
Contextualizing the Role of Osteopontin in the Inflammatory Responses of Alzheimer’s Disease


 , , and
, , and
Abstract
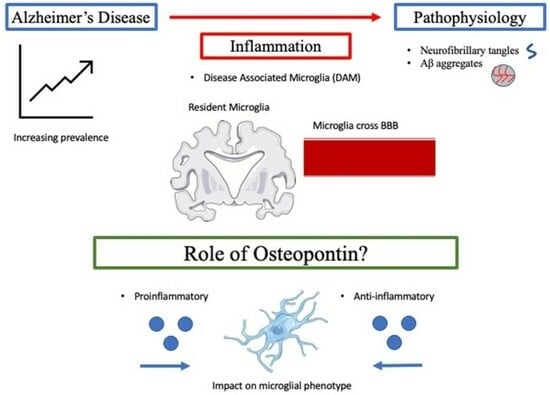
:
1. Introduction
2. Aβ/NFT Pathway
3. Etiology: Molecular Genetic and Epigenetic
4. ApoE and LDLR
5. Inflammation
6. Osteopontin




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Title | Major Findings |
|---|---|---|
| Fujita (2003) [117] | Immunohistochemical examination on intracranial calcification in neurodegenerative diseases | Neurodegenerative diseases have increased intracranial calcification (within basal ganglia and cerebellum) wherein the type 3 capillary calcospherites express increased OPN. |
| Wung (2007) [44] | Increased expression of the remodeling- and tumorigenic-associated factor osteopontin in pyramidal neurons of the Alzheimer’s disease brain | Increased OPN expression in pyramidal neurons of the Ca1 region of the hippocampus are associated with Aβ plaque of symptomatic AD patients. |
| Simonsen (2007) [126] | Novel Panel of Cerebrospinal Fluid Biomarkers for the Prediction of Progression to Alzheimer Dementia in Patients with Mild Cognitive Impairment | The phosphorylated C-terminal thrombin cleavage fragment of OPN is a biomarker of MCI that progresses to AD and is also associated with increased inflammation and gliosis. |
| Wirths (2010) [21] | Inflammatory changes are tightly associated with neurodegeneration in the brain and spinal cord of the APP/PS1KI mouse model of Alzheimer’s Disease (mouse study) | Identifies OPN as a marker of astrocyte and microglial activation and inflammation. |
| Sun (2013) [116] | Elevated osteopontin levels in mild cognitive impairment and Alzheimer’s disease | Elevated OPN levels in CSF are increased in patients with mild cognitive impairment (MCI) that progress to AD, but not in patients with stable MCI. |
| Rentsendorj (2018) [95] | A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models (mouse study) | Elevated OPN expression correlated with protective, anti-inflammatory phenotypes of systemic macrophages that accelerated Aβ fibril clearance. |
| Wang (2020) [133] | Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s Disease model (mouse study) | Antibody-mediated activation of the TREM2 receptor decreases Aβ-induced pathology while being associated with a proliferation of protective microglia and downregulation of OPN. |
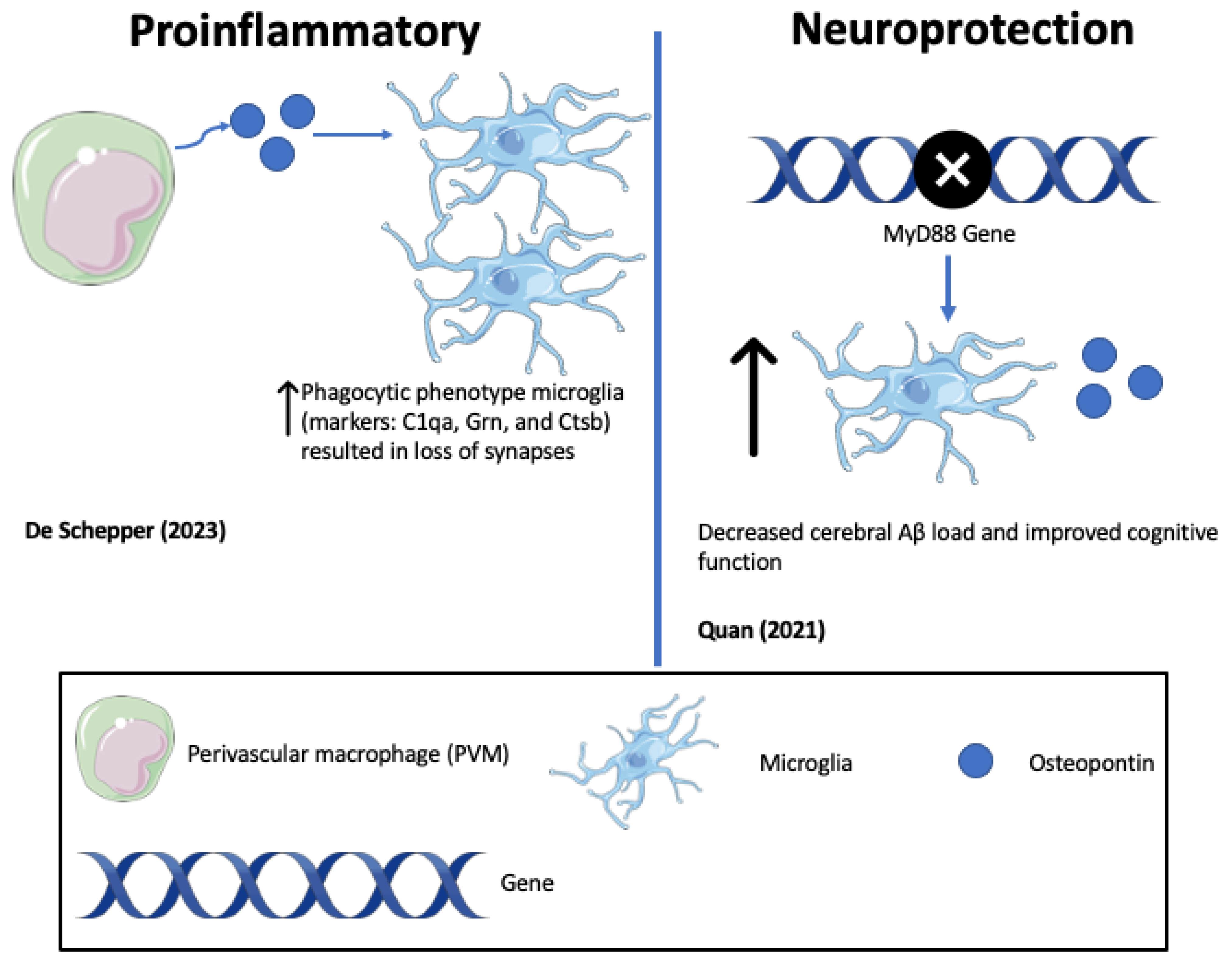
| Quan (2021) [135] | Haploinsufficiency of microglial MyD88 ameliorates Alzheimer’s pathology and vascular disorders in APP/PS1-transgenic mice. (mouse study) | Heterozygous deletion of MyD88 gene decreases cerebral Aβ load and improves cognitive function, while being associated with the upregulation of microglial OPN. |
| De Schepper (2023) [121] | Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease (mouse study) | OPN secreted by hippocampal perivascular macrophages (PVM), the primary responders to toxic agents and pathogens that cross the BBB, transformed microglia into phagocytic cells that engulf synapses in ADtg mice. This may represent phagocytic pathways that become self-destructive and drive AD progression. |
| Qiu (2023) [139] | Definition of the contribution of an Osteopontin-producing CD11c(+) microglial subset to Alzheimer’s disease (mouse study) | Identifies a subset of CD11+ microglia that are the sole source of OPN in ADtg mice:
|
7. Conclusions, Perspectives, and Emerging Treatment Options
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef] [PubMed]
- Rosmus, D.D.; Lange, C.; Ludwig, F.; Ajami, B.; Wieghofer, P. The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives. Biomedicines 2022, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.L.; Chong, J.R.; Raquib, A.R.; Xu, X.; Hilal, S.; Venketasubramanian, N.; Tan, B.Y.; Kumar, A.P.; Sethi, G.; Chen, C.P.; et al. Plasma osteopontin as a biomarker of Alzheimer’s disease and vascular cognitive impairment. Sci. Rep. 2021, 11, 4010. [Google Scholar] [CrossRef]
- Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Waite, L.M. Treatment for Alzheimer’s disease: Has anything changed? Aust. Prescr. 2015, 38, 60–63. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Avramopoulos, D. Genetics of Alzheimer’s disease: Recent advances. Genome Med. 2009, 1, 34. [Google Scholar] [CrossRef]
- van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. J. Am. Med. Assoc. 2023, 330, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J.; Davignon, J.; Bouthillier, D.; Kogan, S.; Bertrand, P.; Gauthier, S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 1993, 342, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Caselli, R.J.; Dueck, A.C.; Osborne, D.; Sabbagh, M.N.; Connor, D.J.; Ahern, G.L.; Baxter, L.C.; Rapcsak, S.Z.; Shi, J.; Woodruff, B.K.; et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N. Engl. J. Med. 2009, 361, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Cras, P.; Kawai, M.; Lowery, D.; Gonzalez-DeWhitt, P.; Greenberg, B.; Perry, G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1991, 88, 7552–7556. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Chau, D.D.; Ng, L.L.; Zhai, Y.; Lau, K.F. Amyloid precursor protein and its interacting proteins in neurodevelopment. Biochem. Soc. Trans. 2023, 51, 1647–1659. [Google Scholar] [CrossRef]
- d’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265. [Google Scholar] [CrossRef]
- Wirths, O.; Breyhan, H.; Marcello, A.; Cotel, M.C.; Brück, W.; Bayer, T.A. Inflammatory changes are tightly associated with neurodegeneration in the brain and spinal cord of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 747–757. [Google Scholar] [CrossRef]
- Ridler, C. Tau seeding starts early in the entorhinal cortex. Nat. Rev. Neurol. 2018, 14, 380. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Kobro-Flatmoen, A.; Lagartos-Donate, M.J.; Aman, Y.; Edison, P.; Witter, M.P.; Fang, E.F. Re-emphasizing early Alzheimer’s disease pathology starting in select entorhinal neurons, with a special focus on mitophagy. Ageing Res. Rev. 2021, 67, 101307. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [CrossRef]
- Sanchez, J.S.; Becker, J.A.; Jacobs, H.I.L.; Hanseeuw, B.J.; Jiang, S.; Schultz, A.P.; Properzi, M.J.; Katz, S.R.; Beiser, A.; Satizabal, C.L.; et al. The cortical origin and initial spread of medial temporal tauopathy in Alzheimer’s disease assessed with positron emission tomography. Sci. Transl. Med. 2021, 13, eabc0655. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Hampel, H.; Au, R.; Mattke, S.; van der Flier, W.M.; Aisen, P.; Apostolova, L.; Chen, C.; Cho, M.; De Santi, S.; Gao, P.; et al. Designing the next-generation clinical care pathway for Alzheimer’s disease. Nat. Aging 2022, 2, 692–703. [Google Scholar] [CrossRef]
- Cummings, J.; Kinney, J. Biomarkers for Alzheimer’s Disease: Context of Use, Qualification, and Roadmap for Clinical Implementation. Medicina 2022, 58, 952. [Google Scholar] [CrossRef] [PubMed]
- Koss, D.J.; Jones, G.; Cranston, A.; Gardner, H.; Kanaan, N.M.; Platt, B. Soluble pre-fibrillar tau and β-amyloid species emerge in early human Alzheimer’s disease and track disease progression and cognitive decline. Acta Neuropathol. 2016, 132, 875–895. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Rothbard, J.J.; Soares, L.; Fathman, C.G.; Steinman, L. Identification of a common immune regulatory pathway induced by small heat shock proteins, amyloid fibrils, and nicotine. Proc. Natl. Acad. Sci. USA 2018, 115, 7081–7086. [Google Scholar] [CrossRef]
- Steinman, L. An intrinsically disordered protein, osteopontin, driving neuropathology in Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2023, 120, e2221816120. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- Crestini, A.; Santilli, F.; Martellucci, S.; Carbone, E.; Sorice, M.; Piscopo, P.; Mattei, V. Prions and Neurodegenerative Diseases: A Focus on Alzheimer’s Disease. J. Alzheimers Dis. 2022, 85, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Bennett, R.E.; Delorme, L.; Robbins, A.B.; Hu, M.; McKenzie, D.; Kirk, M.J.; Schiantarelli, J.; Tunio, N.; Amaral, A.C.; et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019, 5, eaaw6404. [Google Scholar] [CrossRef] [PubMed]
- Wung, J.K.; Perry, G.; Kowalski, A.; Harris, P.L.; Bishop, G.M.; Trivedi, M.A.; Johnson, S.C.; Smith, M.A.; Denhardt, D.T.; Atwood, C.S. Increased expression of the remodeling- and tumorigenic-associated factor osteopontin in pyramidal neurons of the Alzheimer’s disease brain. Curr. Alzheimer Res. 2007, 4, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, D.Y.; Wang, Y.J. Peripheral clearance of brain-derived Aβ in Alzheimer’s disease: Pathophysiology and therapeutic perspectives. Transl. Neurodegener. 2020, 9, 16. [Google Scholar] [CrossRef]
- Parhizkar, S.; Holtzman, D.M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 2022, 59, 101594. [Google Scholar] [CrossRef]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Shi, Y.; Andhey, P.S.; Ising, C.; Wang, K.; Snipes, L.L.; Boyer, K.; Lawson, S.; Yamada, K.; Qin, W.; Manis, M.; et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron 2021, 109, 2413–2426.e7. [Google Scholar] [CrossRef]
- Rabenstein, M.; Vay, S.U.; Flitsch, L.J.; Fink, G.R.; Schroeter, M.; Rueger, M.A. Osteopontin directly modulates cytokine expression of primary microglia and increases their survival. J. Neuroimmunol. 2016, 299, 130–138. [Google Scholar] [CrossRef]
- Tondo, G.; Perani, D.; Comi, C. TAM Receptor Pathways at the Crossroads of Neuroinflammation and Neurodegeneration. Dis. Markers 2019, 2019, 2387614. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Vecchio, D.; Magistrelli, L.; Clemente, N.; Raineri, D.; Barbero Mazzucca, C.; Virgilio, E.; Dianzani, U.; Chiocchetti, A.; Comi, C. The Yin-Yang of osteopontin in nervous system diseases: Damage versus repair. Neural Regen. Res. 2021, 16, 1131–1137. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Pagnon de la Vega, M.; Näslund, C.; Brundin, R.; Lannfelt, L.; Löwenmark, M.; Kilander, L.; Ingelsson, M.; Giedraitis, V. Mutation analysis of disease causing genes in patients with early onset or familial forms of Alzheimer’s disease and frontotemporal dementia. BMC Genom. 2022, 23, 99. [Google Scholar] [CrossRef]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Kim, H.J.; Jang, H.; Weiner, M.W.; DeCarli, C.; Na, D.L.; Seo, S.W. Interaction between Alzheimer’s Disease and Cerebral Small Vessel Disease: A Review Focused on Neuroimaging Markers. Int. J. Mol. Sci. 2022, 23, 10490. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Yang, J.J.; Kwon, H.; Kim, C.; Lee, J.M.; Chun, P.; Kim, Y.J.; Jung, N.Y.; Chin, J.; Kim, S.; et al. Relative impact of amyloid-β, lacunes, and downstream imaging markers on cognitive trajectories. Brain 2016, 139, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Park, J.Y.; Jang, Y.K.; Kim, H.J.; Lee, J.S.; Na, D.L.; Noh, Y.; Lockhart, S.N.; Seong, J.K.; Seo, S.W. Distinct amyloid distribution patterns in amyloid positive subcortical vascular cognitive impairment. Sci. Rep. 2018, 8, 16178. [Google Scholar] [CrossRef]
- Banerjee, G.; Kim, H.J.; Fox, Z.; Jäger, H.R.; Wilson, D.; Charidimou, A.; Na, H.K.; Na, D.L.; Seo, S.W.; Werring, D.J. MRI-visible perivascular space location is associated with Alzheimer’s disease independently of amyloid burden. Brain 2017, 140, 1107–1116. [Google Scholar] [CrossRef]
- Kim, J.; Castellano, J.M.; Jiang, H.; Basak, J.M.; Parsadanian, M.; Pham, V.; Mason, S.M.; Paul, S.M.; Holtzman, D.M. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 2009, 64, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Nyarko, J.N.K.; Quartey, M.O.; Pennington, P.R.; Heistad, R.M.; Dea, D.; Poirier, J.; Baker, G.B.; Mousseau, D.D. Profiles of β-Amyloid Peptides and Key Secretases in Brain Autopsy Samples Differ with Sex and APOE ε4 Status: Impact for Risk and Progression of Alzheimer Disease. Neuroscience 2018, 373, 20–36. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef]
- Fitz, N.F.; Cronican, A.A.; Saleem, M.; Fauq, A.H.; Chapman, R.; Lefterov, I.; Koldamova, R. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J. Neurosci. 2012, 32, 13125–13136. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Remolina Serrano, J.; Hoyle, R.; Holtzman, D.M. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Tognatta, R.; Yan, Z.; Akassoglou, K. ApoE and immunity in Alzheimer’s disease and related tauopathies: Low-density lipoprotein receptor to the rescue. Neuron 2021, 109, 2363–2365. [Google Scholar] [CrossRef]
- Huynh, T.V.; Davis, A.A.; Ulrich, J.D.; Holtzman, D.M. Apolipoprotein E and Alzheimer’s disease: The influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins. J. Lipid Res. 2017, 58, 824–836. [Google Scholar] [CrossRef]
- Farfel, J.M.; Yu, L.; De Jager, P.L.; Schneider, J.A.; Bennett, D.A. Association of APOE with tau-tangle pathology with and without β-amyloid. Neurobiol. Aging 2016, 37, 19–25. [Google Scholar] [CrossRef]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Jiang, H.; Serrano, J.R.; Gonzales, E.R.; Wang, C.; Gratuze, M.; Hoyle, R.; Bien-Ly, N.; Silverman, A.P.; Sullivan, P.M.; et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med. 2021, 13, eabd7522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, W.; Tan, Z.; Zhang, L.; Dong, Z.; Cui, W.; Zhao, K.; Wang, H.; Jing, H.; Cao, R.; et al. A Role of Low-Density Lipoprotein Receptor-Related Protein 4 (LRP4) in Astrocytic Aβ Clearance. J. Neurosci. 2020, 40, 5347–5361. [Google Scholar] [CrossRef]
- Li, Y.; Macyczko, J.R.; Liu, C.C.; Bu, G. ApoE4 reduction: An emerging and promising therapeutic strategy for Alzheimer’s disease. Neurobiol. Aging 2022, 115, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Gu, X.; Song, Q.; Wang, X.; Huang, M.; Hu, M.; Hou, L.; Kang, T.; Chen, J.; Chen, H.; et al. Nanoformulated alpha-mangostin ameliorates Alzheimer’s disease neuropathology by elevating LDLR expression and accelerating amyloid-beta clearance. J. Control. Release 2016, 226, 1–14. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef]
- Merlini, M.; Kirabali, T.; Kulic, L.; Nitsch, R.M.; Ferretti, M.T. Extravascular CD3+ T Cells in Brains of Alzheimer Disease Patients Correlate with Tau but Not with Amyloid Pathology: An Immunohistochemical Study. Neurodegener. Dis. 2018, 18, 49–56. [Google Scholar] [CrossRef]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef]
- Tao, Q.; Ang, T.F.A.; DeCarli, C.; Auerbach, S.H.; Devine, S.; Stein, T.D.; Zhang, X.; Massaro, J.; Au, R.; Qiu, W.Q. Association of Chronic Low-grade Inflammation With Risk of Alzheimer Disease in ApoE4 Carriers. JAMA Netw. Open 2018, 1, e183597. [Google Scholar] [CrossRef]
- Yeung, C.H.C.; Schooling, C.M. Systemic inflammatory regulators and risk of Alzheimer’s disease: A bidirectional Mendelian-randomization study. Int. J. Epidemiol. 2021, 50, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Van Hoecke, L.; Vandenbroucke, R.E. The Impact of Systemic Inflammation on Alzheimer’s Disease Pathology. Front. Immunol. 2021, 12, 796867. [Google Scholar] [CrossRef]
- Rentsendorj, A.; Sheyn, J.; Fuchs, D.T.; Daley, D.; Salumbides, B.C.; Schubloom, H.E.; Hart, N.J.; Li, S.; Hayden, E.Y.; Teplow, D.B.; et al. A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models. Brain Behav. Immun. 2018, 67, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 74. [Google Scholar] [CrossRef] [PubMed]
- Bennett, F.C.; Bennett, M.L.; Yaqoob, F.; Mulinyawe, S.B.; Grant, G.A.; Hayden Gephart, M.; Plowey, E.D.; Barres, B.A. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 2018, 98, 1170–1183.e8. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10. [Google Scholar] [CrossRef]
- Shukla, A.K.; McIntyre, L.L.; Marsh, S.E.; Schneider, C.A.; Hoover, E.M.; Walsh, C.M.; Lodoen, M.B.; Blurton-Jones, M.; Inlay, M.A. CD11a expression distinguishes infiltrating myeloid cells from plaque-associated microglia in Alzheimer’s disease. Glia 2019, 67, 844–856. [Google Scholar] [CrossRef]
- Dubbelaar, M.L.; Kracht, L.; Eggen, B.J.L.; Boddeke, E. The Kaleidoscope of Microglial Phenotypes. Front. Immunol. 2018, 9, 1753. [Google Scholar] [CrossRef]
- Dräger, N.M.; Sattler, S.M.; Huang, C.T.; Teter, O.M.; Leng, K.; Hashemi, S.H.; Hong, J.; Aviles, G.; Clelland, C.D.; Zhan, L.; et al. A CRISPRi/a platform in human iPSC-derived microglia uncovers regulators of disease states. Nat. Neurosci. 2022, 25, 1149–1162. [Google Scholar] [CrossRef]
- Qin, J.; Ma, Z.; Chen, X.; Shu, S. Microglia activation in central nervous system disorders: A review of recent mechanistic investigations and development efforts. Front. Neurol. 2023, 14, 1103416. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagórska, A.; Través, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3, e03385. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Dobri, A.M.; Dudău, M.; Enciu, A.M.; Hinescu, M.E. CD36 in Alzheimer’s Disease: An Overview of Molecular Mechanisms and Therapeutic Targeting. Neuroscience 2021, 453, 301–311. [Google Scholar] [CrossRef]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Kummer, M.P.; Ising, C.; Kummer, C.; Sarlus, H.; Griep, A.; Vieira-Saecker, A.; Schwartz, S.; Halle, A.; Brückner, M.; Händler, K.; et al. Microglial PD-1 stimulation by astrocytic PD-L1 suppresses neuroinflammation and Alzheimer’s disease pathology. EMBO J. 2021, 40, e108662. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Grathwohl, S.A.; Kälin, R.E.; Bolmont, T.; Prokop, S.; Winkelmann, G.; Kaeser, S.A.; Odenthal, J.; Radde, R.; Eldh, T.; Gandy, S.; et al. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 2009, 12, 1361–1363. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yin, X.S.; Guo, H.; Han, R.K.; He, R.D.; Chi, L.J. Elevated osteopontin levels in mild cognitive impairment and Alzheimer’s disease. Mediat. Inflamm. 2013, 2013, 615745. [Google Scholar] [CrossRef]
- Fujita, D.; Terada, S.; Ishizu, H.; Yokota, O.; Nakashima, H.; Ishihara, T.; Kuroda, S. Immunohistochemical examination on intracranial calcification in neurodegenerative diseases. Acta Neuropathol. 2003, 105, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Rittling, S.R. Osteopontin in macrophage function. Expert. Rev. Mol. Med. 2011, 13, e15. [Google Scholar] [CrossRef]
- Shirakawa, K.; Endo, J.; Kataoka, M.; Katsumata, Y.; Yoshida, N.; Yamamoto, T.; Isobe, S.; Moriyama, H.; Goto, S.; Kitakata, H.; et al. IL (Interleukin)-10-STAT3-Galectin-3 Axis Is Essential for Osteopontin-Producing Reparative Macrophage Polarization After Myocardial Infarction. Circulation 2018, 138, 2021–2035. [Google Scholar] [CrossRef]
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; De Prijck, S.; Vanhockerhout, M.; et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 2020, 53, 641–657.e14. [Google Scholar] [CrossRef]
- De Schepper, S.; Ge, J.Z.; Crowley, G.; Ferreira, L.S.S.; Garceau, D.; Toomey, C.E.; Sokolova, D.; Rueda-Carrasco, J.; Shin, S.H.; Kim, J.S.; et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 406–415. [Google Scholar] [CrossRef]
- Comi, C.; Carecchio, M.; Chiocchetti, A.; Nicola, S.; Galimberti, D.; Fenoglio, C.; Cappellano, G.; Monaco, F.; Scarpini, E.; Dianzani, U. Osteopontin is increased in the cerebrospinal fluid of patients with Alzheimer’s disease and its levels correlate with cognitive decline. J. Alzheimers Dis. 2010, 19, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Temmerman, J.; Engelborghs, S.; Bjerke, M.; D’Haeseleer, M. Cerebrospinal fluid inflammatory biomarkers for disease progression in Alzheimer’s disease and multiple sclerosis: A systematic review. Front. Immunol. 2023, 14, 1162340. [Google Scholar] [CrossRef]
- Yao, F.; Hong, X.; Li, S.; Zhang, Y.; Zhao, Q.; Du, W.; Wang, Y.; Ni, J. Urine-Based Biomarkers for Alzheimer’s Disease Identified through Coupling Computational and Experimental Methods. J. Alzheimers Dis. 2018, 65, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Begcevic, I.; Brinc, D.; Brown, M.; Martinez-Morillo, E.; Goldhardt, O.; Grimmer, T.; Magdolen, V.; Batruch, I.; Diamandis, E.P. Brain-related proteins as potential CSF biomarkers of Alzheimer’s disease: A targeted mass spectrometry approach. J. Proteomics 2018, 182, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.H.; McGuire, J.; Hansson, O.; Zetterberg, H.; Podust, V.N.; Davies, H.A.; Waldemar, G.; Minthon, L.; Blennow, K. Novel Panel of Cerebrospinal Fluid Biomarkers for the Prediction of Progression to Alzheimer Dementia in Patients with Mild Cognitive Impairment. Arch. Neurol. 2007, 64, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Månberg, A.; Skene, N.; Sanders, F.; Trusohamn, M.; Remnestål, J.; Szczepińska, A.; Aksoylu, I.S.; Lönnerberg, P.; Ebarasi, L.; Wouters, S.; et al. Altered perivascular fibroblast activity precedes ALS disease onset. Nat. Med. 2021, 27, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Brown, A. Osteopontin: A Key Link Between Immunity, Inflammation and the Central Nervous System. Transl. Neurosci. 2012, 3, 288–293. [Google Scholar] [CrossRef]
- Schmidt-Morgenroth, I.; Michaud, P.; Gasparini, F.; Avrameas, A. Central and Peripheral Inflammation in Mild Cognitive Impairment in the Context of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 10523. [Google Scholar] [CrossRef]
- Carecchio, M.; Comi, C. The role of osteopontin in neurodegenerative diseases. J. Alzheimers Dis. 2011, 25, 179–185. [Google Scholar] [CrossRef]
- Grand Moursel, L.; van der Graaf, L.M.; Bulk, M.; van Roon-Mom, W.M.C.; van der Weerd, L. Osteopontin and phospho-SMAD2/3 are associated with calcification of vessels in D-CAA, an hereditary cerebral amyloid angiopathy. Brain Pathol. 2019, 29, 793–802. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Podleśny-Drabiniok, A.; Marcora, E.; Goate, A.M. Microglial Phagocytosis: A Disease-Associated Process Emerging from Alzheimer’s Disease Genetics. Trends Neurosci. 2020, 43, 965–979. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Luo, Q.; Hao, W.; Tomic, I.; Furihata, T.; Schulz-Schäffer, W.; Menger, M.D.; Fassbender, K.; Liu, Y. Haploinsufficiency of microglial MyD88 ameliorates Alzheimer’s pathology and vascular disorders in APP/PS1-transgenic mice. Glia 2021, 69, 1987–2005. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.P.; Richard, K.L.; Rivest, S. Hematopoietic MyD88-adaptor protein acts as a natural defense mechanism for cognitive deficits in Alzheimer’s disease. Stem Cell Rev. Rep. 2012, 8, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.B.; Jana, M.; Roy, A.; Corbett, G.T.; Kundu, M.; Chandra, S.; Mondal, S.; Dasarathi, S.; Mufson, E.J.; Mishra, R.K.; et al. Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J. Clin. Investig. 2018, 128, 4297–4312. [Google Scholar] [CrossRef] [PubMed]
- Weitz, T.M.; Gate, D.; Rezai-Zadeh, K.; Town, T. MyD88 is dispensable for cerebral amyloidosis and neuroinflammation in APP/PS1 transgenic mice. Am. J. Pathol. 2014, 184, 2855–2861. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Shen, X.; Ravid, O.; Atrakchi, D.; Rand, D.; Wight, A.E.; Kim, H.J.; Liraz-Zaltsman, S.; Cooper, I.; Schnaider Beeri, M.; et al. Definition of the contribution of an Osteopontin-producing CD11c+ microglial subset to Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2218915120. [Google Scholar] [CrossRef] [PubMed]
- Yong, V.W. Microglia in multiple sclerosis: Protectors turn destroyers. Neuron 2022, 110, 3534–3548. [Google Scholar] [CrossRef] [PubMed]
- Koronyo-Hamaoui, M.; Ko, M.K.; Koronyo, Y.; Azoulay, D.; Seksenyan, A.; Kunis, G.; Pham, M.; Bakhsheshian, J.; Rogeri, P.; Black, K.L.; et al. Attenuation of AD-like neuropathology by harnessing peripheral immune cells: Local elevation of IL-10 and MMP-9. J. Neurochem. 2009, 111, 1409–1424. [Google Scholar] [CrossRef] [PubMed]
- Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Li, S.; Ljubimov, V.; Moyseyev, M.; Daley, D.; Fuchs, D.T.; Pham, M.; et al. Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain 2015, 138, 2399–2422. [Google Scholar] [CrossRef]
- Bernstein, K.E.; Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Lopes, D.H.; Shah, K.H.; Bernstein, E.A.; Fuchs, D.T.; Yu, J.J.; et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J. Clin. Investig. 2014, 124, 1000–1012. [Google Scholar] [CrossRef]
- Ueno, M.; Chiba, Y.; Matsumoto, K.; Murakami, R.; Fujihara, R.; Kawauchi, M.; Miyanaka, H.; Nakagawa, T. Blood-brain barrier damage in vascular dementia. Neuropathology 2016, 36, 115–124. [Google Scholar] [CrossRef]
- Zhan, L.; Fan, L.; Kodama, L.; Sohn, P.D.; Wong, M.Y.; Mousa, G.A.; Zhou, Y.; Li, Y.; Gan, L. A MAC2-positive progenitor-like microglial population is resistant to CSF1R inhibition in adult mouse brain. eLife 2020, 9, e51796. [Google Scholar] [CrossRef]
- Zhan, L.; Krabbe, G.; Du, F.; Jones, I.; Reichert, M.C.; Telpoukhovskaia, M.; Kodama, L.; Wang, C.; Cho, S.H.; Sayed, F.; et al. Proximal recolonization by self-renewing microglia re-establishes microglial homeostasis in the adult mouse brain. PLoS Biol. 2019, 17, e3000134. [Google Scholar] [CrossRef]
- Lim, J.Y.; Lee, J.E.; Park, S.A.; Park, S.I.; Yon, J.M.; Park, J.A.; Jeun, S.S.; Kim, S.J.; Lee, H.J.; Kim, S.W.; et al. Protective Effect of Human-Neural-Crest-Derived Nasal Turbinate Stem Cells against Amyloid-β Neurotoxicity through Inhibition of Osteopontin in a Human Cerebral Organoid Model of Alzheimer’s Disease. Cells 2022, 11, 1029. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.K.; Kuan, Y.C.; Lin, H.W.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease: A 2020-2023 update. J. Biomed. Sci. 2023, 30, 83. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Mummery, C.J.; Börjesson-Hanson, A.; Blackburn, D.J.; Vijverberg, E.G.B.; De Deyn, P.P.; Ducharme, S.; Jonsson, M.; Schneider, A.; Rinne, J.O.; Ludolph, A.C.; et al. Tau-targeting antisense oligonucleotide MAPT(Rx) in mild Alzheimer’s disease: A phase 1b, randomized, placebo-controlled trial. Nat. Med. 2023, 29, 1437–1447. [Google Scholar] [CrossRef]
- Rabinovici, G.D. Controversy and Progress in Alzheimer’s Disease—FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Osse, A.M.L.; Kinney, J.W. Alzheimer’s Disease: Novel Targets and Investigational Drugs for Disease Modification. Drugs 2023, 83, 1387–1408. [Google Scholar] [CrossRef] [PubMed]
- Liggins, C.; Snyder, H.M.; Silverberg, N.; Petanceska, S.; Refolo, L.M.; Ryan, L.; Carrillo, M.C. International Alzheimer’s Disease Research Portfolio (IADRP) aims to capture global Alzheimer’s disease research funding. Alzheimers Dement. 2014, 10, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.W.; Lucente, J.D.; Nguyen, H.M.; Singh, V.; Singh, L.; Chavez, M.; Bushong, T.; Wulff, H.; Maezawa, I. Repurposing the KCa3.1 inhibitor senicapoc for Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Boza-Serrano, A.; Ruiz, R.; Sanchez-Varo, R.; García-Revilla, J.; Yang, Y.; Jimenez-Ferrer, I.; Paulus, A.; Wennström, M.; Vilalta, A.; Allendorf, D.; et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019, 138, 251–273. [Google Scholar] [CrossRef] [PubMed]
- Samant, N.P.; Gupta, G.L. Novel therapeutic strategies for Alzheimer’s disease targeting brain cholesterol homeostasis. Eur. J. Neurosci. 2021, 53, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Stamatelopoulos, K.; Bampatsias, D.; Sachse, M.; Zormpas, E.; Vlachogiannis, N.I.; Tual-Chalot, S.; Stellos, K. The Alzheimer’s Disease Amyloid-Beta Hypothesis in Cardiovascular Aging and Disease: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 952–967. [Google Scholar] [CrossRef]
- Ding, W.; Yousefi, K.; Goncalves, S.; Goldstein, B.J.; Sabater, A.L.; Kloosterboer, A.; Ritter, P.; Lambert, G.; Mendez, A.J.; Shehadeh, L.A. Osteopontin deficiency ameliorates Alport pathology by preventing tubular metabolic deficits. JCI Insight 2018, 3, e94818. [Google Scholar] [CrossRef]
- Yousefi, K.; Irion, C.I.; Takeuchi, L.M.; Ding, W.; Lambert, G.; Eisenberg, T.; Sukkar, S.; Granzier, H.L.; Methawasin, M.; Lee, D.I.; et al. Osteopontin Promotes Left Ventricular Diastolic Dysfunction through a Mitochondrial Pathway. J. Am. Coll. Cardiol. 2019, 73, 2705–2718. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalwani, R.C.; Volmar, C.-H.; Wahlestedt, C.; Webster, K.A.; Shehadeh, L.A. Contextualizing the Role of Osteopontin in the Inflammatory Responses of Alzheimer’s Disease. Biomedicines 2023, 11, 3232. https://doi.org/10.3390/biomedicines11123232
Lalwani RC, Volmar C-H, Wahlestedt C, Webster KA, Shehadeh LA. Contextualizing the Role of Osteopontin in the Inflammatory Responses of Alzheimer’s Disease. Biomedicines. 2023; 11(12):3232. https://doi.org/10.3390/biomedicines11123232
Chicago/Turabian StyleLalwani, Roshni C., Claude-Henry Volmar, Claes Wahlestedt, Keith A. Webster, and Lina A. Shehadeh. 2023. "Contextualizing the Role of Osteopontin in the Inflammatory Responses of Alzheimer’s Disease" Biomedicines 11, no. 12: 3232. https://doi.org/10.3390/biomedicines11123232