
Facial Skin Microbiome: Aging-Related Changes and Exploratory Functional Associations with Host Genetic Factors, a Pilot Study
 , , , and
, , , and 
Abstract
:1. Introduction
2. Methods
2.1. Patients Recruitment and Facial Skin Sampling
2.2. Microbial DNA Extraction and Next Generation Sequencing (NGS)
2.3. Bioinformatics for Microbial Community Analysis
2.4. SNPs Genotyping
2.5. Data Availability Statement
3. Results
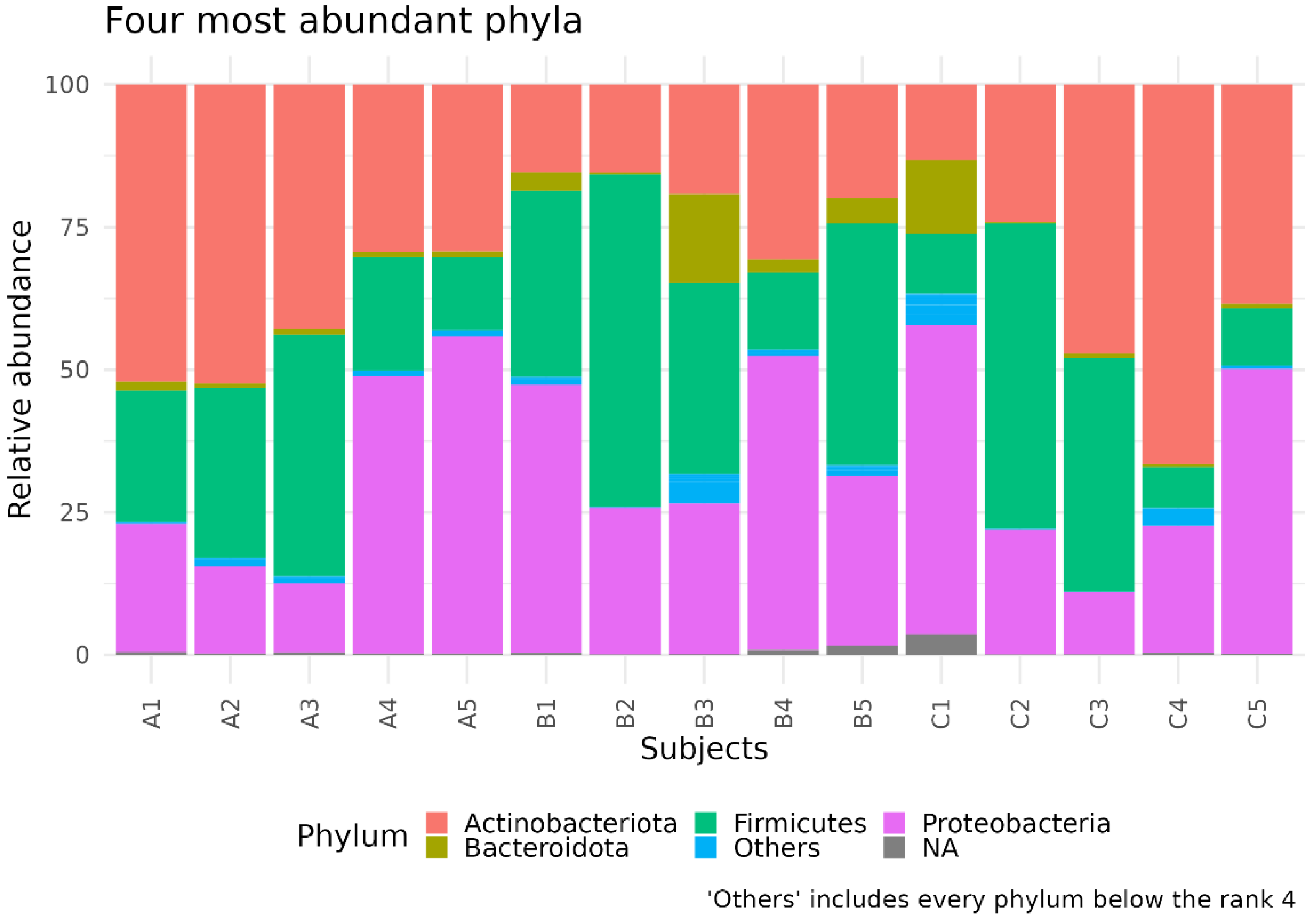
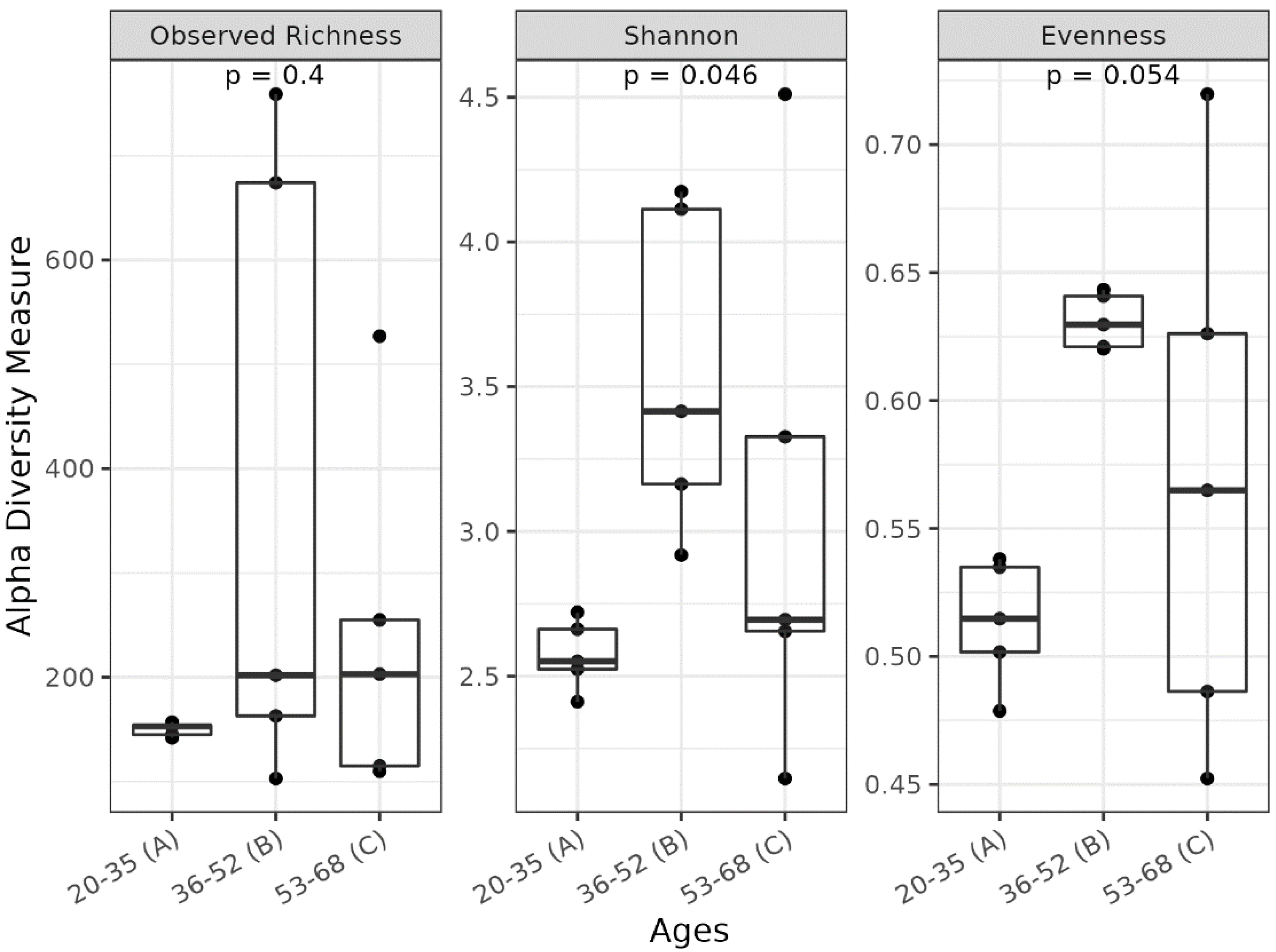
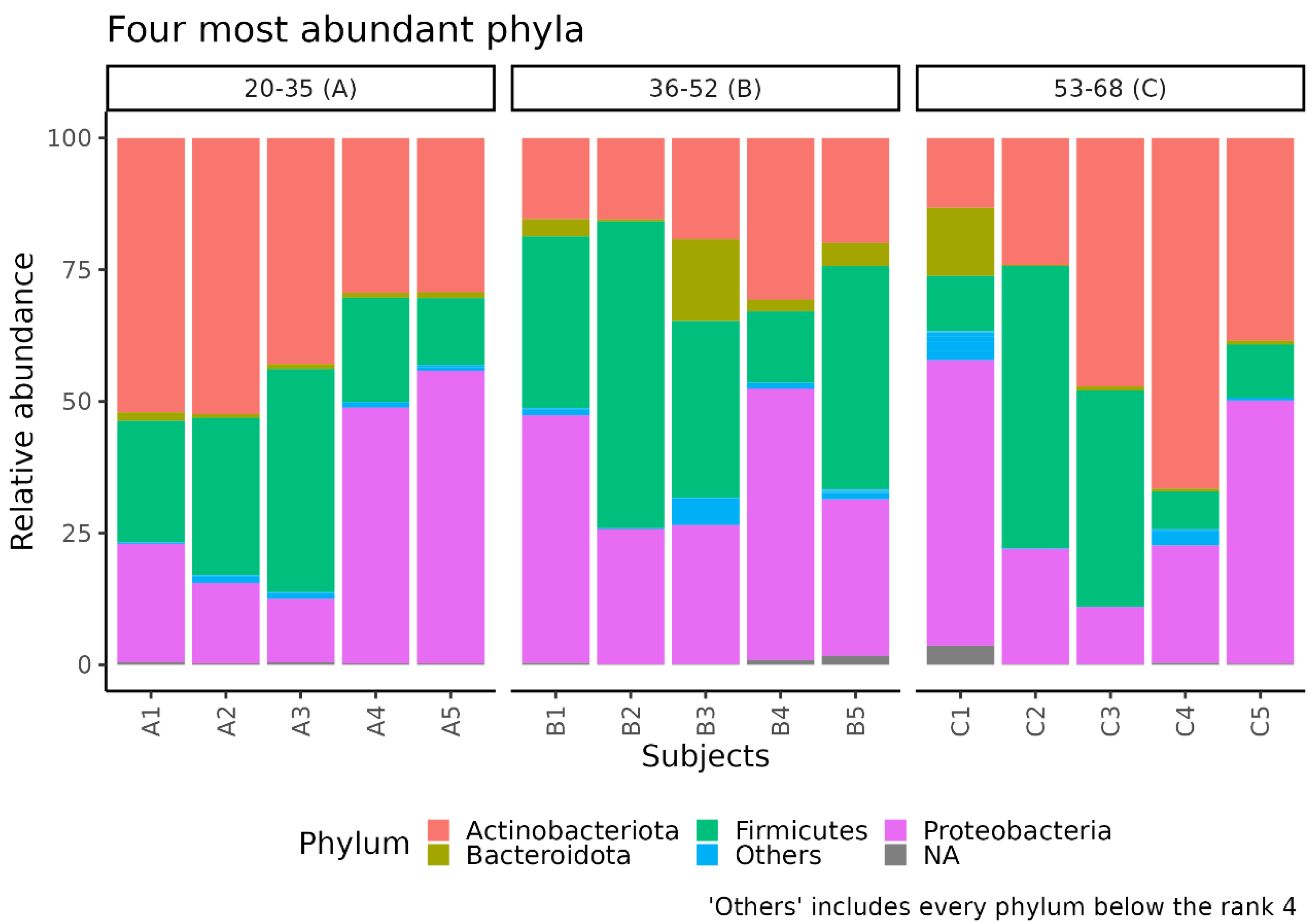
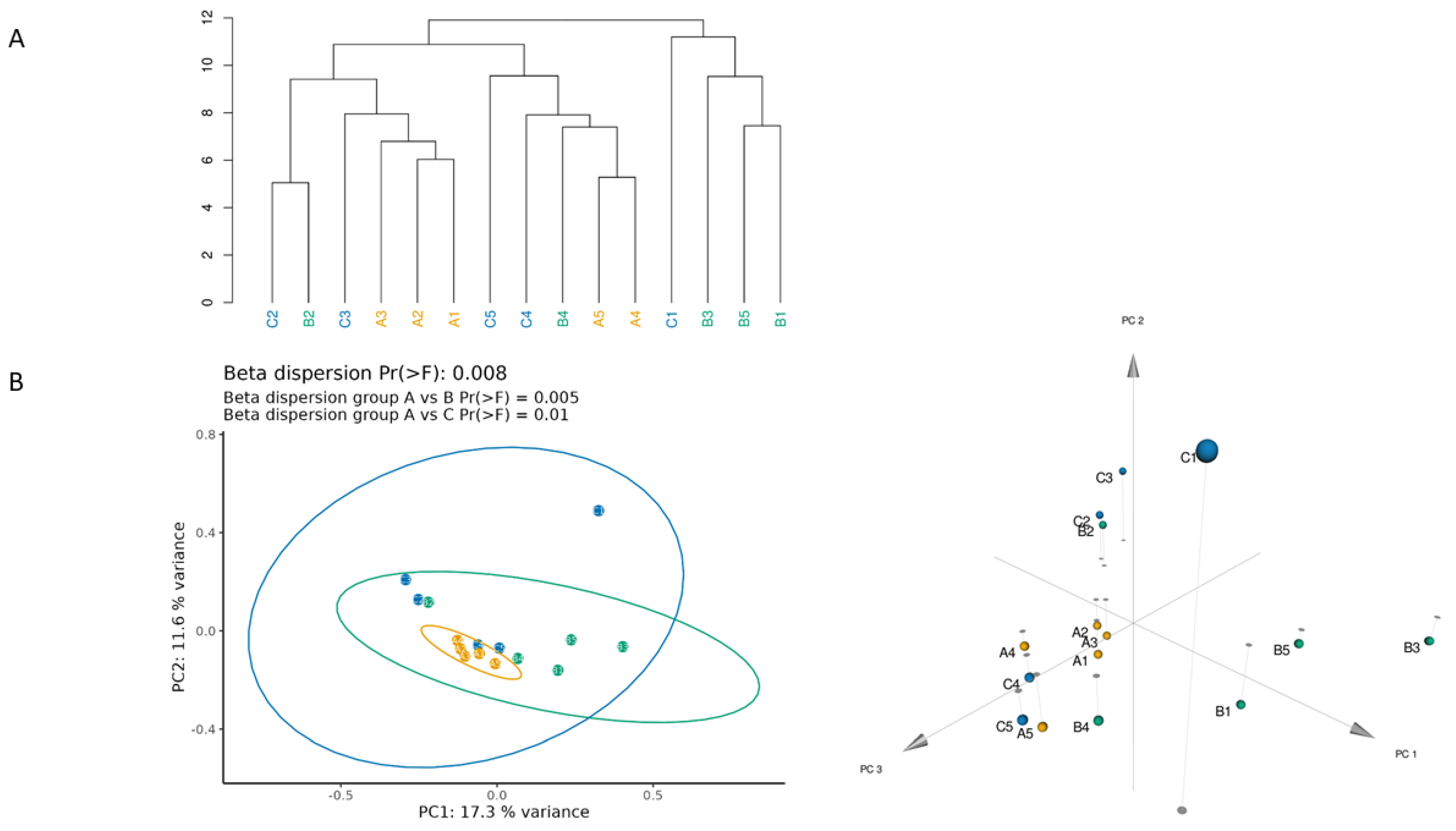
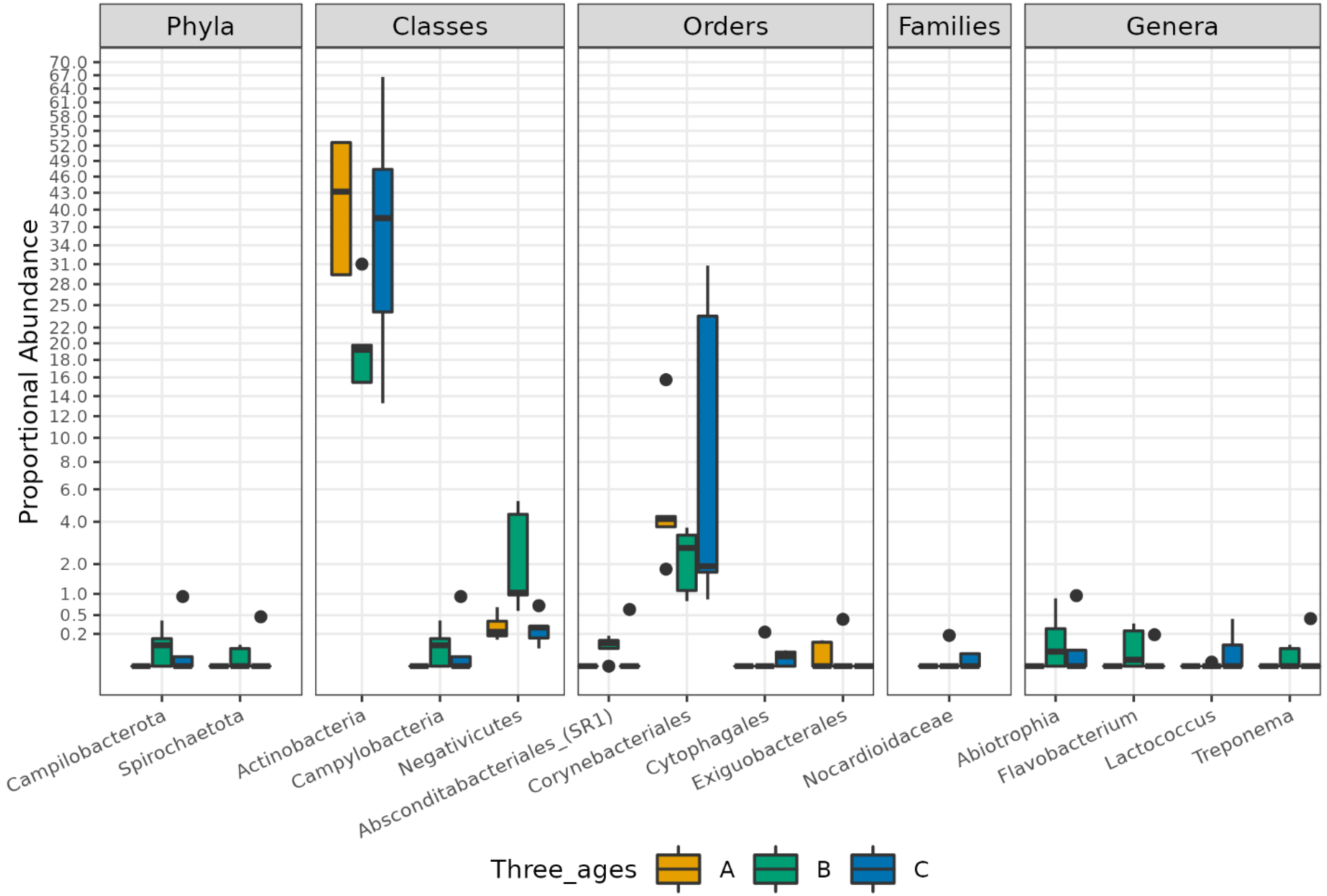
3.1. Different Facial Skin Microbial Signatures Related to Age
3.2. Functional Profiles of Age-Related Facial Skin Microbiota
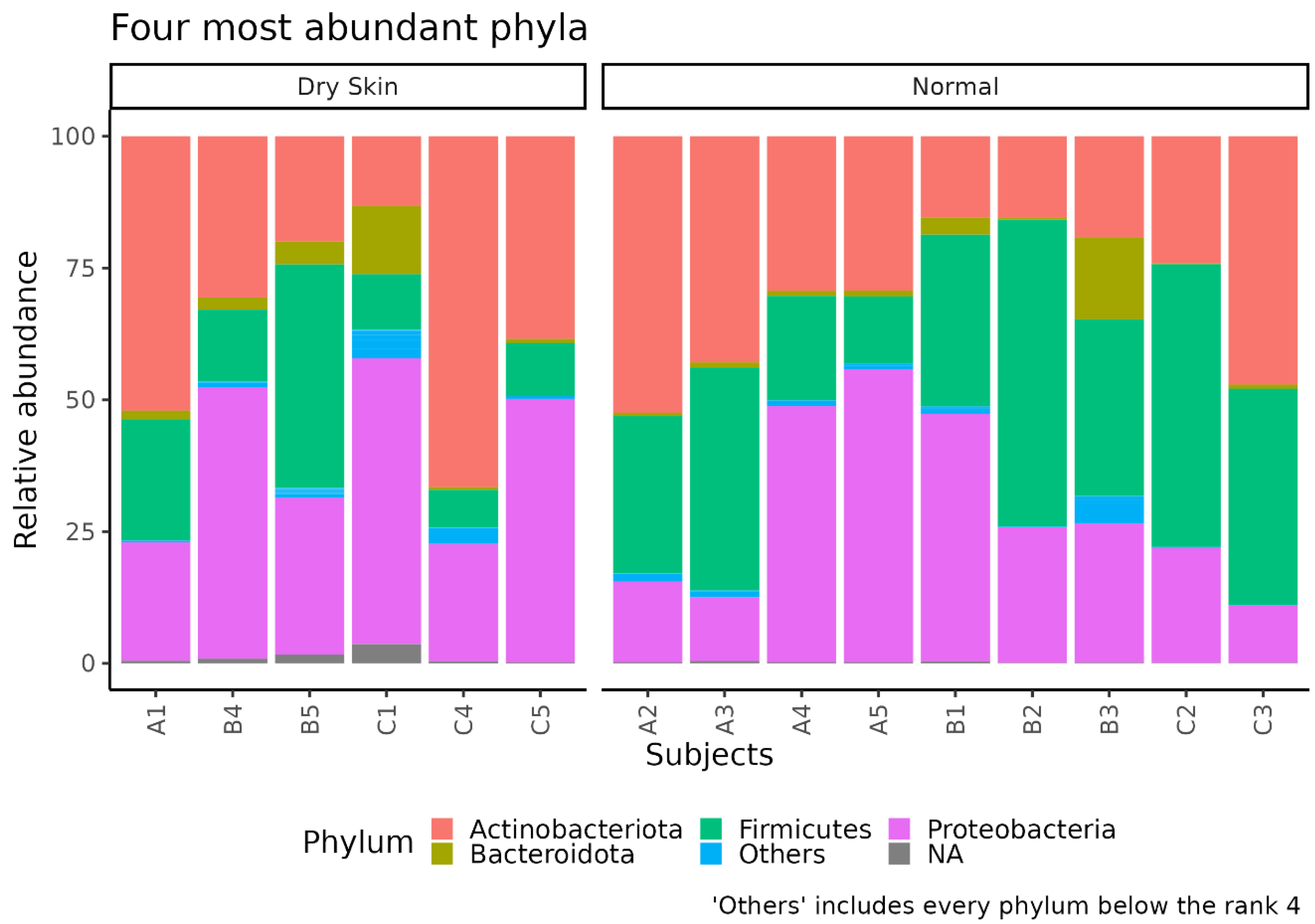
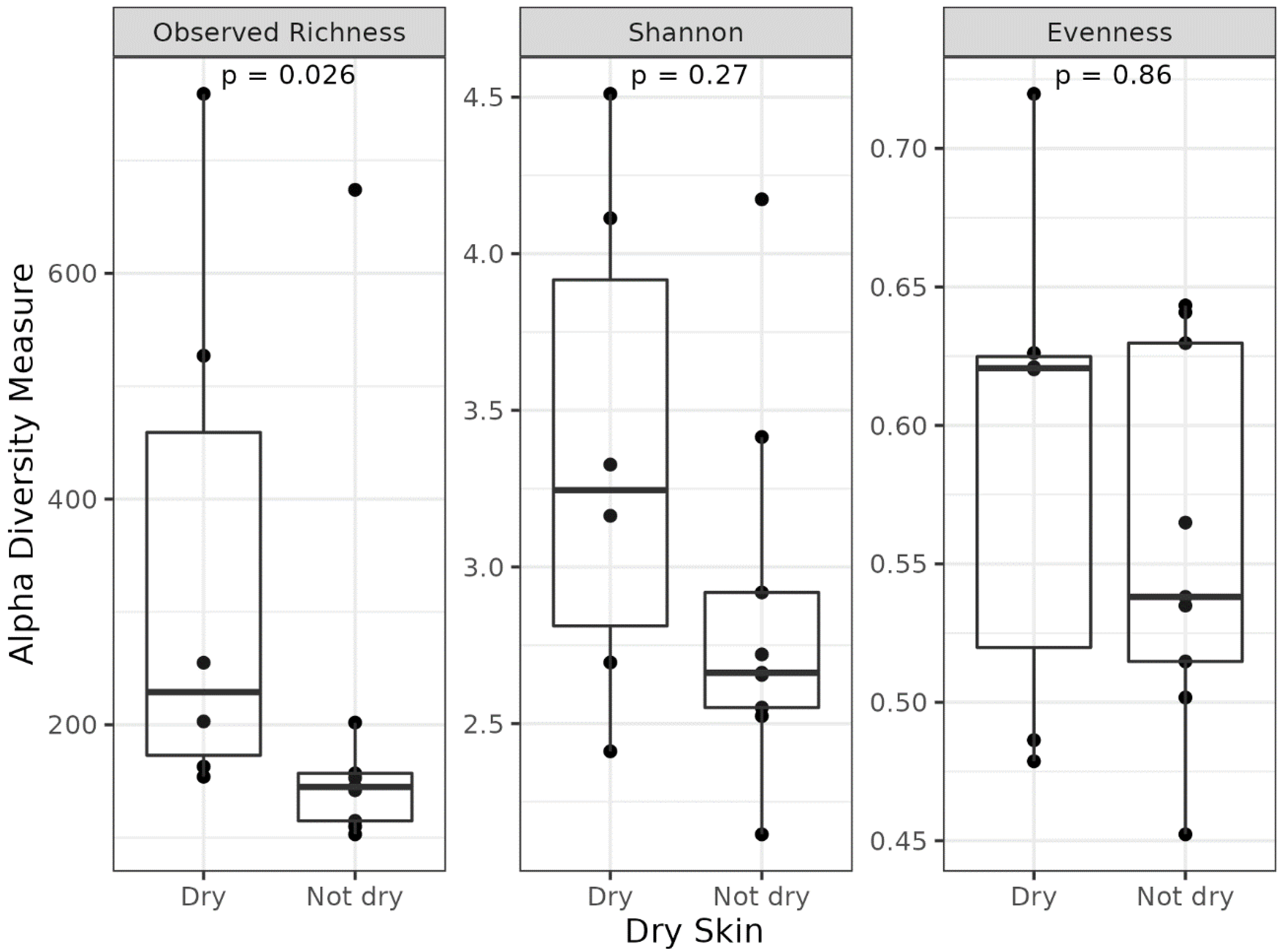
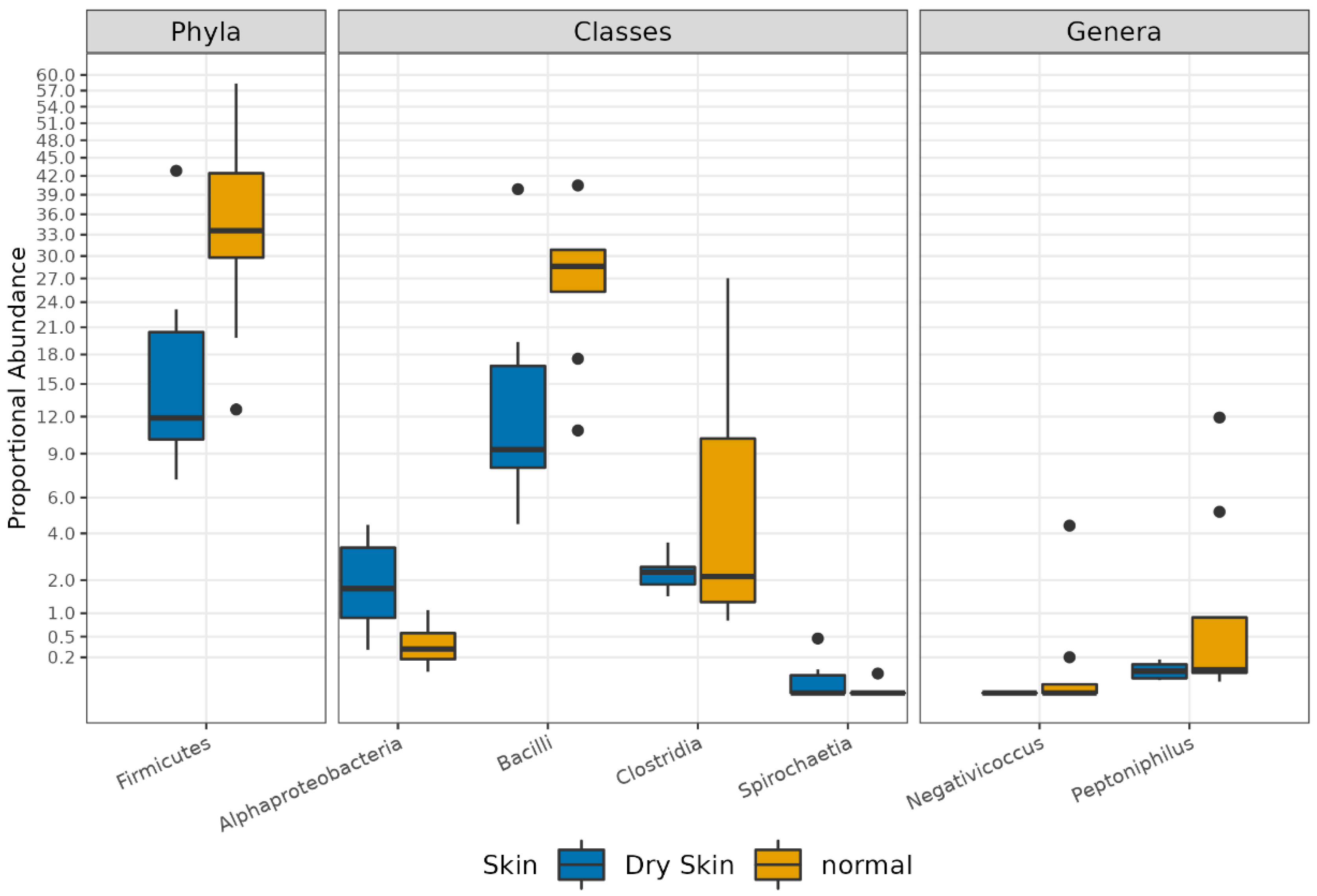
3.3. Different Microbial Signatures Depend on the Hydration Level
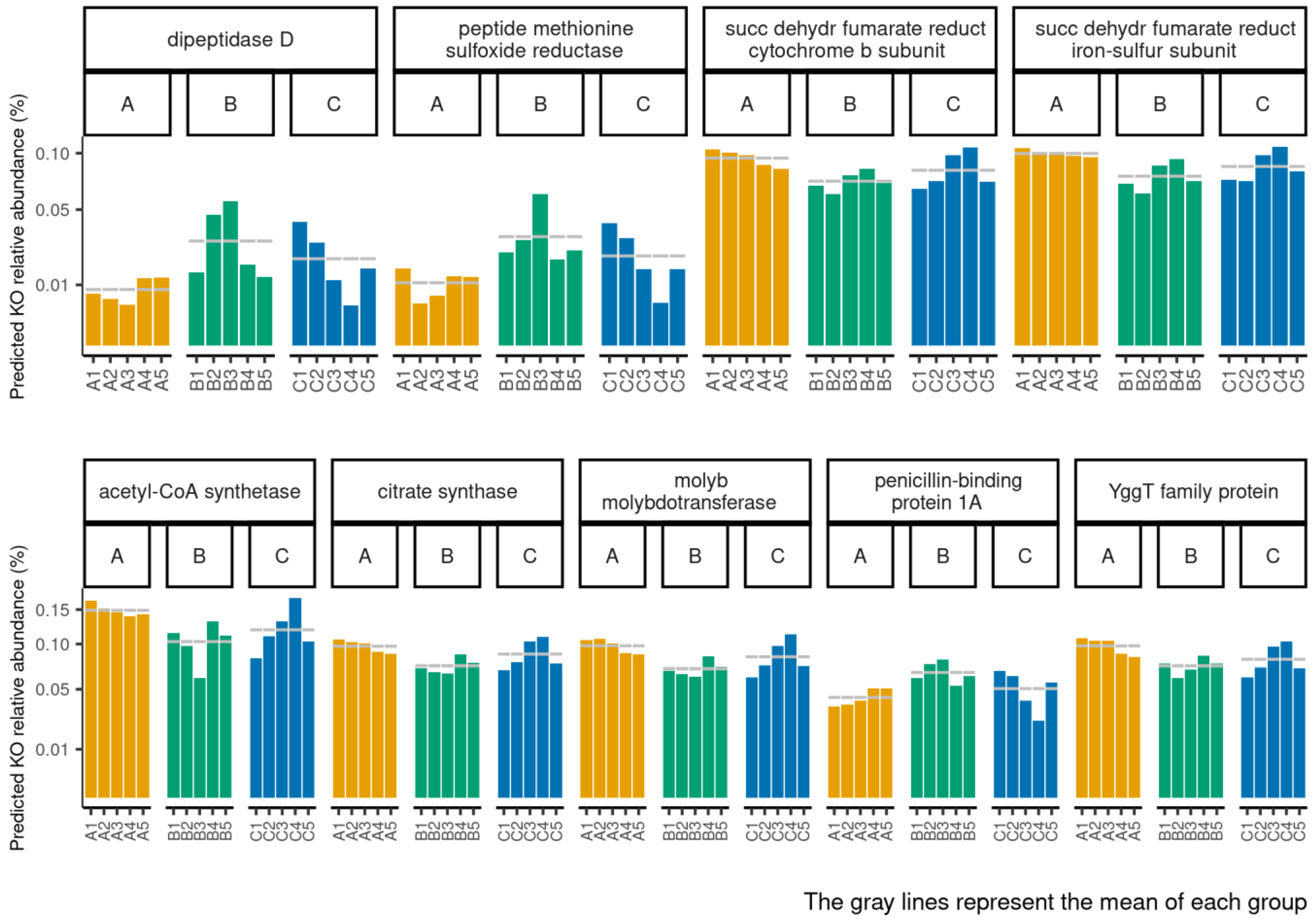
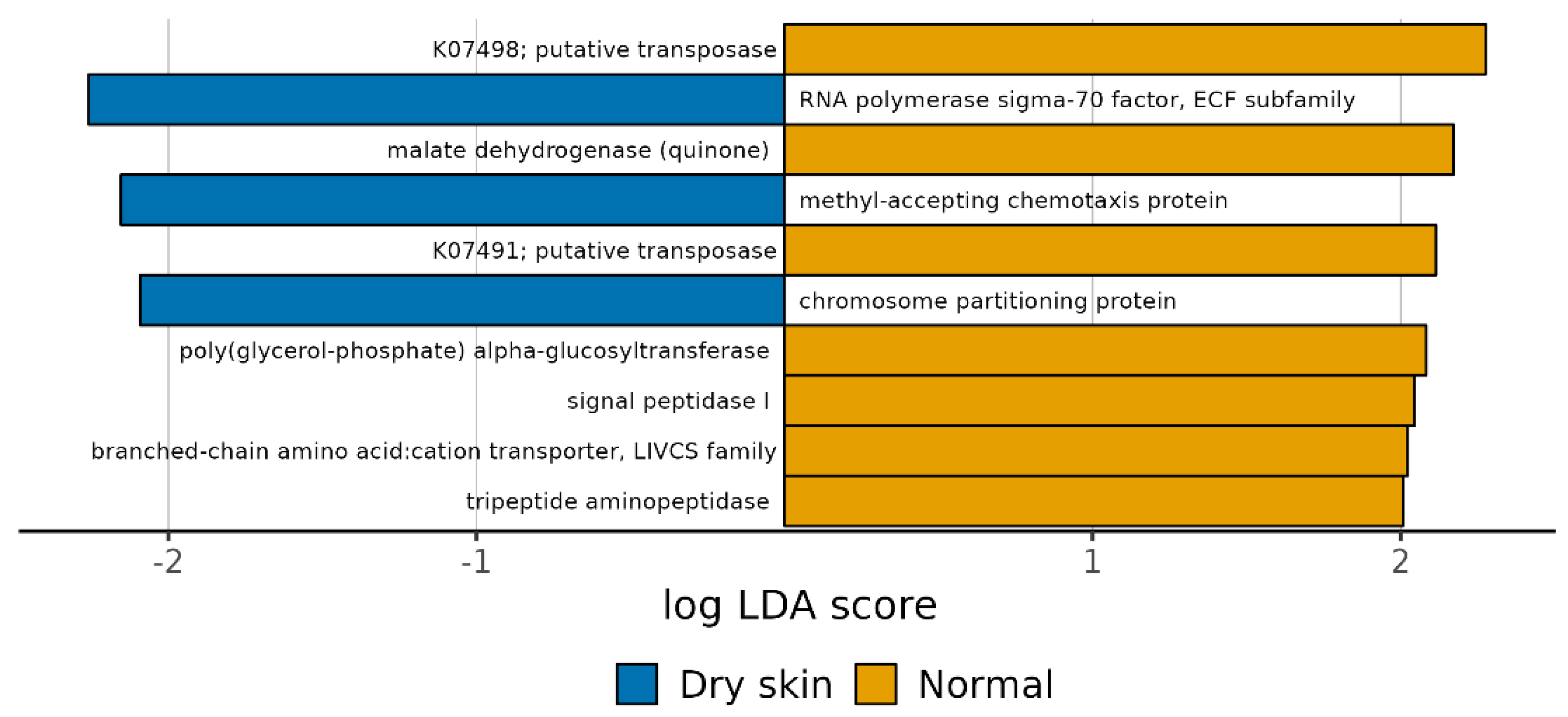
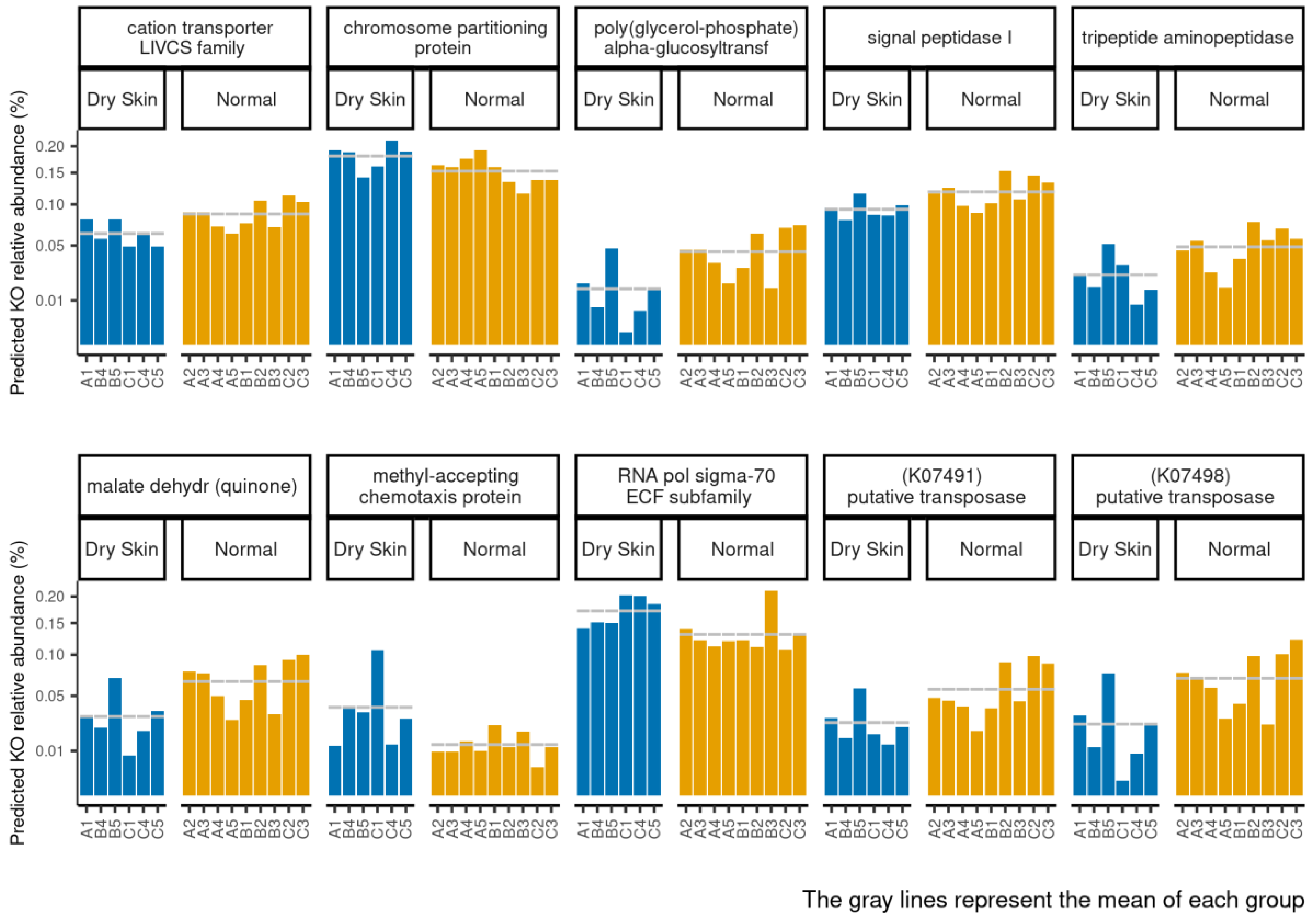
3.4. Functional Profiles of Hydration-Related Facial Skin Microbiota
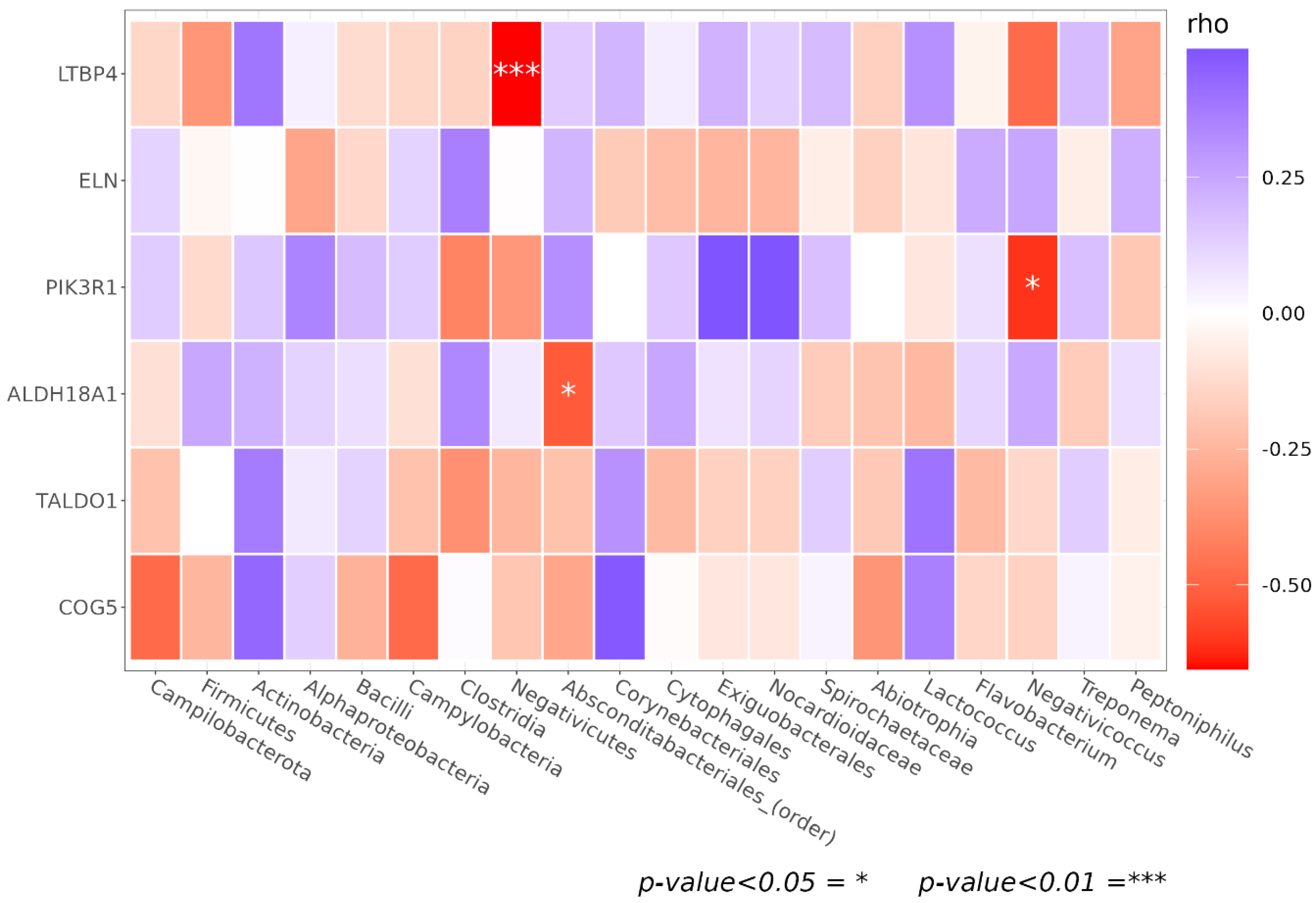
3.5. Genetic Association of Facial Skin Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Program, N.C.S.; Kong, H.H.; Segre, J.A. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meadow, J.F.; Bateman, A.C.; Herkert, K.M.; O’Connor, T.K.; Green, J.L. Significant changes in the skin microbiome mediated by the sport of roller derby. PeerJ 2013, 1, e53. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.; Kim, J.J.; Myeong, N.R.; Kim, T.; Park, T.; Kim, E.; Choi, J.Y.; Lee, J.; An, S.; et al. Fragile skin microbiomes in megacities are assembled by a predominantly niche-based process. Sci. Adv. 2018, 4, e1701581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Freeman, A.F.; Program, N.C.S.; Park, M.; Sokolic, R.; Candotti, F.; Holland, S.M.; Segre, J.A.; Kong, H.H. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013, 23, 2103–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statnikov, A.; Alekseyenko, A.V.; Li, Z.; Henaff, M.; Perez-Perez, G.I.; Blaser, M.J.; Aliferis, C.F. Microbiomic signatures of psoriasis: Feasibility and methodology comparison. Sci. Rep. 2013, 3, 2620. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Tseng, C.H.; Strober, B.E.; Pei, Z.; Blaser, M.J. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE 2008, 3, e2719. [Google Scholar] [CrossRef] [PubMed]
- Alekseyenko, A.V.; Perez-Perez, G.I.; De Souza, A.; Strober, B.; Gao, Z.; Bihan, M.; Li, K.; Methe, B.A.; Blaser, M.J. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome 2013, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Amaro-Ortiz, A.; Yan, B.; D’Orazio, J.A. Ultraviolet radiation, aging and the skin: Prevention of damage by topical cAMP manipulation. Molecules 2014, 19, 6202–6219. [Google Scholar] [CrossRef]
- Gilhar, A.; Ullmann, Y.; Karry, R.; Shalaginov, R.; Assy, B.; Serafimovich, S.; Kalish, R.S. Ageing of human epidermis: The role of apoptosis, Fas and telomerase. Br. J. Dermatol. 2004, 150, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, K.P.; Cua, A.B.; Maibach, H.I. Skin aging. Effect on transepidermal water loss, stratum corneum hydration, skin surface pH, and casual sebum content. Arch. Dermatol. 1991, 127, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Khachatryan, Z.A.; Ktsoyan, Z.A.; Manukyan, G.P.; Kelly, D.; Ghazaryan, K.A.; Aminov, R.I. Predominant role of host genetics in controlling the composition of gut microbiota. PLoS ONE 2008, 3, e3064. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Hamm, C.M.; Gulati, A.S.; Sartor, R.B.; Chen, H.; Wu, X.; Zhang, T.; Rohlf, F.J.; Zhu, W.; Gu, C.; et al. Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PLoS ONE 2012, 7, e26284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; McHardy, I.; Ruegger, P.; Goudarzi, M.; Kashyap, P.C.; Haritunians, T.; Li, X.; Graeber, T.G.; Schwager, E.; Huttenhower, C.; et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J. 2014, 8, 2193–2206. [Google Scholar] [CrossRef] [Green Version]
- Si, J.; Lee, S.; Park, J.M.; Sung, J.; Ko, G. Genetic associations and shared environmental effects on the skin microbiome of Korean twins. BMC Genom. 2015, 16, 992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, U.; Koop, U.; Leneveu-Duchemin, M.C.; Osterrieder, K.; Bielfeldt, S.; Chkarnat, C.; Degwert, J.; Hantschel, D.; Jaspers, S.; Nissen, H.P.; et al. Multicentre comparison of skin hydration in terms of physical-, physiological- and product-dependent parameters by the capacitive method (Corneometer CM 825). Int. J. Cosmet. Sci. 2003, 25, 45–53. [Google Scholar] [CrossRef]
- Pagliai, G.; Russo, E.; Niccolai, E.; Dinu, M.; Di Pilato, V.; Magrini, A.; Bartolucci, G.; Baldi, S.; Menicatti, M.; Giusti, B.; et al. Influence of a 3-month low-calorie Mediterranean diet compared to the vegetarian diet on human gut microbiota and SCFA: The CARDIVEG Study. Eur. J. Nutr. 2020, 59, 2011–2024. [Google Scholar] [CrossRef]
- Russo, E.; Giudici, F.; Ricci, F.; Scaringi, S.; Nannini, G.; Ficari, F.; Luceri, C.; Niccolai, E.; Baldi, S.; D’Ambrosio, M.; et al. Diving into inflammation: A pilot study exploring the dynamics of the immune-microbiota axis in ileal tissue layers of patients with Crohn’s disease. J. Crohns Colitis 2021, 15, 1500–1516. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA); John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Pal, L.R.; Moult, J. Genetic Basis of Common Human Disease: Insight into the Role of Missense SNPs from Genome-Wide Association Studies. J. Mol. Biol. 2015, 427, 2271–2289. [Google Scholar] [CrossRef] [Green Version]
- Wohlrab, J.; Hilpert, K.; Wohlrab, A. Characteristics of aging skin. Hautarzt 2014, 65, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Hinek, A.; Zhang, S.; Smith, A.C.; Callahan, J.W. Impaired elastic-fiber assembly by fibroblasts from patients with either Morquio B disease or infantile GM1-gangliosidosis is linked to deficiency in the 67-kD spliced variant of beta-galactosidase. Am. J. Hum. Genet. 2000, 67, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Krettek, A.; Sukhova, G.K.; Libby, P. Elastogenesis in human arterial disease: A role for macrophages in disordered elastin synthesis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 582–587. [Google Scholar] [CrossRef]
- Chen, Y.E.; Fischbach, M.A.; Belkaid, Y. Skin microbiota-host interactions. Nature 2018, 553, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Huang, S.; Leng, F.; Bei, Y.; Chen, Y.; Chen, M.; Hu, Y.; Huang, Y.; Xiang, Q. Analysis of the Bacterial Flora of Sensitive Facial Skin Among Women in Guangzhou. Clin. Cosmet. Investig. Dermatol. 2021, 14, 655–664. [Google Scholar] [CrossRef]
- Holland, K.T.; Bojar, R.A. Cosmetics: What is their influence on the skin microflora? Am. J. Clin. Dermatol. 2002, 3, 445–449. [Google Scholar] [CrossRef]
- Li, Z.; Bai, X.; Peng, T.; Yi, X.; Luo, L.; Yang, J.; Liu, J.; Wang, Y.; He, T.; Wang, X.; et al. New Insights into the Skin Microbial Communities and Skin Aging. Front. Microbiol. 2020, 11, 565549. [Google Scholar] [CrossRef]
- Shibagaki, N.; Suda, W.; Clavaud, C.; Bastien, P.; Takayasu, L.; Iioka, E.; Kurokawa, R.; Yamashita, N.; Hattori, Y.; Shindo, C.; et al. Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 2017, 7, 10567. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, J.J.; Myeong, N.R.; Kim, T.; Kim, D.; An, S.; Kim, H.; Park, T.; Jang, S.I.; Yeon, J.H.; et al. Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 2019, 9, 16748. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Son, S.M.; Park, H.; Kim, B.K.; Choi, I.S.; Kim, H.; Huh, C.S. Taxonomic profiling of skin microbiome and correlation with clinical skin parameters in healthy Koreans. Sci. Rep. 2021, 11, 16269. [Google Scholar] [CrossRef]
- Perez Perez, G.I.; Gao, Z.; Jourdain, R.; Ramirez, J.; Gany, F.; Clavaud, C.; Demaude, J.; Breton, L.; Blaser, M.J. Body Site Is a More Determinant Factor than Human Population Diversity in the Healthy Skin Microbiome. PLoS ONE 2016, 11, e0151990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutka, K.; Zychowska, M.; Reich, A. Diversity and Composition of the Skin, Blood and Gut Microbiome in Rosacea-A Systematic Review of the Literature. Microorganisms 2020, 8, 1756. [Google Scholar] [CrossRef]
- Antunes, L.C.; Poppleton, D.; Klingl, A.; Criscuolo, A.; Dupuy, B.; Brochier-Armanet, C.; Beloin, C.; Gribaldo, S. Phylogenomic analysis supports the ancestral presence of LPS-outer membranes in the Firmicutes. Elife 2016, 5, e14589. [Google Scholar] [CrossRef]
- Barbul, A. Proline precursors to sustain Mammalian collagen synthesis. J. Nutr. 2008, 138, 2021S–2024S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surazynski, A.; Miltyk, W.; Palka, J.; Phang, J.M. Prolidase-dependent regulation of collagen biosynthesis. Amino Acids 2008, 35, 731–738. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative stress in aging human skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasnacht, M.; Polacek, N. Oxidative Stress in Bacteria and the Central Dogma of Molecular Biology. Front. Mol. Biosci. 2021, 8, 671037. [Google Scholar] [CrossRef]
- Leff, J.W.; Fierer, N. Bacterial communities associated with the surfaces of fresh fruits and vegetables. PLoS ONE 2013, 8, e59310. [Google Scholar] [CrossRef] [Green Version]
- Schwer, B.; Bunkenborg, J.; Verdin, R.O.; Andersen, J.S.; Verdin, E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA 2006, 103, 10224–10229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, S.L.; Larcombe, D.L.; Logan, A.C.; West, C.; Burks, W.; Caraballo, L.; Levin, M.; Etten, E.V.; Horwitz, P.; Kozyrskyj, A.; et al. The skin microbiome: Impact of modern environments on skin ecology, barrier integrity, and systemic immune programming. World Allergy Organ. J. 2017, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Hospodsky, D.; Pickering, A.J.; Julian, T.R.; Miller, D.; Gorthala, S.; Boehm, A.B.; Peccia, J. Hand bacterial communities vary across two different human populations. Microbiology 2014, 160, 1144–1152. [Google Scholar] [CrossRef] [Green Version]
- Leung, M.H.; Wilkins, D.; Lee, P.K. Insights into the pan-microbiome: Skin microbial communities of Chinese individuals differ from other racial groups. Sci. Rep. 2015, 5, 11845. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Wu, X.; Zhou, J.; Li, X.; Huang, X.; Li, J.; Wu, J.; Bian, Q.; Wang, Y.; Tian, Y. NuRD mediates mitochondrial stress-induced longevity via chromatin remodeling in response to acetyl-CoA level. Sci. Adv. 2020, 6, eabb2529. [Google Scholar] [CrossRef] [PubMed]
- Flynn, T.C.; Petros, J.; Clark, R.E.; Viehman, G.E. Dry skin and moisturizers. Clin. Dermatol. 2001, 19, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, M.; Xu, Y.; Muhamadali, H.; White, I.; Wilkinson, M.; Hollywood, K.; Baguneid, M.; Goodacre, R.; Bayat, A. A microbiome and metabolomic signature of phases of cutaneous healing identified by profiling sequential acute wounds of human skin: An exploratory study. PLoS ONE 2020, 15, e0229545. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Female Subjects | Codes | Age (Years) | Hydration Levels (A.U.) * | Daily Sun Exposition | Daily Use of Sunscreen | Smoke ** | Skin Care Routine *** |
|---|---|---|---|---|---|---|---|
| 1 | A1 | 25 | >45 | No | No | Yes | No |
| 2 | A2 | 26 | <45 | Yes | No | No | Yes |
| 3 | A3 | 28 | <45 | Yes | Yes | Yes | No |
| 4 | A4 | 33 | <45 | No | No | No | Yes |
| 5 | A5 | 35 | <45 | Yes | No | No | Yes |
| 6 | B1 | 36 | <45 | No | No | Ex | No |
| 7 | B2 | 41 | <45 | Yes | Yes | Yes | Yes |
| 8 | B3 | 41 | <45 | No | No | No | No |
| 9 | B4 | 50 | >45 | No | No | No | Yes |
| 10 | B5 | 52 | >45 | Yes | Yes | No | Yes |
| 11 | C1 | 53 | >45 | Yes | Yes | No | Yes |
| 12 | C2 | 54 | <45 | No | Yes | Yes | Yes |
| 13 | C3 | 62 | <45 | Yes | Yes | No | Yes |
| 14 | C4 | 63 | >45 | No | No | No | Yes |
| 15 | C5 | 68 | >45 | No | No | No | No |
| Total | Assigned Taxonomy | % | |
|---|---|---|---|
| ASV | 2647 | 2202 | 83.19% |
| Genus | 506 | 487 | 96.24% |
| Family | 221 | 216 | 97.74% |
| Order | 117 | 115 | 98.29% |
| Class | 43 | 42 | 97.67% |
| Phylum | 22 | 21 | 95.45% |
| Gene Symbol | SNPs | Chr | SNP chr Position | Minor Allele in Caucasian | Mutation Aminoacid [Nucleotide] | Associated Taxon | p-Value |
|---|---|---|---|---|---|---|---|
| LTBP4 | rs1051303 | 19 | 41117806–41118306 | G | T [ACC] > A [GCC] | Negativicutes | 0.032 |
| rs1131620 | 19 | 41117619–41118119 | G | T [ACT] > A [GCT] | |||
| rs2303729 | 19 | 41110819–41111319 | A | I [ATA] > V [GTA] | |||
| ALDH18A1 | rs2275272 | 10 | 97387912–97388412 | A | T [ACC] > I [ATC] (minus strand) | Absconditabacteriales (SR1) | 0.045 |
| PIK3R1 | rs3730089 | 5 | 67587898–67588398 | A | M [ATG] > I [ATA] | Negativicoccus | 0.016 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, E.; Di Gloria, L.; Cerboneschi, M.; Smeazzetto, S.; Baruzzi, G.P.; Romano, F.; Ramazzotti, M.; Amedei, A. Facial Skin Microbiome: Aging-Related Changes and Exploratory Functional Associations with Host Genetic Factors, a Pilot Study. Biomedicines 2023, 11, 684. https://doi.org/10.3390/biomedicines11030684
Russo E, Di Gloria L, Cerboneschi M, Smeazzetto S, Baruzzi GP, Romano F, Ramazzotti M, Amedei A. Facial Skin Microbiome: Aging-Related Changes and Exploratory Functional Associations with Host Genetic Factors, a Pilot Study. Biomedicines. 2023; 11(3):684. https://doi.org/10.3390/biomedicines11030684
Chicago/Turabian StyleRusso, Edda, Leandro Di Gloria, Matteo Cerboneschi, Serena Smeazzetto, Gian Paolo Baruzzi, Francesca Romano, Matteo Ramazzotti, and Amedeo Amedei. 2023. "Facial Skin Microbiome: Aging-Related Changes and Exploratory Functional Associations with Host Genetic Factors, a Pilot Study" Biomedicines 11, no. 3: 684. https://doi.org/10.3390/biomedicines11030684