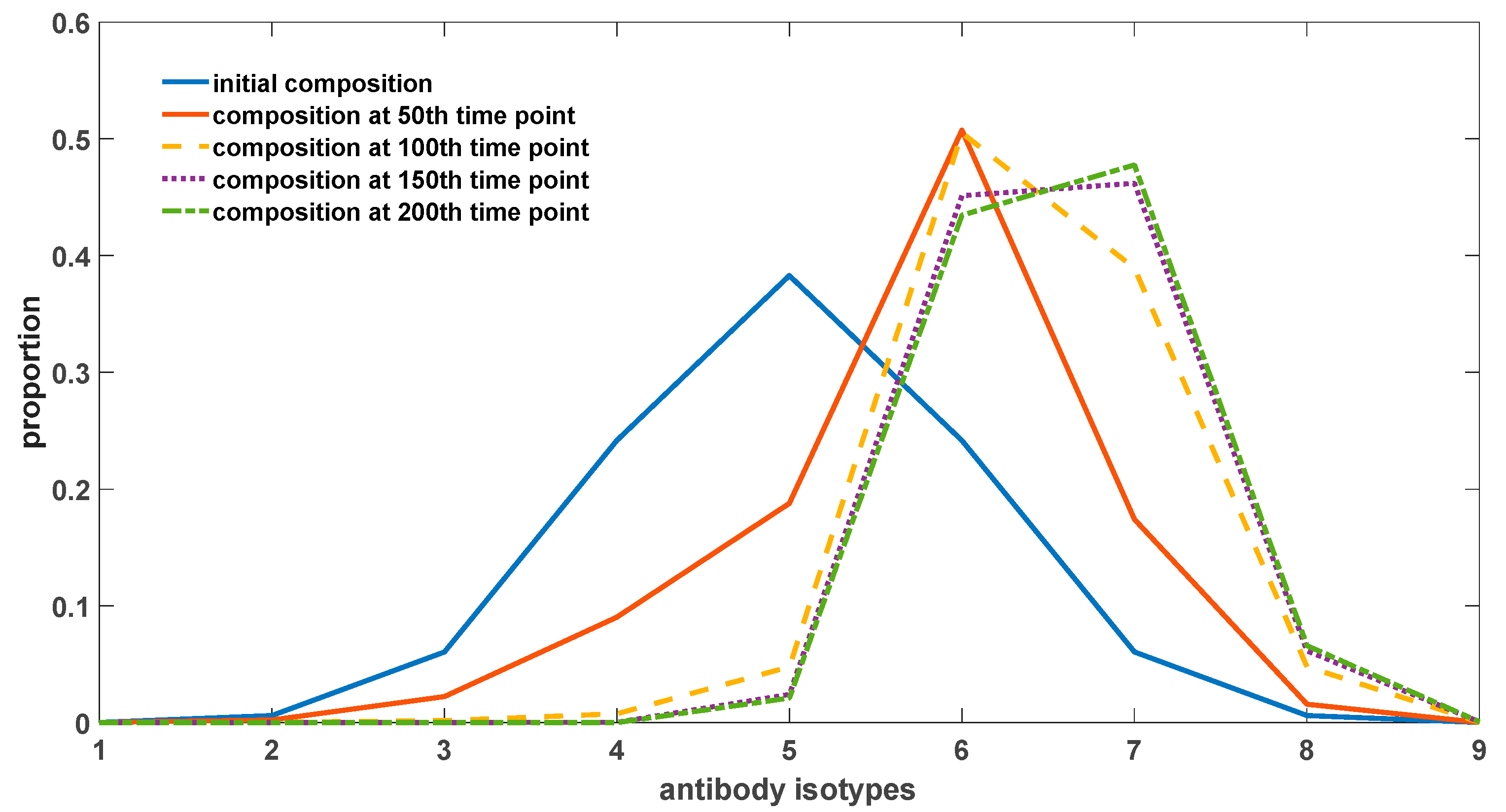
The primary content illustrated in
Figure 8 pertains to the temporal changes in the proportion of antibodies within the organism. Antibodies exhibiting a strong binding affinity to self-antigens are observed to constitute a higher proportion, resulting in a right-skewed overall distribution as depicted in the figure. In the case of different antibodies, specifically antibodies 1 to 9, their respective affinities to self-antigens progressively increase. Initially, these antibodies originate from newly generated B-cells and have not yet undergone environmental selection. As a consequence, the overall distribution adheres to a normal distribution, characterized by a prevalence of antibodies with intermediate binding affinity, while those with high or low affinities remain less prevalent. With the passage of time, this distribution undergoes a shift, wherein the proportion of high-affinity antibodies gradually escalates. This phenomenon encompasses antibodies 7, 8, and 9. However, due to the presence of somatic hypermutation in somatic cells, it is plausible that antibody 7 exhibits the highest proportion within the depicted figure. Antibodies 8 and 9 possess a considerable likelihood of transforming into antibodies with diminished binding affinities, thereby impeding their capacity to dominate the distribution.
The evolutionary inclination of antibodies towards increased affinity to self-antigens stems from the reciprocal interactions between antigens and antibodies. The regeneration and proliferation of antibodies necessitate stimulation by antigen–antibody complexes. High-affinity antibodies engender a greater generation of antigen–antibody complexes vis-à-vis competition with low-affinity antibodies, thus triggering the production of additional cognate antibodies. Consequently, this drives the evolutionary trajectory of antibodies towards a heightened binding affinity to self-antigens. Notably, the existence of somatic hypermutation in somatic cells serves to temper this process, as hypermutation events occurring in high-affinity antibodies manifest a substantial probability of favoring lower-affinity outcomes. This mechanism ensures that antibodies with exceedingly strong affinities, such as antibodies 8 and 9, do not assert dominance within the distribution.
In order to better depict the evolutionary trend of antibodies, we employed self-antigens that have a strong stimulatory effect on antibody regeneration. Specifically, we increased the value of parameter
to 0.7. However, it is important to note that this value remains significantly smaller than the stimulating effects induced by exogenous antigens. For instance, in
Figure 5b, this value equals 1.1. We used a total antibody concentration (
y(0)) of 10
6, following a normal distribution as outlined in
Figure 2. From this distribution, we calculated the initial concentrations for each isotype.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









