
Collagens Regulating Adipose Tissue Formation and Functions

 , and
, and
Abstract
:1. Adipose Tissues
1.1. Pathological Conditions in AT
1.1.1. Obesity and Type 2 Diabetes
1.1.2. AT Fibrosis
1.1.3. Lipodystropy
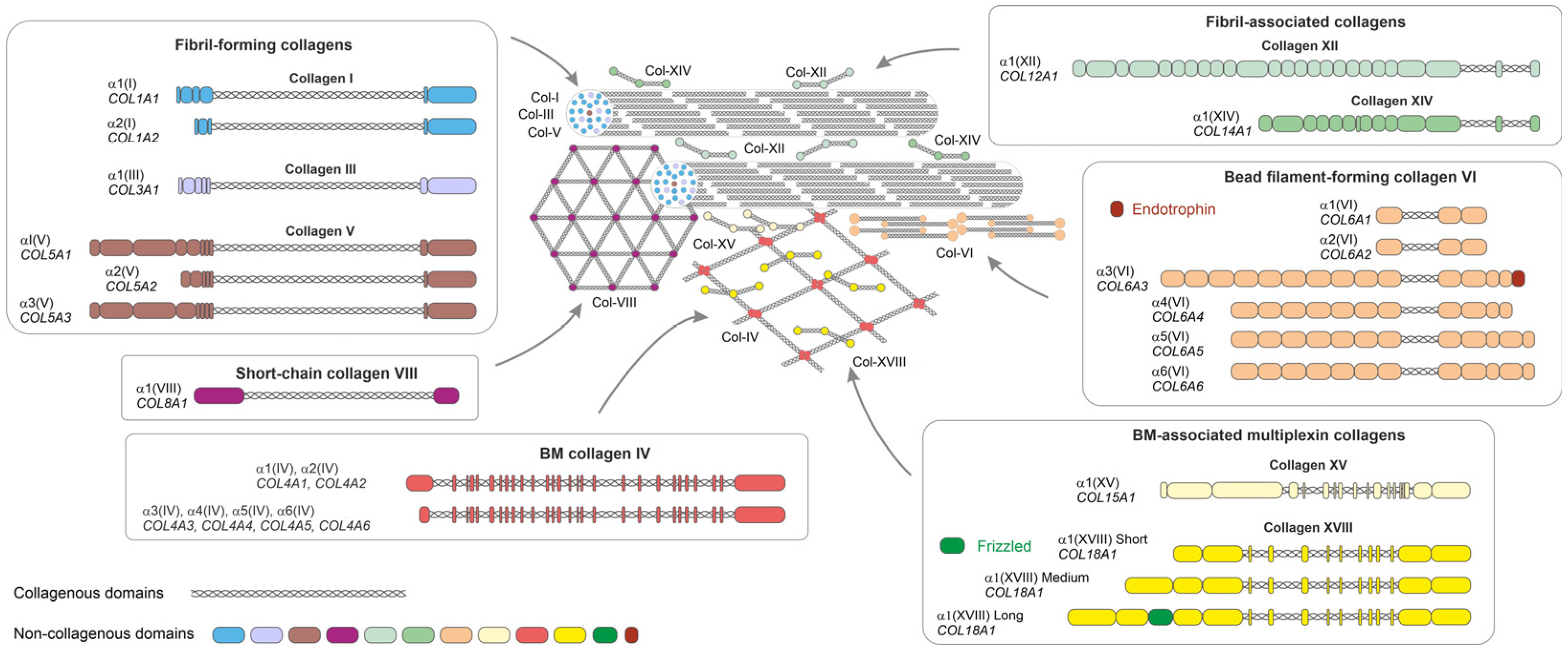
2. Extracellular Matrix in AT
3. AT Collagens and Their Roles in Adipogenic Differentiation and Dysfunctional AT
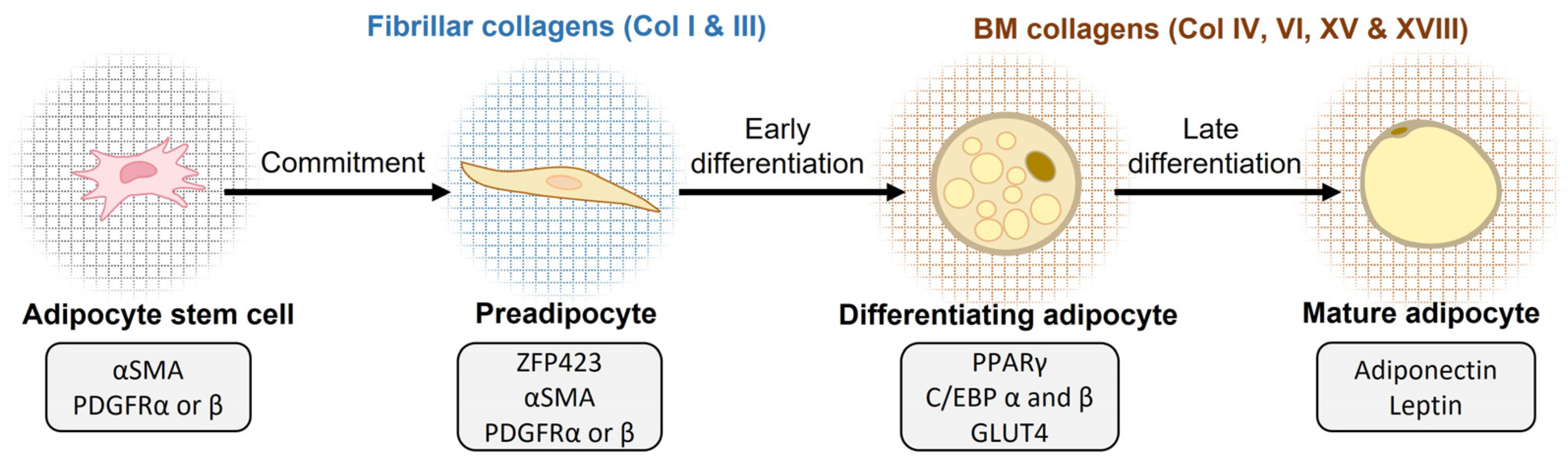
3.1. Collagens in Adipogenesis
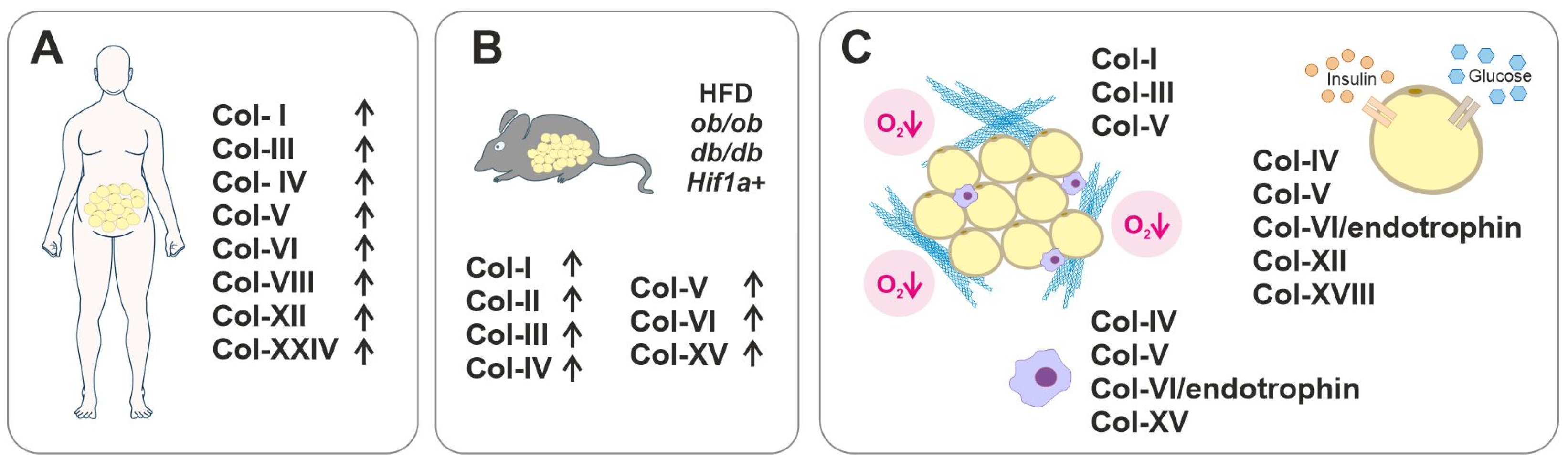
3.2. Collagens in Dysfunctional AT and Metabolic Diseases
3.3. Collagen I
3.4. Collagen III
3.5. Collagen IV
3.6. Collagen V
3.7. Collagen VI
3.8. Multiplexin Collagens XV and XVIII
3.9. Other Collagens
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zorena, K.; Jachimowicz-Duda, O.; Ślęzak, D.; Robakowska, M.; Mrugacz, M. Adipokines and Obesity. Potential Link to Metabolic Disorders and Chronic Complications. Int. J. Mol. Sci. 2020, 21, 3570. [Google Scholar] [CrossRef] [PubMed]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M. Subcutaneous and Visceral Adipose Tissue: Structural and Functional Differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Smorlesi, A.; Frontini, A.; Barbatelli, G.; Cinti, S. White, Brown and Pink Adipocytes: The Extraordinary Plasticity of the Adipose Organ. Eur. J. Endocrinol. 2014, 170, R159–R171. [Google Scholar] [CrossRef] [PubMed]
- Wronska, A.; Kmiec, Z. Structural and Biochemical Characteristics of Various White Adipose Tissue Depots. Acta Physiol. 2012, 205, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Froy, O.; Garaulet, M. The Circadian Clock in White and Brown Adipose Tissue: Mechanistic, Endocrine, and Clinical Aspects. Endocr. Rev. 2018, 39, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Mulya, A.; Kirwan, J.P. Brown and Beige Adipose Tissue. Endocrinol Metab. Clin. N. Am. 2016, 45, 605–621. [Google Scholar] [CrossRef]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.-J.; Enerbäck, S.; et al. Functional Brown Adipose Tissue in Healthy Adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.A.F.L.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J.J. Cold-Activated Brown Adipose Tissue in Healthy Men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and Importance of Brown Adipose Tissue in Adult Humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Giordano, A.; Frontini, A.; Cinti, S. Convertible Visceral Fat as a Therapeutic Target to Curb Obesity. Nat. Rev. Drug. Discov. 2016, 15, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Cinti, F.; Canese, R.; Carpinelli, G.; Colleluori, G.; Di Vincenzo, A.; Palombelli, G.; Severi, I.; Moretti, M.; Redaelli, C.; et al. The Adipose Organ Is a Unitary Structure in Mice and Humans. Biomedicines 2022, 10, 2275. [Google Scholar] [CrossRef] [PubMed]
- Harb, E.; Kheder, O.; Poopalasingam, G.; Rashid, R.; Srinivasan, A.; Izzi-Engbeaya, C. Brown Adipose Tissue and Regulation of Human Body Weight. Diabetes Metab. Res. Rev. 2023, 39, e3594. [Google Scholar] [CrossRef] [PubMed]
- Armani, A.; Feraco, A.; Camajani, E.; Gorini, S.; Lombardo, M.; Caprio, M. Nutraceuticals in Brown Adipose Tissue Activation. Cells 2022, 11, 3996. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose Tissue Inflammation and Metabolic Dysfunction in Obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and Cancer: Inflammation Bridges the Two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and Cancer Risk: Emerging Biological Mechanisms and Perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and Nonalcoholic Fatty Liver Disease: From Pathophysiology to Therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Fujii, H.; Kawada, N. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef]
- Bloksgaard, M.; Lindsey, M.; Martinez-Lemus, L.A. Extracellular Matrix in Cardiovascular Pathophysiology. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1687–H1690. [Google Scholar] [CrossRef]
- Elagizi, A.; Kachur, S.; Carbone, S.; Lavie, C.J.; Blair, S.N. A Review of Obesity, Physical Activity, and Cardiovascular Disease. Curr. Obes. Rep. 2020, 9, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and Metabolic Health. Nat. Rev. Mol. Cell. Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, L.; Facey, A.; Omoruyi, F. Diabetes Mellitus and Its Metabolic Complications: The Role of Adipose Tissues. Int. J. Mol. Sci. 2021, 22, 7644. [Google Scholar] [CrossRef]
- Crewe, C.; An, Y.A.; Scherer, P.E. The Ominous Triad of Adipose Tissue Dysfunction: Inflammation, Fibrosis, and Impaired Angiogenesis. J. Clin. Investig. 2017, 127, 74–82. [Google Scholar] [CrossRef]
- Herold, J.; Kalucka, J. Angiogenesis in Adipose Tissue: The Interplay Between Adipose and Endothelial Cells. Front. Physiol. 2021, 11, 624903. [Google Scholar] [CrossRef]
- Lee, M.-J. Transforming Growth Factor Beta Superfamily Regulation of Adipose Tissue Biology in Obesity. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1160–1171. [Google Scholar] [CrossRef]
- Debari, M.K.; Abbott, R.D. Adipose Tissue Fibrosis: Mechanisms, Models, and Importance. Int. J. Mol. Sci. 2020, 21, 6030. [Google Scholar] [CrossRef]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose Tissue Transcriptomic Signature Highlights the Pathological Relevance of Extracellular Matrix in Human Obesity. Genome Biol. 2008, 9, R14. [Google Scholar] [CrossRef]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-Inducible Factor 1α Induces Fibrosis and Insulin Resistance in White Adipose Tissue. Mol. Cell. Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef]
- Anvari, G.; Bellas, E. Hypoxia Induces Stress Fiber Formation in Adipocytes in the Early Stage of Obesity. Sci. Rep. 2021, 11, 21473. [Google Scholar] [CrossRef] [PubMed]
- Pastel, E.; Price, E.; Sjöholm, K.; McCulloch, L.J.; Rittig, N.; Liversedge, N.; Knight, B.; Møller, N.; Svensson, P.-A.; Kos, K. Lysyl Oxidase and Adipose Tissue Dysfunction. Metabolism 2018, 78, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.W.; Mehta, K. Transglutaminase Regulation of Cell Function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Muise, E.S.; Iyengar, P.; Wang, Z.V.; Chandalia, M.; Abate, N.; Zhang, B.B.; Bonaldo, P.; Chua, S.; Scherer, P.E. Metabolic Dysregulation and Adipose Tissue Fibrosis: Role of Collagen VI. Mol. Cell. Biol. 2009, 29, 1575–1591. [Google Scholar] [CrossRef] [PubMed]
- Lackey, D.E.; Burk, D.H.; Ali, M.R.; Mostaedi, R.; Smith, W.H.; Park, J.; Scherer, P.E.; Seay, S.A.; McCoin, C.S.; Bonaldo, P.; et al. Contributions of Adipose Tissue Architectural and Tensile Properties toward Defining Healthy and Unhealthy Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E233–E246. [Google Scholar] [CrossRef]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef]
- Divoux, A.; Tordjman, J.; Lacasa, D.; Veyrie, N.; Hugol, D.; Aissat, A.; Basdevant, A.; Guerre-Millo, M.; Poitou, C.; Zucker, J.-D.; et al. Fibrosis in Human Adipose Tissue: Composition, Distribution, and Link with Lipid Metabolism and Fat Mass Loss. Diabetes 2010, 59, 2817–2825. [Google Scholar] [CrossRef]
- Spencer, M.; Unal, R.; Zhu, B.; Rasouli, N.; McGehee, R.E.; Peterson, C.A.; Kern, P.A. Adipose Tissue Extracellular Matrix and Vascular Abnormalities in Obesity and Insulin Resistance. J. Clin. Endocrinol. Metab. 2011, 96, E1990–E1998. [Google Scholar] [CrossRef]
- Lee, S.G.; Kim, J.S.; Kim, H.-J.; Schlaepfer, D.D.; Kim, I.-S.; Nam, J.-O. Endothelial Angiogenic Activity and Adipose Angiogenesis Is Controlled by Extracellular Matrix Protein TGFBI. Sci. Rep. 2021, 11, 9644. [Google Scholar] [CrossRef]
- Sun, K.; Li, X.; Scherer, P.E. Extracellular Matrix (ECM) and Fibrosis in Adipose Tissue: Overview and Perspectives. Compr. Physiol. 2023, 13, 4387–4407. [Google Scholar] [CrossRef]
- Jones, J.E.C.; Rabhi, N.; Orofino, J.; Gamini, R.; Perissi, V.; Vernochet, C.; Farmer, S.R. The Adipocyte Acquires a Fibroblast-Like Transcriptional Signature in Response to a High Fat Diet. Sci. Rep. 2020, 10, 2380. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Z.; Rabhi, N.; Farmer, S.R. Myocardin-Related Transcription Factor A Promotes Recruitment of ITGA5+ Profibrotic Progenitors during Obesity-Induced Adipose Tissue Fibrosis. Cell Rep. 2018, 23, 1977–1987. [Google Scholar] [CrossRef]
- Iwayama, T.; Steele, C.; Yao, L.; Dozmorov, M.G.; Karamichos, D.; Wren, J.D.; Olson, L.E. PDGFRα Signaling Drives Adipose Tissue Fibrosis by Targeting Progenitor Cell Plasticity. Genes Dev. 2015, 29, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, G.; Ferreira, A.; Liu, Y.; Atlan, M.; Aron-Wisnewsky, J.; Pelloux, V.; Botbol, Y.; Ambrosini, M.; Fradet, M.; Rouault, C.; et al. A PDGFRα-Mediated Switch toward CD9high Adipocyte Progenitors Controls Obesity-Induced Adipose Tissue Fibrosis. Cell Metab. 2017, 25, 673–685. [Google Scholar] [CrossRef]
- Shen, H.; Huang, X.; Zhao, Y.; Wu, D.; Xue, K.; Yao, J.; Wang, Y.; Tang, N.; Qiu, Y. The Hippo Pathway Links Adipocyte Plasticity to Adipose Tissue Fibrosis. Nat. Commun. 2022, 13, 6030. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Caron-Debarle, M.; Le Dour, C.; Magré, J.; Capeau, J. Molecular Mechanisms of Human Lipodystrophies: From Adipocyte Lipid Droplet to Oxidative Stress and Lipotoxicity. Int. J. Biochem. Cell Biol. 2011, 43, 862–876. [Google Scholar] [CrossRef]
- Kaartinen, M.T.; Hang, A.; Barry, A.; Arora, M.; Heinonen, S.; Lundbom, J.; Hakkarainen, A.; Lundholm, N.; Rissanen, A.; Kaprio, J.; et al. Matrisome Alterations in Obesity—Adipose Tissue Transcriptome Study on Monozygotic Weight-Discordant Twins. Matrix Biol. 2022, 108, 1–19. [Google Scholar] [CrossRef]
- Kaartinen, M.T.; Arora, M.; Heinonen, S.; Rissanen, A.; Kaprio, J.; Pietiläinen, K.H. Transglutaminases and Obesity in Humans: Association of F13A1 to Adipocyte Hypertrophy and Adipose Tissue Immune Response. Int. J. Mol. Sci. 2020, 21, 8289. [Google Scholar] [CrossRef]
- Kaartinen, M.T.; Arora, M.; Heinonen, S.; Hang, A.; Barry, A.; Lundbom, J.; Hakkarainen, A.; Lundholm, N.; Rissanen, A.; Kaprio, J.; et al. F13A1 Transglutaminase Expression in Human Adipose Tissue Increases in Acquired Excess Weight and Associates with Inflammatory Status of Adipocytes. Int. J. Obes. 2021, 45, 577–587. [Google Scholar] [CrossRef]
- Myneni, V.D.; Melino, G.; Kaartinen, M.T. Transglutaminase 2—A Novel Inhibitor of Adipogenesis. Cell Death Dis. 2015, 6, e1868. [Google Scholar] [CrossRef]
- Myneni, V.D.; Mousa, A.; Kaartinen, M.T. Factor XIII-A Transglutaminase Deficient Mice Show Signs of Metabolically Healthy Obesity on High Fat Diet. Sci. Rep. 2016, 6, 35574. [Google Scholar] [CrossRef]
- Myneni, V.D.; Hitomi, K.; Kaartinen, M.T. Factor XIII-A Transglutaminase Acts as a Switch between Preadipocyte Proliferation and Differentiation. Blood 2014, 124, 1344–1353. [Google Scholar] [CrossRef]
- Soták, M.; Rajan, M.R.; Clark, M.; Biörserud, C.; Wallenius, V.; Hagberg, C.E.; Börgeson, E. Healthy Subcutaneous and Omental Adipose Tissue Is Associated with High Expression of Extracellular Matrix Components. Int. J. Mol. Sci. 2022, 23, 520. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Muir, L.A.; Washabaugh, A.R.; Neeley, C.K.; Chen, S.Y.P.; Flesher, C.G.; Vorwald, J.; Finks, J.F.; Ghaferi, A.A.; Mulholland, M.W.; et al. Diabetes-Specific Regulation of Adipocyte Metabolism by the Adipose Tissue Extracellular Matrix. J. Clin. Endocrinol. Metab. 2017, 102, 1032–1043. [Google Scholar] [CrossRef]
- Strieder-Barboza, C.; Baker, N.A.; Flesher, C.G.; Karmakar, M.; Patel, A.; Lumeng, C.N.; O’Rourke, R.W. Depot-Specific Adipocyte-Extracellular Matrix Metabolic Crosstalk in Murine Obesity. Adipocyte 2020, 9, 189–196. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Mendez-Gutierrez, A.; Aguilera, C.M.; Plaza-Diaz, J. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef]
- Pozzi, A.; Yurchenco, P.D.; Iozzo, R.V. The Nature and Biology of Basement Membranes. Matrix Biol. 2017, 57–58, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mariman, E.C.; Wang, P. Adipocyte Extracellular Matrix Composition, Dynamics and Role in Obesity. Cell. Mol. Life Sci. 2010, 67, 1277–1292. [Google Scholar] [CrossRef]
- Lin, D.; Chun, T.-H.; Kang, L. Adipose Extracellular Matrix Remodelling in Obesity and Insulin Resistance. Biochem. Pharmacol. 2016, 119, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A Guide to the Composition and Functions of the Extracellular Matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The Extracellular Matrix at a Glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Mosher, D.F.; Fogerty, F.J.; Chernousov, M.A.; Barry, E.L.R. Assembly of Fibronectin into Extracellular Matrix. Ann. N. Y. Acad. Sci. 1991, 614, 167–180. [Google Scholar] [CrossRef]
- Schwarzbauer, J.E.; DeSimone, D.W. Fibronectins, Their Fibrillogenesis, and In Vivo Functions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005041. [Google Scholar] [CrossRef]
- Mezzenga, R.; Mitsi, M. The Molecular Dance of Fibronectin: Conformational Flexibility Leads to Functional Versatility. Biomacromolecules 2019, 20, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Fogelgren, B.; Polgár, N.; Szauter, K.M.; Újfaludi, Z.; Laczkó, R.; Fong, K.S.K.; Csiszar, K. Cellular Fibronectin Binds to Lysyl Oxidase with High Affinity and Is Critical for Its Proteolytic Activation. J. Biol. Chem. 2005, 280, 24690–24697. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.T.; Schwarzbauer, J.E. Fibronectin Matrix as a Scaffold for Procollagen Proteinase Binding and Collagen Processing. Mol. Biol. Cell 2019, 30, 2218–2226. [Google Scholar] [CrossRef]
- Huang, G.; Zhang, Y.; Kim, B.; Ge, G.; Annis, D.S.; Mosher, D.F.; Greenspan, D.S. Fibronectin Binds and Enhances the Activity of Bone Morphogenetic Protein 1. J. Biol. Chem. 2009, 284, 25879–25888. [Google Scholar] [CrossRef]
- Sottile, J.; Hocking, D.C. Fibronectin Polymerization Regulates the Composition and Stability of Extracellular Matrix Fibrils and Cell-Matrix Adhesions. Mol. Biol. Cell 2002, 13, 3546–3559. [Google Scholar] [CrossRef]
- Velling, T.; Risteli, J.; Wennerberg, K.; Mosher, D.F.; Johansson, S. Polymerization of Type I and III Collagens Is Dependent On Fibronectin and Enhanced By Integrins A11β1and A2β1. J. Biol. Chem. 2002, 277, 37377–37381. [Google Scholar] [CrossRef]
- Kinsey, R.; Williamson, M.R.; Chaudhry, S.; Mellody, K.T.; McGovern, A.; Takahashi, S.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 Microfibril Deposition Is Dependent on Fibronectin Assembly. J. Cell Sci. 2008, 121, 2696–2704. [Google Scholar] [CrossRef]
- Sabatier, L.; Chen, D.; Fagotto-Kaufmann, C.; Hubmacher, D.; McKee, M.D.; Annis, D.S.; Mosher, D.F.; Reinhardt, D.P. Fibrillin Assembly Requires Fibronectin. Mol. Biol. Cell 2009, 20, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Pankov, R.; Yamada, K.M. Fibronectin at a Glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef]
- To, W.S.; Midwood, K.S. Plasma and Cellular Fibronectin: Distinct and Independent Functions during Tissue Repair. Fibrogenesis Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Moretti, F.A.; Chauhan, A.K.; Iaconcig, A.; Porro, F.; Baralle, F.E.; Muro, A.F. A Major Fraction of Fibronectin Present in the Extracellular Matrix of Tissues Is Plasma-Derived. J. Biol. Chem. 2007, 282, 28057–28062. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Ginty, C.A. Fibronectin Modulation of Cell Shape and Lipogenic Gene Expression in 3t3-Adipocytes. Cell 1983, 35, 657–666. [Google Scholar] [CrossRef]
- Fukai, F.; Iso, T.; Sekiguchi, K.; Miyatake, N.; Tsugita, A.; Katayama, T. An Amino-Terminal Fibronectin Fragment Stimulates the Differentiation of ST-13 Preadipocytes. Biochemistry 1993, 32, 5746–5751. [Google Scholar] [CrossRef]
- Kamiya, S.; Kato, R.; Wakabayashi, M.; Tohyama, T.; Enami, I.; Ueki, M.; Yajima, H.; Ishii, T.; Nakamura, H.; Katayama, T.; et al. Fibronectin Peptides Derived from Two Distinct Regions Stimulate Adipocyte Differentiation by Preventing Fibronectin Matrix Assembly. Biochemistry 2002, 41, 3270–3277. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, L.; Smas, C.; Sul, H.S. Pref-1 Interacts with Fibronectin To Inhibit Adipocyte Differentiation. Mol. Cell. Biol. 2010, 30, 3480–3492. [Google Scholar] [CrossRef]
- Mosher, D.F. Cross-Linking of Fibronectin to Collagenous Proteins. Mol. Cell. Biochem. 1984, 58, 63–68. [Google Scholar] [CrossRef]
- Barry, E.L.; Mosher, D.F. Factor XIIIa-Mediated Cross-Linking of Fibronectin in Fibroblast Cell Layers. Cross-Linking of Cellular and Plasma Fibronectin and of Amino-Terminal Fibronectin Fragments. J. Biol. Chem. 1989, 264, 4179–4185. [Google Scholar] [CrossRef]
- Rajak, S.; Hussain, Y.; Singh, K.; Tiwari, S.; Ahmad, B.; Bharti, S.; Prakash, P. Cellular Fibronectin Containing Extra Domain A Causes Insulin Resistance via Toll-like Receptor 4. Sci. Rep. 2020, 10, 9102. [Google Scholar] [CrossRef]
- Dejgaard, A.; Andersen, T.; Christoffersen, P.; Clemmensen, I.; Gluud, C. Plasma Fibronectin Concentrations in Morbidly Obese Patients. Scand. J. Clin. Lab. Investig. 1984, 44, 207–210. [Google Scholar] [CrossRef]
- Cucuianu, M.; Bodizs, G.; Duncea, I.; Colhon, D. Plasma Fibronectin in Overweight Men and Women: Correlation with Serum Triglyceride Levels and Serum Cholinesterase Activity. Blood Coagul. Fibrinolysis 1996, 7, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Aouadi, M.; Tencerova, M.; Vangala, P.; Yawe, J.C.; Nicoloro, S.M.; Amano, S.U.; Cohen, J.L.; Czech, M.P. Gene Silencing in Adipose Tissue Macrophages Regulates Whole-Body Metabolism in Obese Mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8278–8283. [Google Scholar] [CrossRef]
- Gómez-Ambrosi, J.; Catalán, V.; Ramírez, B.; Rodríguez, A.; Colina, I.; Silva, C.; Rotellar, F.; Mugueta, C.; Gil, M.J.; Cienfuegos, J.A.; et al. Plasma Osteopontin Levels and Expression in Adipose Tissue Are Increased in Obesity. J. Clin. Endocrinol. Metab. 2007, 92, 3719–3727. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Yan, X.; Shinmura, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yamamoto, T.; Anzai, A.; Isobe, S.; Yoshida, N.; et al. Obesity Accelerates T Cell Senescence in Murine Visceral Adipose Tissue. J. Clin. Investig. 2016, 126, 4626–4639. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, T.; Perez-Tilve, D.; Ogawa, D.; Gizard, F.; Zhao, Y.; Heywood, E.B.; Jones, K.L.; Kawamori, R.; Cassis, L.A.; Tschöp, M.H.; et al. Osteopontin Mediates Obesity-Induced Adipose Tissue Macrophage Infiltration and Insulin Resistance in Mice. J. Clin. Investig. 2007, 117, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Lancha, A.; Rodríguez, A.; Catalán, V.; Becerril, S.; Sáinz, N.; Ramírez, B.; Burrell, M.A.; Salvador, J.; Frühbeck, G.; Gómez-Ambrosi, J. Osteopontin Deletion Prevents the Development of Obesity and Hepatic Steatosis via Impaired Adipose Tissue Matrix Remodeling and Reduced Inflammation and Fibrosis in Adipose Tissue and Liver in Mice. PLoS ONE 2014, 9, e98398. [Google Scholar] [CrossRef]
- Naor, D.; Sionov, R.V.; Ish-Shalom, D. CD44: Structure, Function and Association with the Malignant Process. Adv Cancer Res 1997, 71, 241–319. [Google Scholar] [CrossRef]
- Han, C.Y.; Subramanian, S.; Chan, C.K.; Omer, M.; Chiba, T.; Wight, T.N.; Chait, A. Adipocyte-Derived Serum Amyloid A3 and Hyaluronan Play a Role in Monocyte Recruitment and Adhesion. Diabetes 2007, 56, 2260–2273. [Google Scholar] [CrossRef]
- Ji, E.; Jung, M.Y.; Park, J.H.; Kim, S.; Seo, C.R.; Park, K.W.; Lee, E.K.; Yeom, C.H.; Lee, S. Inhibition of Adipogenesis in 3T3-L1 Cells and Suppression of Abdominal Fat Accumulation in High-Fat Diet-Feeding C57BL/6J Mice after Downregulation of Hyaluronic Acid. Int. J. Obes. 2014, 38, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Kruglikov, I.L.; Akgul, Y.; Scherer, P.E. Hyaluronan in Adipogenesis, Adipose Tissue Physiology and Systemic Metabolism. Matrix Biol. 2019, 78–79, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.; Steadman, R.; Muller, I.; Draman, M.; Rees, D.A.; Taylor, P.; Dayan, C.M.; Ludgate, M.; Zhang, L. Role of Hyaluronan in Human Adipogenesis: Evidence from in-Vitro and in-Vivo Studies. Int. J. Mol. Sci. 2019, 20, 2675. [Google Scholar] [CrossRef]
- Kang, L.; Lantier, L.; Kennedy, A.; Bonner, J.S.; Mayes, W.H.; Bracy, D.P.; Bookbinder, L.H.; Hasty, A.H.; Thompson, C.B.; Wasserman, D.H. Hyaluronan Accumulates with High-Fat Feeding and Contributes to Insulin Resistance. Diabetes 2013, 62, 1888–1896. [Google Scholar] [CrossRef]
- Li, Y.; Tong, X.; Rumala, C.; Clemons, K.; Wang, S. Thrombospondin1 Deficiency Reduces Obesity-Associated Inflammation and Improves Insulin Sensitivity in a Diet-Induced Obese Mouse Model. PLoS ONE 2011, 6, e26656. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.; Yao-Borengasser, A.; Bodles, A.M.; Rasouli, N.; Phanavanh, B.; Nolen, G.T.; Kern, E.M.; Nagarajan, R.; Spencer, H.J.; Lee, M.-J.; et al. Thrombospondin-1 Is an Adipokine Associated with Obesity, Adipose Inflammation, and Insulin Resistance. Diabetes 2008, 57, 432–439. [Google Scholar] [CrossRef]
- García-Bernal, D.; García-Arranz, M.; Yáñez, R.M.; Hervás-Salcedo, R.; Cortés, A.; Fernández-García, M.; Hernando-Rodríguez, M.; Quintana-Bustamante, Ó.; Bueren, J.A.; García-Olmo, D.; et al. The Current Status of Mesenchymal Stromal Cells: Controversies, Unresolved Issues and Some Promising Solutions to Improve Their Therapeutic Efficacy. Front. Cell Dev. Biol. 2021, 9, 650664. [Google Scholar] [CrossRef]
- Hepler, C.; Vishvanath, L.; Gupta, R.K. Sorting out Adipocyte Precursors and Their Role in Physiology and Disease. Genes Dev. 2017, 31, 127–140. [Google Scholar] [CrossRef]
- Kuri-Harcuch, W.; Velez-delValle, C.; Vazquez-Sandoval, A.; Hernández-Mosqueira, C.; Fernandez-Sanchez, V. A Cellular Perspective of Adipogenesis Transcriptional Regulation. J. Cell. Physiol. 2019, 234, 1111–1129. [Google Scholar] [CrossRef]
- Abuhattum, S.; Gefen, A.; Weihs, D. Ratio of Total Traction Force to Projected Cell Area Is Preserved in Differentiating Adipocytes. Integr. Biol. 2015, 7, 1212–1217. [Google Scholar] [CrossRef]
- Gupta, R.K.; Arany, Z.; Seale, P.; Mepani, R.J.; Ye, L.; Conroe, H.M.; Roby, Y.A.; Kulaga, H.; Reed, R.R.; Spiegelman, B.M. Transcriptional Control of Preadipocyte Determination by Zfp423. Nature 2010, 464, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the Global Map of Adipogenesis and Beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ Is Required for the Differentiation of Adipose Tissue In Vivo and In Vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Napolitano, L. The Differentiation of White Adipose Cells. J. Cell Biol. 1963, 18, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Mor-Yossef Moldovan, L.; Lustig, M.; Naftaly, A.; Mardamshina, M.; Geiger, T.; Gefen, A.; Benayahu, D. Cell Shape Alteration during Adipogenesis Is Associated with Coordinated Matrix Cues. J. Cell. Physiol. 2019, 234, 3850–3863. [Google Scholar] [CrossRef]
- Huang, G.; Greenspan, D.S. ECM Roles in the Function of Metabolic Tissues. Trends Endocrinol. Metab. 2012, 23, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Reggio, S.; Rouault, C.; Poitou, C.; Bichet, J.-C.; Prifti, E.; Bouillot, J.-L.; Rizkalla, S.; Lacasa, D.; Tordjman, J.; Clément, K. Increased Basement Membrane Components in Adipose Tissue During Obesity: Links With TGFβ and Metabolic Phenotypes. J. Clin. Endocrinol. Metab. 2016, 101, 2578–2587. [Google Scholar] [CrossRef]
- Mauney, J.; Volloch, V. Human Bone Marrow-Derived Stromal Cells Show Highly Efficient Stress-Resistant Adipogenesis on Denatured Collagen IV Matrix but Not on Its Native Counterpart: Implications for Obesity. Matrix Biol. 2010, 29, 9–14. [Google Scholar] [CrossRef]
- Johnston, E.K.; Abbott, R.D. Adipose Tissue Development Relies on Coordinated Extracellular Matrix Remodeling, Angiogenesis, and Adipogenesis. Biomedicines 2022, 10, 2227. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Arkkila, P.E.T.; Rönnemaa, T.; Koskinen, P.J.; Kantola, I.M.; Seppänen, E.; Viikari, J.S.A. Biochemical Markers of Type III and I Collagen: Association with Retinopathy and Neuropathy in Type 1 Diabetic Subjects. Diabet. Med. 2001, 18, 816–821. [Google Scholar] [CrossRef]
- Gupta, R.K.; Mepani, R.J.; Kleiner, S.; Lo, J.C.; Khandekar, M.J.; Cohen, P.; Frontini, A.; Bhowmick, D.C.; Ye, L.; Cinti, S.; et al. Zfp423 Expression Identifies Committed Preadipocytes and Localizes to Adipose Endothelial and Perivascular Cells. Cell Metab. 2012, 15, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, E.; Nishizawa, Y.; Shoji, S.; Morii, H. Serum Type III, IV Collagens and TIMP in Patients with Type II Diabetes Mellitus. Life Sci. 1996, 58, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Hirai, S.; Ohyane, C.; Kim, Y.-I.; Lin, S.; Goto, T.; Takahashi, N.; Kim, C.-S.; Kang, J.; Yu, R.; Kawada, T. Involvement of Mast Cells in Adipose Tissue Fibrosis. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E247–E255. [Google Scholar] [CrossRef]
- Huang, G.; Ge, G.; Wang, D.; Gopalakrishnan, B.; Butz, D.H.; Colman, R.J.; Nagy, A.; Greenspan, D.S. A3(V) Collagen Is Critical for Glucose Homeostasis in Mice Due to Effects in Pancreatic Islets and Peripheral Tissues. J. Clin. Investig. 2011, 121, 769–783. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.J.; Rawling, T.J.; Sjöholm, K.; Franck, N.; Dankel, S.N.; Price, E.J.; Knight, B.; Liversedge, N.H.; Mellgren, G.; Nystrom, F.; et al. COL6A3 Is Regulated by Leptin in Human Adipose Tissue and Reduced in Obesity. Endocrinology 2015, 156, 134–146. [Google Scholar] [CrossRef]
- Oh, J.; Kim, C.S.; Kim, M.; Jo, W.; Sung, Y.H.; Park, J. Type VI Collagen and Its Cleavage Product, Endotrophin, Cooperatively Regulate the Adipogenic and Lipolytic Capacity of Adipocytes. Metabolism 2021, 114, 154430. [Google Scholar] [CrossRef]
- Spencer, M.; Yao-Borengasser, A.; Unal, R.; Rasouli, N.; Gurley, C.M.; Zhu, B.; Peterson, C.A.; Kern, P.A. Adipose Tissue Macrophages in Insulin-Resistant Subjects Are Associated with Collagen VI and Fibrosis and Demonstrate Alternative Activation. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1016–E1027. [Google Scholar] [CrossRef]
- Dankel, S.N.; Svärd, J.; Matthä, S.; Claussnitzer, M.; Klöting, N.; Glunk, V.; Fandalyuk, Z.; Grytten, E.; Solsvik, M.H.; Nielsen, H.J.; et al. COL6A3 Expression in Adipocytes Associates with Insulin Resistance and Depends on PPARγ and Adipocyte Size. Obesity 2014, 22, 1807–1813. [Google Scholar] [CrossRef]
- Williams, L.M.; McCann, F.E.; Cabrita, M.A.; Layton, T.; Cribbs, A.; Knezevic, B.; Fang, H.; Knight, J.; Zhang, M.; Fischer, R.; et al. Identifying Collagen VI as a Target of Fibrotic Diseases Regulated by CREBBP/EP300. Proc. Natl. Acad. Sci. USA 2020, 117, 20753–20763. [Google Scholar] [CrossRef]
- Williams, L.; Layton, T.; Yang, N.; Feldmann, M.; Nanchahal, J. Collagen VI as a Driver and Disease Biomarker in Human Fibrosis. FEBS J. 2022, 289, 3603–3629. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, M.; Xu, Y.; Wu, S.; Saeed, M.; Sun, C. ColXV Promotes Adipocyte Differentiation via Inhibiting DNA Methylation and CAMP/PKA Pathway in Mice. Oncotarget 2017, 8, 60135–60148. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Shen, Z.; Cai, J.; Pan, M.; Sun, C. ColXV Aggravates Adipocyte Apoptosis by Facilitating Abnormal Extracellular Matrix Remodeling in Mice. Int. J. Mol. Sci. 2020, 21, 959. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, Y.; Li, Y.; Tai, R.; Sun, Z.; Wu, Q.; Liu, Y.; Sun, C. Collagen XV Promotes ER Stress-Induced Inflammation through Activating Integrin Β1/FAK Signaling Pathway and M1 Macrophage Polarization in Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 9997. [Google Scholar] [CrossRef]
- Bishop, J.R.; Passos-Bueno, M.R.; Fong, L.; Stanford, K.I.; Gonzales, J.C.; Yeh, E.; Young, S.G.; Bensadoun, A.; Witztum, J.L.; Esko, J.D.; et al. Deletion of the Basement Membrane Heparan Sulfate Proteoglycan Type XVIII Collagen Causes Hypertriglyceridemia in Mice and Humans. PLoS ONE 2010, 5, e13919. [Google Scholar] [CrossRef] [PubMed]
- Petäistö, T.; Vicente, D.; Mäkelä, K.A.; Finnilä, M.A.; Miinalainen, I.; Koivunen, J.; Izzi, V.; Aikio, M.; Karppinen, S.; Devarajan, R.; et al. Lack of Collagen XVIII Leads to Lipodystrophy and Perturbs Hepatic Glucose and Lipid Homeostasis. J. Physiol. 2020, 598, 3373–3393. [Google Scholar] [CrossRef]
- Aikio, M.; Elamaa, H.; Vicente, D.; Izzi, V.; Kaur, I.; Seppinen, L.; Speedy, H.E.; Kaminska, D.; Kuusisto, S.; Sormunen, R.; et al. Specific Collagen XVIII Isoforms Promote Adipose Tissue Accrual via Mechanisms Determining Adipocyte Number and Affect Fat Deposition. Proc. Natl. Acad. Sci. USA 2014, 111, 3043. [Google Scholar] [CrossRef]
- Peloso, G.M.; Auer, P.L.; Bis, J.C.; Voorman, A.; Morrison, A.C.; Stitziel, N.O.; Brody, J.A.; Khetarpal, S.A.; Crosby, J.R.; Fornage, M.; et al. Association of Low-Frequency and Rare Coding-Sequence Variants with Blood Lipids and Coronary Heart Disease in 56,000 Whites and Blacks. Am. J. Hum. Genet. 2014, 94, 223–232. [Google Scholar] [CrossRef]
- Errera, F.I.; Canani, L.H.; Yeh, E.; Kague, E.; Armelin-Correa, L.M.; Suzuki, O.T.; Tschiedel, B.; Silva, M.E.; Sertie, A.L.; Passos-Bueno, M.R. COL18A1 Is Highly Expressed during Human Adipocyte Differentiation and the SNP c.1136C T in Its “Frizzled” Motif Is Associated with Obesity in Diabetes Type 2 Patients. An Acad. Bras. Cienc. 2008, 80, 167–177. [Google Scholar] [CrossRef]
- Kaur, I.; Ruskamo, S.; Koivunen, J.; Heljasvaara, R.; Lackman, J.J.; Izzi, V.; Petaja-Repo, U.E.; Kursula, P.; Pihlajaniemi, T. The N-Terminal Domain of Unknown Function (DUF959) in Collagen XVIII Is Intrinsically Disordered and Highly O-Glycosylated. Biochem. J. 2018, 475, 3577–3593. [Google Scholar] [CrossRef]
- Weng, X.; Lin, D.; Huang, J.T.J.; Stimson, R.H.; Wasserman, D.H.; Kang, L. Collagen 24 α1 Is Increased in Insulin-Resistant Skeletal Muscle and Adipose Tissue. Int. J. Mol. Sci. 2020, 21, 5738. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Kiuchi, S.; Ouchi, A.; Hase, T.; Murase, T. Characteristic Expression of Extracellular Matrix in Subcutaneous Adipose Tissue Development and Adipogenesis; Comparison with Visceral Adipose Tissue. Int. J. Biol. Sci. 2014, 10, 825–833. [Google Scholar] [CrossRef]
- Ojima, K.; Oe, M.; Nakajima, I.; Muroya, S.; Nishimura, T. Dynamics of Protein Secretion during Adipocyte Differentiation. FEBS Open Bio 2016, 6, 816–826. [Google Scholar] [CrossRef]
- Chun, T.H.; Hotary, K.B.; Sabeh, F.; Saltiel, A.R.; Allen, E.D.; Weiss, S.J. A Pericellular Collagenase Directs the 3-Dimensional Development of White Adipose Tissue. Cell 2006, 125, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.; Löffler, M.; Bilban, M.; Reimers, M.; Kadl, A.; Todoric, J.; Zeyda, M.; Geyeregger, R.; Schreiner, M.; Weichhart, T.; et al. Prevention of High-Fat Diet-Induced Adipose Tissue Remodeling in Obese Diabetic Mice by n-3 Polyunsaturated Fatty Acids. Int. J. Obes. 2007, 31, 1004–1013. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Q.; Liu, W.; Yao, G.; Zhao, Y.; Xu, F.; Hayashi, T.; Fujisaki, H.; Hattori, S.; Tashiro, S.; et al. Enhanced Migration of Murine Fibroblast-like 3T3-L1 Preadipocytes on Type I Collagen-Coated Dish Is Reversed by Silibinin Treatment. Mol. Cell. Biochem. 2018, 441, 35–62. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, X.; Liu, W.; Hayashi, T.; Yamato, M.; Fujisaki, H.; Hattori, S.; Tashiro, S.; Onodera, S.; Ikejima, T. Type I Collagen-Induced YAP Nuclear Expression Promotes Primary Cilia Growth and Contributes to Cell Migration in Confluent Mouse Embryo Fibroblast 3T3-L1 Cells. Mol. Cell. Biochem. 2019, 450, 87–96. [Google Scholar] [CrossRef]
- Liu, X.; Long, X.; Gao, Y.; Liu, W.; Hayashi, T.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Ogura, T.; Onodera, S.; et al. Type I Collagen Inhibits Adipogenic Differentiation via YAP Activation in Vitro. J. Cell. Physiol. 2020, 235, 1821–1837. [Google Scholar] [CrossRef]
- Gusinjac, A.; Gagnon, A.; Sorisky, A. Effect of Collagen I and Aortic Carboxypeptidase-like Protein on 3T3-L1 Adipocyte Differentiation. Metabolism 2011, 60, 782–788. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Tinahones, A.; El Bekay, R.; Malagón, M.M.; Tinahones, F.J. The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health. Metabolites 2020, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ma, K.; Kang, Y.; Liu, W.; Liu, X.; Long, X.; Hayashi, T.; Hattori, S.; Mizuno, K.; Fujisaki, H.; et al. Type I Collagen Reduces Lipid Accumulation during Adipogenesis of Preadipocytes 3T3-L1 via the YAP-MTOR-Autophagy Axis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159181. [Google Scholar] [CrossRef]
- Al Hasan, M.; Martin, P.E.; Shu, X.; Patterson, S.; Bartholomew, C. Type III Collagen Is Required for Adipogenesis and Actin Stress Fibre Formation in 3T3-L1 Preadipocytes. Biomolecules 2021, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- de Winter, T.J.J.; Nusse, R. Running Against the Wnt: How Wnt/β-Catenin Suppresses Adipogenesis. Front. Cell Dev. Biol. 2021, 9, 627429. [Google Scholar] [CrossRef] [PubMed]
- Aratani, Y.; Kitagawa, Y. Enhanced Synthesis and Secretion of Type IV Collagen and Entactin during Adipose Conversion of 3T3-L1 Cells and Production of Unorthodox Laminin Complex. J. Biol. Chem. 1988, 263, 16163–16169. [Google Scholar] [CrossRef]
- Tajima, R.; Kawaguchi, N.; Horino, Y.; Takahashi, Y.; Toriyama, K.; Inou, K.; Torii, S.; Kitagawa, Y. Hypoxic Enhancement of Type IV Collagen Secretion Accelerates Adipose Conversion of 3T3-L1 Fibroblasts. Biochim. Biophys. Acta Mol. Cell Res. 2001, 1540, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.M.; Png, C.Y.M.; Lee, D.J. Type V Collagen in Health, Disease, and Fibrosis. Anat. Rec. 2016, 299, 613–629. [Google Scholar] [CrossRef]
- Nakajima, I.; Muroya, S.; Tanabe, R.-I.; Chikuni, K. Extracellular Matrix Development during Differentiation into Adipocytes with a Unique Increase in Type V and VI Collagen. Biol. Cell 2002, 94, 197–203. [Google Scholar] [CrossRef]
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type V Collagen Controls the Initiation of Collagen Fibril Assembly. J. Biol. Chem. 2004, 279, 53331–53337. [Google Scholar] [CrossRef]
- Park, A.C.; Phan, N.; Massoudi, D.; Liu, Z.; Kernien, J.F.; Adams, S.M.; Davidson, J.M.; Birk, D.E.; Liu, B.; Greenspan, D.S. Deficits in Col5a2 Expression Result in Novel Skin and Adipose Abnormalities and Predisposition to Aortic Aneurysms and Dissections. Am. J. Pathol. 2017, 187, 2300–2311. [Google Scholar] [CrossRef]
- Imamura, Y.; Scott, I.C.; Greenspan, D.S. The Pro-A3(V) Collagen Chain: Complete Primary Structure, Expression Domains in Adult and Developing Tissues, and Comparison to the Structures and Expression Domains of the Other Types V and XI Procollagen Chains. J. Biol. Chem. 2000, 275, 8749–8759. [Google Scholar] [CrossRef]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a Glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef]
- Divoux, A.; Clément, K. Architecture and the Extracellular Matrix: The Still Unappreciated Components of the Adipose Tissue. Obes. Rev. 2011, 12, e494–e503. [Google Scholar] [CrossRef]
- Pasarica, M.; Gowronska-Kozak, B.; Burk, D.; Remedios, I.; Hymel, D.; Gimble, J.; Ravussin, E.; Bray, G.A.; Smith, S.R. Adipose Tissue Collagen VI in Obesity. J. Clin. Endocrinol. Metab. 2009, 94, 5155–5162. [Google Scholar] [CrossRef]
- Zhao, Y.; Gu, X.; Zhang, N.; Kolonin, M.G.; An, Z.; Sun, K. Divergent Functions of Endotrophin on Different Cell Populations in Adipose Tissue. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E952–E963. [Google Scholar] [CrossRef]
- Sun, K.; Park, J.; Gupta, O.T.; Holland, W.L.; Auerbach, P.; Zhang, N.; Marangoni, R.G.; Nicoloro, S.M.; Czech, M.P.; Varga, J.; et al. Endotrophin Triggers Adipose Tissue Fibrosis and Metabolic Dysfunction. Nat. Commun. 2014, 5, 3485. [Google Scholar] [CrossRef]
- Staunstrup, L.M.; Bager, C.L.; Frederiksen, P.; Helge, J.W.; Brunak, S.; Christiansen, C.; Karsdal, M. Endotrophin Is Associated with Chronic Multimorbidity and All-Cause Mortality in a Cohort of Elderly Women. EBioMedicine 2021, 68, 103391. [Google Scholar] [CrossRef] [PubMed]
- Yoldemir, S.A.; Arman, Y.; Akarsu, M.; Altun, O.; Ozcan, M.; Tukek, T. Correlation of Glycemic Regulation and Endotrophin in Patients with Type 2 Diabetes; Pilot Study. Diabetol. Metab. Syndr. 2021, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Bretaud, S.; Guillon, E.; Karppinen, S.-M.; Pihlajaniemi, T.; Ruggiero, F. Collagen XV, a Multifaceted Multiplexin Present across Tissues and Species. Matrix Biol. Plus 2020, 6–7, 100023. [Google Scholar] [CrossRef]
- Heljasvaara, R.; Aikio, M.; Ruotsalainen, H.; Pihlajaniemi, T. Collagen XVIII in Tissue Homeostasis and Dysregulation—Lessons Learned from Model Organisms and Human Patients. Matrix Biol. 2017, 57–58, 55–75. [Google Scholar] [CrossRef] [PubMed]
- North, K.E.; Miller, M.B.; Coon, H.; Martin, L.J.; Peacock, J.M.; Arnett, D.; Zhang, B.; Province, M.; Oberman, A.; Blangero, J.; et al. Evidence for a Gene Influencing Fasting LDL Cholesterol and Triglyceride Levels on Chromosome 21q. Atherosclerosis 2005, 179, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Gordts, P.L.S.M.; Esko, J.D. The Heparan Sulfate Proteoglycan Grip on Hyperlipidemia and Atherosclerosis. Matrix Biol. 2018, 71–72, 262–282. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; John, C.; Schaltenberg, N.; Berbée, J.F.P.; Worthmann, A.; Cherradi, M.L.; Schlein, C.; Piepenburg, J.; Boon, M.R.; Rinninger, F.; et al. Thermogenic Adipocytes Promote HDL Turnover and Reverse Cholesterol Transport. Nat. Commun. 2017, 8, 15010. [Google Scholar] [CrossRef]
- Berbée, J.F.P.; Boon, M.R.; Khedoe, P.P.S.J.; Bartelt, A.; Schlein, C.; Worthmann, A.; Kooijman, S.; Hoeke, G.; Mol, I.M.; John, C.; et al. Brown Fat Activation Reduces Hypercholesterolaemia and Protects from Atherosclerosis Development. Nat. Commun. 2015, 6, 6356. [Google Scholar] [CrossRef]
- Shuttleworth, C.A. Type VIII Collagen. Int. J. Biochem. Cell Biol. 1997, 29, 1145–1148. [Google Scholar] [CrossRef]
- Ullah, M.; Sittinger, M.; Ringe, J. Extracellular Matrix of Adipogenically Differentiated Mesenchymal Stem Cells Reveals a Network of Collagen Filaments, Mostly Interwoven by Hexagonal Structural Units. Matrix Biol. 2013, 32, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Ruehl, M.; Erben, U.; Schuppan, D.; Wagner, C.; Zeller, A.; Freise, C.; Al-Hasani, H.; Loesekann, M.; Notter, M.; Wittig, B.M.; et al. The Elongated First Fibronectin Type III Domain of Collagen XIV Is an Inducer of Quiescence and Differentiation in Fibroblasts and Preadipocytes. J. Biol. Chem. 2005, 280, 38537–38543. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Collagen | Pathological Condition | Expression/Manifestation | References |
|---|---|---|---|
| I | Fibrotic AT T1D | Increased expression in obese AT compared to lean subjects Decreased level of crosslinked telopeptide in the serum of T1D patients with retinopathy | [34,37,111,112] |
| II | T2D | Increased expression in epididymal AT in diabetic (db/db) mice | [34] |
| III | Fibrotic AT T1D T2D | Increased expression in obese AT compared to lean subjects and in patients with T1D with retinopathy Increased levels of procollagen aminopeptide in patients with T2D and progressing diabetic nephropathy | [37,111,113] |
| IV | T2D | Increased Col4a1 and Col4a2 expression in the WAT of diabetic mice Downregulation of COL4A1 in SAT after gastric bypass and improvement of HOMA-IR | [34,107] |
| V | Fibrotic AT Impaired glucose metabolism Insulin resistance | Increased expression in the WAT of diabetic mice Increased expression in obesity; accumulation in fibrotic areas, especially around large blood vessels. Fibrotic promotion causes insulin resistance Lack of Col5a3 leads to impaired glucose metabolism | [34,38,114,115] |
| VI | Fibrotic AT Altered glucose metabolism | Increased expression in obese patients associates with pericellular fibrosis Biomarker in AT fibrosis Conflicting results in glucose metabolism; insulin resistance vs. improved glucose metabolism Increased expression in obese/diabetic mice while downregulated in obese humans | [34,37,116,117,118,119,120,121] |
| VIII | Obesity | In the twins study, an increased expression of COL8A2 was found in the heavier twin | [47] |
| XII | Insulin resistance | In the twins study, COL12A1 expression positively associated with LDL cholesterol, and low expression associated with increased insulin sensitivity | [47] |
| XV | Obesity | Increased expression in AT in HFD-induced obesity in mice Regulates adipocyte apoptosis and inflammation in AT | [122,123,124] |
| XVIII | Visceral obesity in T2D Dyslipidemia Lipodystrophy | Specific SNPs associate with obesity in patients with T2D (c.1136C > T) and with abnormal circulating lipid content (c.331G > A, p.Gly111Arg) Patients with Knobloch syndrome due to COL18A1 null mutation have fasting hypertriglyceridemia Lack of Col18a1 in mice causes lipodystrophy, T2D, and increased serum triglyceride levels Expression of long isoforms of collagen XVIII in visceral fat positively correlates with free fatty acid levels in the plasma | [125,126,127,128,129,130] |
| XXIV | T2D | Increased expression in insulin-resistant obese VAT | [131] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jääskeläinen, I.; Petäistö, T.; Mirzarazi Dahagi, E.; Mahmoodi, M.; Pihlajaniemi, T.; Kaartinen, M.T.; Heljasvaara, R. Collagens Regulating Adipose Tissue Formation and Functions. Biomedicines 2023, 11, 1412. https://doi.org/10.3390/biomedicines11051412
Jääskeläinen I, Petäistö T, Mirzarazi Dahagi E, Mahmoodi M, Pihlajaniemi T, Kaartinen MT, Heljasvaara R. Collagens Regulating Adipose Tissue Formation and Functions. Biomedicines. 2023; 11(5):1412. https://doi.org/10.3390/biomedicines11051412
Chicago/Turabian StyleJääskeläinen, Iida, Tiina Petäistö, Elahe Mirzarazi Dahagi, Mahdokht Mahmoodi, Taina Pihlajaniemi, Mari T. Kaartinen, and Ritva Heljasvaara. 2023. "Collagens Regulating Adipose Tissue Formation and Functions" Biomedicines 11, no. 5: 1412. https://doi.org/10.3390/biomedicines11051412