
Novel Therapeutic Targets for the Treatment of Atopic Dermatitis
 , and
, and
Abstract
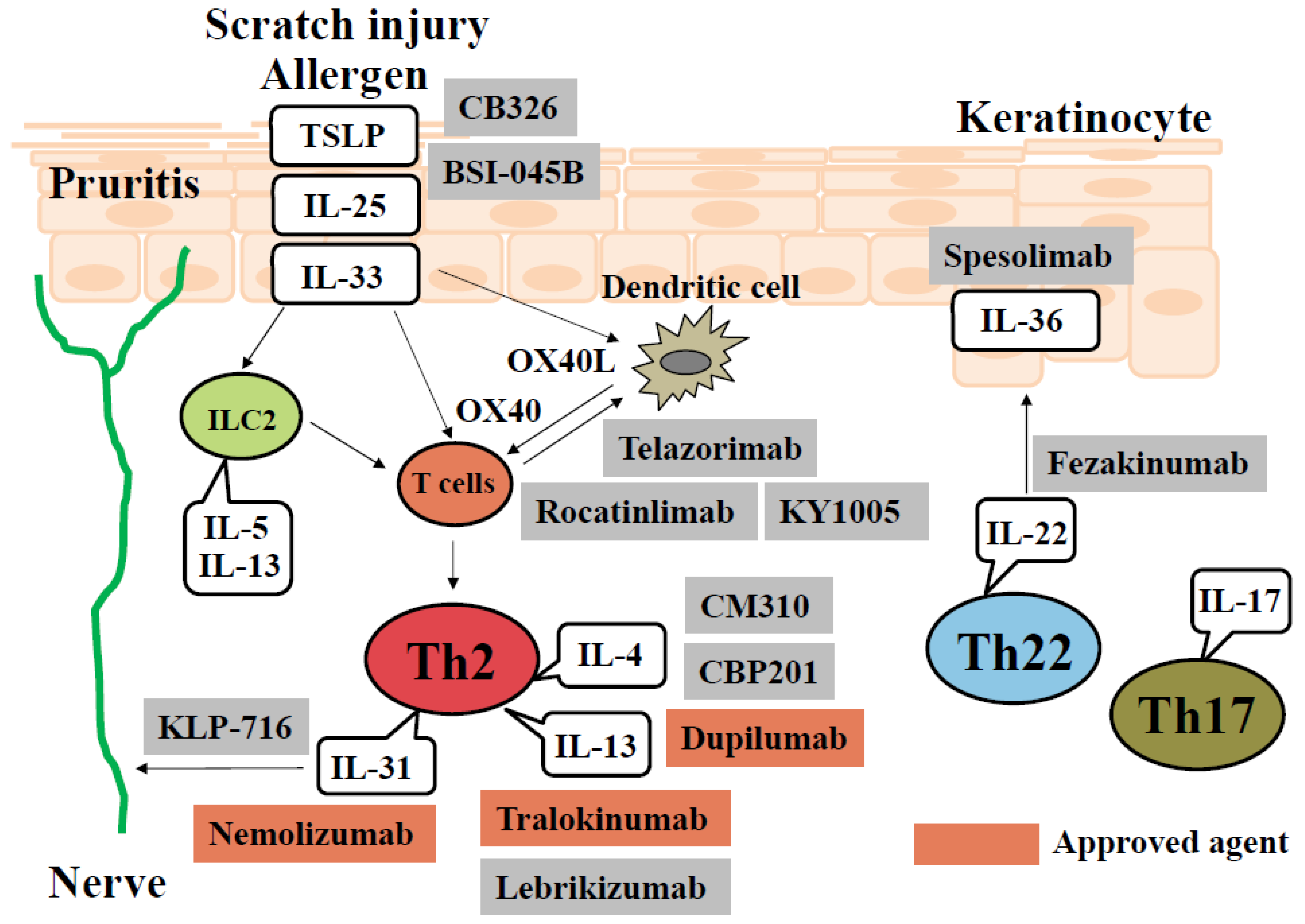
:1. Introduction
2. Cytokines
2.1. IL-4/IL-13
2.2. IL-22
2.3. IL-31
2.4. IL-23/IL-17 Axis and IL-36
2.5. IL-33
2.6. TSLP (Thymic Stromal Lymphopoietin)
3. Janus Kinase (JAK)—Signal Transduction and Activator of Transcription (STAT) Pathway
4. OX40-OX40L Interaction
5. Histamine H4 Receptor (H4R)
6. Aryl Hydrocarbon Receptor (AHR)
7. Phosphodiesterase 4 (PDE4)
8. Microbiome
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and recent advances in the pathophysiology of atopic dermatitis. J. Dermatol. 2021, 48, 130–139. [Google Scholar] [CrossRef]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y. ILC2s in skin disorders. Allergol. Int. 2023, 72, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell-mediated allergic inflammation. J. Allergy Clin. Immunol. 2007, 120, 238–244, quiz 245–246. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.A.; Cork, M.J.; Amagai, M.; De Benedetto, A.; Kabashima, K.; Hamilton, J.D.; Rossi, A.B. Type 2 inflammation contributes to skin barrier dysfunction in atopic dermatitis. JID Innov. 2022, 2, 100131. [Google Scholar] [CrossRef] [PubMed]
- Tokura, Y.; Hayano, S. Subtypes of atopic dermatitis: From phenotype to endotype. Allergol. Int. 2022, 71, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; de Bruin-Weller, M.; Gooderham, M.; Cather, J.C.; Weisman, J.; Pariser, D.; Simpson, E.L.; Papp, K.A.; Hong, H.C.; Rubel, D.; et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): A 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet 2017, 389, 2287–2303. [Google Scholar] [CrossRef]
- Trier, A.M.; Kim, B.S. Insights into atopic dermatitis pathogenesis lead to newly approved systemic therapies. Br. J. Dermatol. 2022, ljac016. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic dermatitis is an IL-13-dominant disease with greater molecular heterogeneity compared to psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef]
- Koppes, S.A.; Brans, R.; Ljubojevic Hadzavdic, S.; Frings-Dresen, M.H.; Rustemeyer, T.; Kezic, S. Stratum corneum tape stripping: Monitoring of inflammatory mediators in atopic dermatitis patients using topical therapy. Int. Arch. Allergy Immunol. 2016, 170, 187–193. [Google Scholar] [CrossRef]
- Szegedi, K.; Lutter, R.; Res, P.C.; Bos, J.D.; Luiten, R.M.; Kezic, S.; Middelkamp-Hup, M.A. Cytokine profiles in interstitial fluid from chronic atopic dermatitis skin. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 2136–2144. [Google Scholar] [CrossRef] [PubMed]
- Ungar, B.; Garcet, S.; Gonzalez, J.; Dhingra, N.; Correa da Rosa, J.; Shemer, A.; Krueger, J.G.; Suarez-Farinas, M.; Guttman-Yassky, E. An integrated model of atopic dermatitis biomarkers highlights the systemic nature of the disease. J. Investig. Dermatol. 2017, 137, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Langley, R.G.; Lacour, J.P.; Toth, D.; Laquer, V.; Beissert, S.; Wollenberg, A.; Herranz, P.; Pink, A.E.; Peris, K.; et al. Long-term 2-year safety and efficacy of tralokinumab in adults with moderate-to-severe atopic dermatitis: Interim analysis of the ECZTEND open-label extension trial. J. Am. Acad. Dermatol. 2022, 87, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Flohr, C.; Eichenfield, L.F.; Bieber, T.; Sofen, H.; Taïeb, A.; Owen, R.; Putnam, W.; Castro, M.; DeBusk, K.; et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: A randomized, placebo-controlled phase II trial (TREBLE). J. Am. Acad. Dermatol. 2018, 78, 863–871.e11. [Google Scholar] [CrossRef]
- Furue, M.; Ulzii, D.; Nakahara, T.; Tsuji, G.; Furue, K.; Hashimoto-Hachiya, A.; Kido-Nakahara, M. Implications of IL-13Rα2 in atopic skin inflammation. Allergol. Int. 2020, 69, 412–416. [Google Scholar] [CrossRef]
- Furue, M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic implications in atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 5382. [Google Scholar] [CrossRef]
- Lou, H.; Lu, J.; Choi, E.B.; Oh, M.H.; Jeong, M.; Barmettler, S.; Zhu, Z.; Zheng, T. Expression of IL-22 in the skin causes Th2-biased immunity, epidermal barrier dysfunction, and pruritus via stimulating epithelial Th2 cytokines and the GRP pathway. J. Immunol. 2017, 198, 2543–2555. [Google Scholar] [CrossRef]
- Hayashida, S.; Uchi, H.; Takeuchi, S.; Esaki, H.; Moroi, Y.; Furue, M. Significant correlation of serum IL-22 levels with CCL17 levels in atopic dermatitis. J. Dermatol. Sci. 2011, 61, 78–79. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Brunner, P.M.; Neumann, A.U.; Khattri, S.; Pavel, A.B.; Malik, K.; Singer, G.K.; Baum, D.; Gilleaudeau, P.; Sullivan-Whalen, M.; et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase-blind study. A randomized, double-blind, phase 2a trial. J. Am. Acad. Dermatol. 2018, 78, 872–881.e6. [Google Scholar] [CrossRef]
- Schallreuter, K.; Levenig, C.; Berger, J.; Umbert, J.; Winkelmann, R.; Wegener, L.; Correia, O.; Chosidow, O.; Saiag, P.; Bastuji-Garin, S.; et al. Severity scoring of atopic dermatitis: The SCORAD index. Consensus Report of the European Task Force on Atopic Dermatitis. Dermatology 1993, 186, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Yamamura, K.; Kido-Nakahara, M.; Nakahara, T.; Fukui, Y. Emerging role of interleukin-31 and interleukin-31 receptor in pruritus in atopic dermatitis. Allergy 2018, 73, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Orfali, R.L.; Aoki, V. Blockage of the IL-31 pathway as a potential target therapy for atopic dermatitis. Pharmaceutics 2023, 15, 577. [Google Scholar] [CrossRef] [PubMed]
- Arai, I.; Tsuji, M.; Takeda, H.; Akiyama, N.; Saito, S. A single dose of interleukin-31 (IL-31) causes continuous itch-associated scratching behaviour in mice. Exp. Dermatol. 2013, 22, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Feld, M.; Garcia, R.; Buddenkotte, J.; Katayama, S.; Lewis, K.; Muirhead, G.; Hevezi, P.; Plesser, K.; Schrumpf, H.; Krjutskov, K.; et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J. Allergy Clin. Immunol. 2016, 138, 500–508.e24. [Google Scholar] [CrossRef]
- Oyama, S.; Kitamura, H.; Kuramochi, T.; Higuchi, Y.; Matsushita, H.; Suzuki, T.; Goto, M.; Adachi, H.; Kasutani, K.; Sakamoto, A.; et al. Cynomolgus monkey model of interleukin-31-induced scratching depicts blockade of human interleukin-31 receptor A by a humanized monoclonal antibody. Exp. Dermatol. 2018, 27, 14–21. [Google Scholar] [CrossRef]
- Nemmer, J.M.; Kuchner, M.; Datsi, A.; Oláh, P.; Julia, V.; Raap, U.; Homey, B. Interleukin-31 signaling bridges the gap between immune cells, the nervous system and epithelial tissues. Front. Med. 2021, 8, 639097. [Google Scholar] [CrossRef]
- Kabashima, K.; Matsumura, T.; Komazaki, H.; Kawashima, M.; Nemolizumab JP01 and JP02 Study Group. Nemolizumab plus topical agents in patients with atopic dermatitis (AD) and moderate-to-severe pruritus provide improvement in pruritus and signs of AD for up to 68 weeks: Results from two phase III, long-term studies. Br. J. Dermatol. 2022, 186, 642–651. [Google Scholar] [CrossRef]
- Ständer, S.; Yosipovitch, G.; Lacour, J.P.; Legat, F.J.; Paul, C.; Reich, A.; Chaouche, K.; Ahmad, F.; Piketty, C. Nemolizumab efficacy in prurigo nodularis: Onset of action on itch and sleep disturbances. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1820–1825. [Google Scholar] [CrossRef]
- Mikhak, Z.; Bissonnette, R.; Siri, D.; Tyring, S.; Tessari, E.; Gandhi, R.; Fang, F.; Paolini, J. KPL-716, anti-oncostatin M receptor beta antibody, reduced pruritus in atopic dermatitis. J. Investig. Dermatol. 2019, 139, S96. [Google Scholar] [CrossRef]
- Ungar, B.; Pavel, A.B.; Li, R.; Kimmel, G.; Nia, J.; Hashim, P.; Kim, H.J.; Chima, M.; Vekaria, A.S.; Estrada, Y.; et al. Phase 2 randomized, double-blind study of IL-17 targeting with secukinumab in atopic dermatitis. J. Allergy Clin. Immunol. 2021, 147, 394–397. [Google Scholar] [CrossRef]
- Tyring, S.K.; Rich, P.; Tada, Y.; Beeck, S.; Messina, I.; Liu, J.; Huang, X.; Shumack, S. Risankizumab in patients with moderate-to-severe atopic dermatitis: A phase 2, randomized, double-blind, placebo-controlled study. Dermatol. Ther. 2023, 13, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Hanifin, J.M.; Thurston, M.; Omoto, M.; Cherill, R.; Tofte, S.J.; Graeber, M.; EASI Evaluator Group. The eczema area and severity index (EASI): Assessment of reliability in atopic dermatitis. Exp. Dermatol. 2001, 10, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Morita, A.; Tsai, T.F.; Yee, E.Y.W.; Okubo, Y.; Imafuku, S.; Zheng, M.; Li, L.; Quaresma, M.; Thoma, C.; Choon, S.E. Efficacy and safety of spesolimab in Asian patients with a generalized pustular psoriasis flare: Results from the randomized, double-blind, placebo-controlled Effisayil™ 1 study. J. Dermatol. 2023, 50, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Abramovits, W.; Saint-Cyr Proulx, É.; Lee, P.; Guttman-Yassky, E.; Zovko, E.; Sigmund, R.; Willcox, J.; Bieber, T. Spesolimab, an anti-interleukin-36 receptor antibody, in patients with moderate-to-severe atopic dermatitis: Results from a multicentre, randomized, double-blind, placebo-controlled, phase IIa study. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimäki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in atopic dermatitis: Expression profiles and modulation by triggering factors. J. Investig. Dermatol. 2012, 132, 1392–1400. [Google Scholar] [CrossRef]
- Tamagawa-Mineoka, R.; Okuzawa, Y.; Masuda, K.; Katoh, N. Increased serum levels of interleukin 33 in patients with atopic dermatitis. J. Am. Acad. Dermatol. 2014, 70, 882–888. [Google Scholar] [CrossRef]
- Yang, N.; Chen, Z.; Zhang, X.; Shi, Y. Novel targeted biological agents for the treatment of atopic dermatitis. BioDrugs 2021, 35, 401–415. [Google Scholar] [CrossRef]
- Laquer, V.; Parra, V.; Lacour, J.P.; Takahashi, H.; Knorr, J.; Okragly, A.J.; James, D.E.; Sims, J.T.; Chang, C.Y.; Chao, J.; et al. Interleukin-33 antibody fails to demonstrate benefit in a phase II, double-blind, randomized, placebo-controlled study in adult patients with moderate-to-severe atopic dermatitis. Br. J. Dermatol. 2022, 187, 599–602. [Google Scholar] [CrossRef]
- Maurer, M.; Cheung, D.S.; Theess, W.; Yang, X.; Dolton, M.; Guttman, A.; Choy, D.F.; Dash, A.; Grimbaldeston, M.A.; Soong, W. Phase 2 randomized clinical trial of astegolimab in patients with moderate to severe atopic dermatitis. J. Allergy Clin. Immunol. 2022, 150, 1517–1524. [Google Scholar] [CrossRef]
- Liu, Y.J. Thymic stromal lymphopoietin: Master switch for allergic inflammation. J. Exp. Med. 2006, 203, 269–273. [Google Scholar] [CrossRef]
- Simpson, E.L.; Parnes, J.R.; She, D.; Crouch, S.; Rees, W.; Mo, M.; van der Merwe, R. Tezepelumab, an anti-thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J. Am. Acad. Dermatol. 2019, 80, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Tsiogka, A.; Kyriazopoulou, M.; Kontochristopoulos, G.; Nicolaidou, E.; Stratigos, A.; Rigopoulos, D.; Gregoriou, S. The JAK/STAT pathway and its selective inhibition in the treatment of atopic dermatitis: A systematic review. J. Clin. Med. 2022, 11, 4431. [Google Scholar] [CrossRef] [PubMed]
- Traidl, S.; Freimooser, S.; Werfel, T. Janus kinase inhibitors for the therapy of atopic dermatitis. Allergol. Sel. 2021, 5, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Teixeira, H.D.; Simpson, E.L.; Costanzo, A.; De Bruin-Weller, M.; Barbarot, S.; Prajapati, V.H.; Lio, P.; Hu, X.; Wu, T.; et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: A randomized clinical trial. JAMA Dermatol. 2021, 157, 1047–1055, Erratum in JAMA Dermatol. 2022, 158, 219. [Google Scholar] [CrossRef]
- Reich, K.; Teixeira, H.D.; de Bruin-Weller, M.; Bieber, T.; Soong, W.; Kabashima, K.; Werfel, T.; Zeng, J.; Huang, X.; Hu, X.; et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): Results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2169–2181, Erratum in Lancet 2021, 397, 2336; Erratum in Lancet 2021, 398, 746. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Fabbrocini, G.; Genco, L.; Martora, F.; Potestio, L.; Patruno, C. Rapid improvement in pruritus in atopic dermatitis patients treated with upadacitinib: A real-life experience. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1497–1498. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Teixeira, H.D.; Simpson, E.L.; Papp, K.A.; Pangan, A.L.; Blauvelt, A.; Thaçi, D.; Chu, C.Y.; Hong, H.C.; Katoh, N.; et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): Results from two replicate double-blind, randomised controlled phase 3 trials. Lancet 2021, 397, 2151–2168, Erratum in Lancet 2021, 397, 2150. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Sinclair, R.; Forman, S.; Wollenberg, A.; Aschoff, R.; Cork, M.; Bieber, T.; Thyssen, J.P.; Yosipovitch, G.; Flohr, C.; et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet 2020, 396, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Bieber, T.; Simpson, E.L.; Silverberg, J.I.; Thaçi, D.; Paul, C.; Pink, A.E.; Kataoka, Y.; Chu, C.Y.; DiBonaventura, M.; Rojo, R.; et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N. Engl. J. Med. 2021, 384, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Lacour, J.P.; Spelman, L.; Galimberti, R.; Eichenfield, L.F.; Bissonnette, R.; King, B.A.; Thyssen, J.P.; Silverberg, J.I.; Bieber, T.; et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: Results from two randomized monotherapy phase III trials. Br. J. Dermatol. 2020, 183, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Bieber, T.; Reich, K.; Paul, C.; Tsunemi, Y.; Augustin, M.; Lacour, J.P.; Ghislain, P.D.; Dutronc, Y.; Liao, R.; Yang, F.E.; et al. Efficacy and safety of baricitinib in combination with topical corticosteroids in patients with moderate-to-severe atopic dermatitis with inadequate response, intolerance or contraindication to ciclosporin: Results from a randomized, placebo-controlled, phase III clinical trial (BREEZE-AD4). Br. J. Dermatol. 2022, 187, 338–352. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cheng, J.; Yang, H.; Tu, W.; Zhang, Y.; Luo, X.; Wang, H. The efficacy and safety of Janus kinase inhibitors in patients with atopic dermatitis: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2022, 87, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Szepietowski, J.C.; Kircik, L.; Toth, D.; Eichenfield, L.F.; Forman, S.B.; Kuligowski, M.E.; Kallender, H.; Sun, K.; Ren, H.; et al. Long-term safety and disease control with ruxolitinib cream in atopic dermatitis: Results from two phase 3 studies. J. Am. Acad. Dermatol. 2022, 88, 1008–1016. [Google Scholar] [CrossRef]
- Nakagawa, H.; Nemoto, O.; Igarashi, A.; Saeki, H.; Kabashima, K.; Oda, M.; Nagata, T. Delgocitinib ointment in pediatric patients with atopic dermatitis: A phase 3, randomized, double-blind, vehicle-controlled study and a subsequent open-label, long-term study. J. Am. Acad. Dermatol. 2021, 85, 854–862. [Google Scholar] [CrossRef]
- Nakagawa, H.; Nemoto, O.; Igarashi, A.; Saeki, H.; Kaino, H.; Nagata, T. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: A phase 3, randomized, double-blind, vehicle-controlled study and an open-label, long-term extension study. J. Am. Acad. Dermatol. 2020, 82, 823–831, Erratum in J. Am. Acad. Dermatol. 2021, 85, 1069. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Papp, K.A.; Poulin, Y.; Gooderham, M.; Raman, M.; Mallbris, L.; Wang, C.; Purohit, V.; Mamolo, C.; Papacharalambous, J.; et al. Topical tofacitinib for atopic dermatitis: A phase IIa randomized trial. Br. J. Dermatol. 2016, 175, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Piscitelli, S.C.; Pavel, A.B.; McHale, K.; Jett, J.E.; Collins, J.; Gillmor, D.; Tabolt, G.; Li, R.; Song, T.; Zhang, N.; et al. A phase 1b, randomized, single-center trial of topical cerdulatinib (DMVT-502) in patients with mild-to-moderate atopic dermatitis. J. Investig. Dermatol. 2021, 141, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Redmond, W.L. Current clinical trial landscape of OX40 agonists. Curr. Oncol. Rep. 2022, 24, 951–960. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Pavel, A.B.; Zhou, L.; Estrada, Y.D.; Zhang, N.; Xu, H.; Peng, X.; Wen, H.C.; Govas, P.; Gudi, G.; et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 482–493.e7. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Simpson, E.L.; Reich, K.; Kabashima, K.; Igawa, K.; Suzuki, T.; Mano, H.; Matsui, T.; Esfandiari, E.; Furue, M. An anti-OX40 antibody to treat moderate-to-severe atopic dermatitis: A multicentre, double-blind, placebo-controlled phase 2b study. Lancet 2023, 401, 204–214. [Google Scholar] [CrossRef]
- Weidinger, S.; Bieber, T.; Cork, M. 34312 Treatment with amlitelimab (KY1005, SAR445229): A novel nondepleting anti-OX40Ligand (OX40L) mAb reduces IL-13 serum levels in a phase 2a randomized placebo-controlled trial in patients with moderate to severe atopic dermatitis. J. Am. Acad. Dermatol. 2022, 87, AB123. [Google Scholar] [CrossRef]
- Branco, A.C.C.C.; Yoshikawa, F.S.Y.; Pietrobon, A.J.; Sato, M.N. Role of histamine in modulating the immune response and inflammation. Mediat. Inflamm. 2018, 2018, 9524075. [Google Scholar] [CrossRef] [PubMed]
- Yamaura, K.; Shigemori, A.; Suwa, E.; Ueno, K. Expression of the histamine H4 receptor in dermal and articular tissues. Life Sci. 2013, 92, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, Y.; Hirasawa, N. The role of histamine H1 and H4 receptors in atopic dermatitis: From basic research to clinical study. Allergol. Int. 2014, 63, 533–542. [Google Scholar] [CrossRef]
- Murata, Y.; Song, M.; Kikuchi, H.; Hisamichi, K.; Xu, X.L.; Greenspan, A.; Kato, M.; Chiou, C.F.; Kato, T.; Guzzo, C.; et al. Phase 2a, randomized, double-blind, placebo-controlled, multicenter, parallel-group study of an H4 R-antagonist (JNJ-39758979) in Japanese adults with moderate atopic dermatitis. J. Dermatol. 2015, 42, 129–139. [Google Scholar] [CrossRef]
- Werfel, T.; Layton, G.; Yeadon, M.; Whitlock, L.; Osterloh, I.; Jimenez, P.; Liu, W.; Lynch, V.; Asher, A.; Tsianakas, A.; et al. Efficacy and safety of the histamine H4 receptor antagonist ZPL-3893787 in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 1830–1837.e4. [Google Scholar] [CrossRef]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Aryl hydrocarbon receptor in atopic dermatitis and psoriasis. Int. J. Mol. Sci. 2019, 20, 5424. [Google Scholar] [CrossRef]
- Napolitano, M.; Fabbrocini, G.; Martora, F.; Picone, V.; Morelli, P.; Patruno, C. Role of Aryl Hydrocarbon Receptor Activation in Inflammatory Chronic Skin Diseases. Cells 2021, 10, 3559. [Google Scholar] [CrossRef]
- Dale, B.A.; Presland, R.B.; Lewis, S.P.; Underwood, R.A.; Fleckman, P. Transient expression of epidermal filaggrin in cultured cells causes collapse of intermediate filament networks with alteration of cell shape and nuclear integrity. J. Investig. Dermatol. 1997, 108, 179–187. [Google Scholar] [CrossRef]
- Nomura, T.; Sandilands, A.; Akiyama, M.; Liao, H.; Evans, A.T.; Sakai, K.; Ota, M.; Sugiura, H.; Yamamoto, K.; Sato, H.; et al. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J. Allergy Clin. Immunol. 2007, 119, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Illig, T.; Baurecht, H.; Irvine, A.D.; Rodriguez, E.; Diaz-Lacava, A.; Klopp, N.; Wagenpfeil, S.; Zhao, Y.; Liao, H.; et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J. Allergy Clin. Immunol. 2006, 118, 214–219, Erratum in J. Allergy Clin. Immunol. 2006, 118, 922; Erratum in J. Allergy Clin. Immunol. 2006, 118, 724. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Kypriotou, M.; Huber, M.; Hohl, D. The human epidermal differentiation complex: Cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp. Dermatol. 2012, 21, 643–649. [Google Scholar] [CrossRef]
- Takei, K.; Mitoma, C.; Hashimoto-Hachiya, A.; Takahara, M.; Tsuji, G.; Nakahara, T.; Furue, M. Galactomyces fermentation filtrate prevents T helper 2-mediated reduction of filaggrin in an aryl hydrocarbon receptor-dependent manner. Clin. Exp. Dermatol. 2015, 40, 786–793. [Google Scholar] [CrossRef]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Antioxidative phytochemicals accelerate epidermal terminal differentiation via the AHR-OVOL1 pathway: Implications for atopic dermatitis. Acta Derm. Venereol. 2018, 98, 918–923. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Kiyomatsu-Oda, M.; Takemura, M.; Ohno, F.; Ito, T.; Morino-Koga, S.; Mitoma, C.; Nakahara, T.; Uchi, H.; et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. 2017, 8, e2931. [Google Scholar] [CrossRef]
- Hashimoto-Hachiya, A.; Tsuji, G.; Murai, M.; Yan, X.; Furue, M. Upregulation of FLG, LOR, and IVL expression by Rhodiola renulate root extract via aryl hydrocarbon receptor: Differential involvement of OVOL. Int. J. Mol. Sci. 2018, 19, 1654. [Google Scholar] [CrossRef]
- Hirano, A.; Goto, M.; Mitsui, T.; Hashimoto-Hachiya, A.; Tsuji, G.; Furue, M. Antioxidant Artemisia princeps extract enhances the expression of filaggrin and loricrin via the AHR/OVOL1 pathway. Int. J. Mol. Sci. 2017, 18, 1948. [Google Scholar] [CrossRef]
- van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schröder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Investig. 2013, 123, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.; Teng, A.; Bilanchone, V.; Agrawal, A.; Li, B.; Dai, X. Ovol1 regulates the growth arrest of embryonic epidermal progenitor cells and represses c-myc transcription. J. Cell Biol. 2006, 173, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Takahara, M.; Uchi, H.; Matsuda, T.; Chiba, T.; Takeuchi, S.; Yasukawa, F.; Moroi, Y.; Furue, M. Identification of ketoconazole as an AhR-Nrf2 activator in cultured human keratinocytes: The basis of its anti-inflammatory effect. J. Investig. Dermatol. 2012, 132, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Newton, E.M.; Hsiao, J.; Shi, V.Y. Aryl hydrocarbon receptor/nuclear factor E2-related factor 2 (AHR/NRF2) signalling: A novel therapeutic target for atopic dermatitis. Exp. Dermatol. 2022, 31, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Edamitsu, T.; Taguchi, K.; Okuyama, R.; Yamamoto, M. AHR and NRF2 in skin homeostasis and atopic dermatitis. Antioxidants 2022, 11, 227. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Hidaka, T.; Ogawa, E.; Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Fujimura, T.; Aiba, S.; Nakayama, K.; Okuyama, R.; et al. The aryl hydrocarbon receptor AhR links atopic dermatitis and air pollution via induction of the neurotrophic factor artemin. Nat. Immunol. 2017, 18, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Edamitsu, T.; Taguchi, K.; Kobayashi, E.H.; Okuyama, R.; Yamamoto, M. Aryl Hydrocarbon Receptor Directly Regulates Artemin Gene Expression. Mol. Cell. Biol. 2019, 39, e00190-19. [Google Scholar] [CrossRef]
- Abolhasani, R.; Araghi, F.; Tabary, M.; Aryannejad, A.; Mashinchi, B.; Robati, R.M. The impact of air pollution on skin and related disorders: A comprehensive review. Dermatol. Ther. 2021, 34, e14840. [Google Scholar] [CrossRef]
- Roberts, W. Air pollution and skin disorders. Int. J. Women’s Dermatol. 2021, 7, 91–97. [Google Scholar] [CrossRef]
- Kim, J.; Kim, E.H.; Oh, I.; Jung, K.; Han, Y.; Cheong, H.K.; Ahn, K. Symptoms of atopic dermatitis are influenced by outdoor air pollution. J. Allergy Clin. Immunol. 2013, 132, 495–498.e491. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lee, K.; Lee, Y.M.; Lee, J.H.; Lee, S.I.; Yu, S.D.; Paek, D. Acute health effects of urban fine and ultrafine particles on children with atopic dermatitis. Environ. Res. 2011, 111, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Kim, J.; Goleva, E.; Berdyshev, E.; Lee, J.; Vang, K.A.; Lee, U.H.; Han, S.; Leung, S.; Hall, C.F.; et al. Particulate matter causes skin barrier dysfunction. JCI Insight 2021, 6, e145185. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Uibel, S.; Braun, M.; Klingelhoefer, D.; Takemura, M.; Groneberg, D.A. Tobacco smoke particles and indoor air quality (ToPIQ)-the protocol of a new study. J. Occup. Med. Toxicol. 2011, 6, 35. [Google Scholar] [CrossRef]
- Bissonnette, R.; Stein Gold, L.; Rubenstein, D.S.; Tallman, A.M.; Armstrong, A. Tapinarof in the treatment of psoriasis: A review of the unique mechanism of action of a novel therapeutic aryl hydrocarbon receptor-modulating agent. J. Am. Acad. Dermatol. 2021, 84, 1059–1067. [Google Scholar] [CrossRef]
- Strober, B.; Stein Gold, L.; Bissonnette, R.; Armstrong, A.W.; Kircik, L.; Tyring, S.K.; Piscitelli, S.C.; Brown, P.M.; Rubenstein, D.S.; Tallman, A.M.; et al. One-year safety and efficacy of tapinarof cream for the treatment of plaque psoriasis: Results from the PSOARING 3 trial. J. Am. Acad. Dermatol. 2022, 87, 800–806. [Google Scholar] [CrossRef]
- Peppers, J.; Paller, A.S.; Maeda-Chubachi, T.; Wu, S.; Robbins, K.; Gallagher, K.; Kraus, J.E. A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 89–98.e3. [Google Scholar] [CrossRef]
- Smith, S.H.; Jayawickreme, C.; Rickard, D.J.; Nicodeme, E.; Bui, T.; Simmons, C.; Coquery, C.M.; Neil, J.; Pryor, W.M.; Mayhew, D.; et al. Tapinarof Is a Natural AhR Agonist that Resolves Skin Inflammation in Mice and Humans. J. Investig. Dermatol. 2017, 137, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Vu, Y.H.; Hashimoto-Hachiya, A.; Takemura, M.; Yumine, A.; Mitamura, Y.; Nakahara, T.; Furue, M.; Tsuji, G. IL-24 Negatively Regulates Keratinocyte Differentiation Induced by Tapinarof, an Aryl Hydrocarbon Receptor Modulator: Implication in the Treatment of Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 9412. [Google Scholar] [CrossRef]
- Schafer, P.H.; Adams, M.; Horan, G.; Truzzi, F.; Marconi, A.; Pincelli, C. Apremilast normalizes gene expression of inflammatory mediators in human keratinocytes and reduces antigen-induced atopic dermatitis in mice. Drugs R&D 2019, 19, 329–338. [Google Scholar] [CrossRef]
- Volf, E.M.; Au, S.C.; Dumont, N.; Scheinman, P.; Gottlieb, A.B. A phase 2, open-label, investigator-initiated study to evaluate the safety and efficacy of apremilast in subjects with recalcitrant allergic contact or atopic dermatitis. J. Drugs Dermatol. 2012, 11, 341–346. [Google Scholar] [CrossRef]
- Paller, A.S.; Tom, W.L.; Lebwohl, M.G.; Blumenthal, R.L.; Boguniewicz, M.; Call, R.S.; Eichenfield, L.F.; Forsha, D.W.; Rees, W.C.; Simpson, E.L.; et al. Efficacy and safety of crisaborole ointment, a novel, nonsteroidal phosphodiesterase 4 (PDE4) inhibitor for the topical treatment of atopic dermatitis (AD) in children and adults. J. Am. Acad. Dermatol. 2016, 75, 494–503.e6, Erratum in J. Am. Acad. Dermatol. 2017, 76, 777. [Google Scholar] [CrossRef] [PubMed]
- Saeki, H.; Ito, K.; Yokota, D.; Tsubouchi, H. Difamilast ointment in adult patients with atopic dermatitis: A phase 3 randomized, double-blind, vehicle-controlled trial. J. Am. Acad. Dermatol. 2022, 86, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Thurston, A.W., Jr.; Osborne, D.W.; Snyder, S.; Higham, R.C.; Burnett, P.; Berk, D.R. Pharmacokinetics of roflumilast cream in chronic plaque psoriasis: Data from phase I to phase III studies. Am. J. Clin. Dermatol. 2023, 24, 315–324. [Google Scholar] [CrossRef]
- Gooderham, M.; Kircik, L.; Zirwas, M.; Lee, M.; Kempers, S.; Draelos, Z.; Ferris, L.; Jones, T.; Saint-Cyr Proulx, E.; Bissonnette, R.; et al. The safety and efficacy of roflumilast cream 0.15% and 0.05% in patients with atopic dermatitis: Randomized, double-blind, phase 2 proof of concept study. J. Drugs Dermatol. 2023, 22, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, R.D.; Bandier, J.; Skov, L.; Engstrand, L.; Johansen, J.D. The role of the skin microbiome in atopic dermatitis: A systematic review. Br. J. Dermatol. 2017, 177, 1272–1278. [Google Scholar] [CrossRef]
- Natarelli, N.; Gahoonia, N.; Sivamani, R.K. Bacteriophages and the microbiome in dermatology: The role of the phageome and a potential therapeutic strategy. Int. J. Mol. Sci. 2023, 24, 2695. [Google Scholar] [CrossRef]
- Khadka, V.D.; Key, F.M.; Romo-González, C.; Martínez-Gayosso, A.; Campos-Cabrera, B.L.; Gerónimo-Gallegos, A.; Lynn, T.C.; Durán-McKinster, C.; Coria-Jiménez, R.; Lieberman, T.D.; et al. The skin microbiome of patients with atopic dermatitis normalizes gradually during treatment. Front. Cell Infect. Microbiol. 2021, 11, 720674. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Hata, T.R.; Tong, Y.; Cheng, J.Y.; Shafiq, F.; Butcher, A.M.; Salem, S.S.; Brinton, S.L.; Rudman Spergel, A.K.; Johnson, K.; et al. Development of a human skin commensal microbe for bacteriotherapy of atopic dermatitis and use in a phase 1 randomized clinical trial. Nat. Med. 2021, 27, 700–709. [Google Scholar] [CrossRef]
- Maura, D.; Elmekki, N.; Goddard, C.A. The ammonia oxidizing bacterium Nitrosomonas eutropha blocks T helper 2 cell polarization via the anti-inflammatory cytokine IL-10. Sci. Rep. 2021, 11, 14162. [Google Scholar] [CrossRef]


{kind=link}
| Phase I/II | Phase III | Approved | |
|---|---|---|---|
| IL-4/IL-13 | CM310 CBP201 | Lebrikizumab | * Tralokinumab |
| IL-22 | Fezakinumab | ||
| IL-31RA/OSMRβ | KPL-716 | ** Nemolizumab | |
| IL-23·IL-36 | Spesolimab | ||
| TSLP | CM326 BSI-045B | ||
| JAK-STAT | Tofacitinib Cerdulatinib | ** Delgocitinib *** Ruxocitinib | |
| OX40-OX40L | Telazorlimab Rocatinlimab KY1005 | ||
| H4R | LEO152020 | ||
| AHR | Tapinarof | ||
| PDE4 | Roflumilast | *** Crisaborole * Difamilast | |
| Microbiome | ShA9 B244 | ||
| * USA and Europe only | |||
| ** Japan only | |||
| *** USA only |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuji, G.; Yamamura, K.; Kawamura, K.; Kido-Nakahara, M.; Ito, T.; Nakahara, T. Novel Therapeutic Targets for the Treatment of Atopic Dermatitis. Biomedicines 2023, 11, 1303. https://doi.org/10.3390/biomedicines11051303
Tsuji G, Yamamura K, Kawamura K, Kido-Nakahara M, Ito T, Nakahara T. Novel Therapeutic Targets for the Treatment of Atopic Dermatitis. Biomedicines. 2023; 11(5):1303. https://doi.org/10.3390/biomedicines11051303
Chicago/Turabian StyleTsuji, Gaku, Kazuhiko Yamamura, Koji Kawamura, Makiko Kido-Nakahara, Takamichi Ito, and Takeshi Nakahara. 2023. "Novel Therapeutic Targets for the Treatment of Atopic Dermatitis" Biomedicines 11, no. 5: 1303. https://doi.org/10.3390/biomedicines11051303