
PPARα in the Epigenetic Driver Seat of NAFLD: New Therapeutic Opportunities for Epigenetic Drugs?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Peroxisome Proliferator Activated Receptor Alpha—PPARα
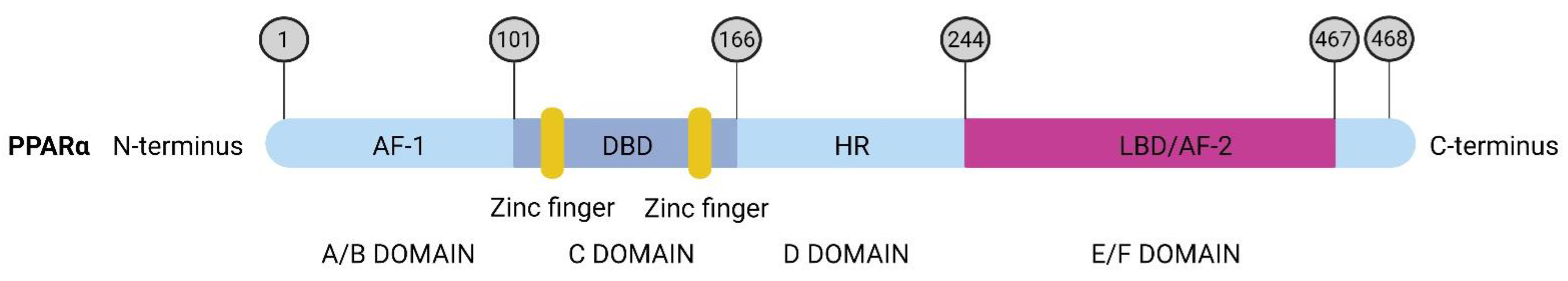
Structure and Regulation of PPARα
3. Epigenetic Regulation of PPARα in NAFLD
3.1. Epigenetics
3.2. Methylation State of PPARα Is a Biomarker of NAFLD Development
3.3. Histone Modifications at the Promoter Region of PPARα Related to the Development of NAFLD
3.4. PPARα-Targeting microRNAs Contribute to NAFLD Development
4. Epigenetic Interaction Partners in Crime in PPARα-Dependent Liver Pathologies
4.1. PPARα Interactions with Histone Modifying Enzymes
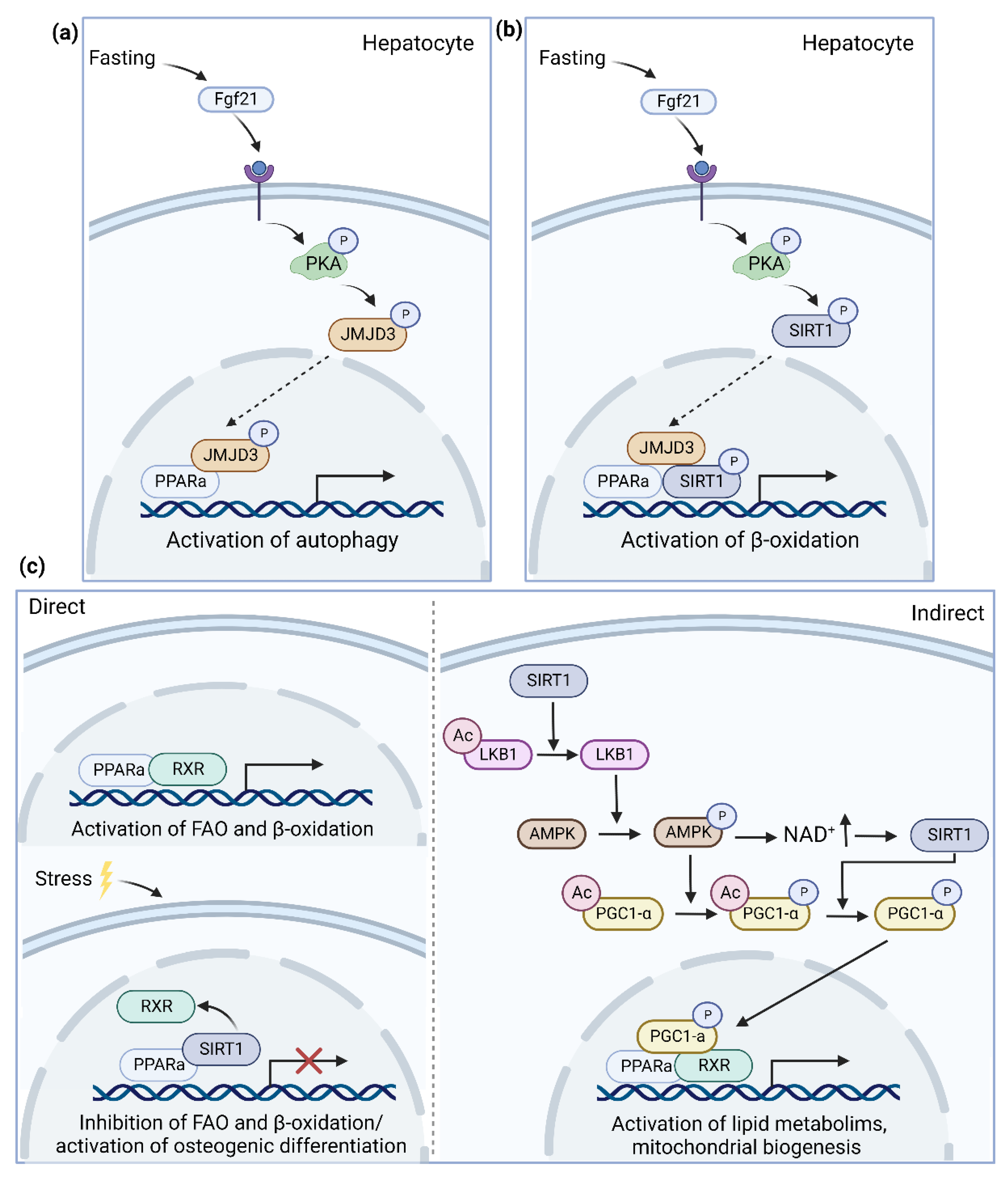
4.1.1. SIRT1
4.1.2. JMJD3
4.2. PPARα Interactions with DNA-Modifying Enzymes
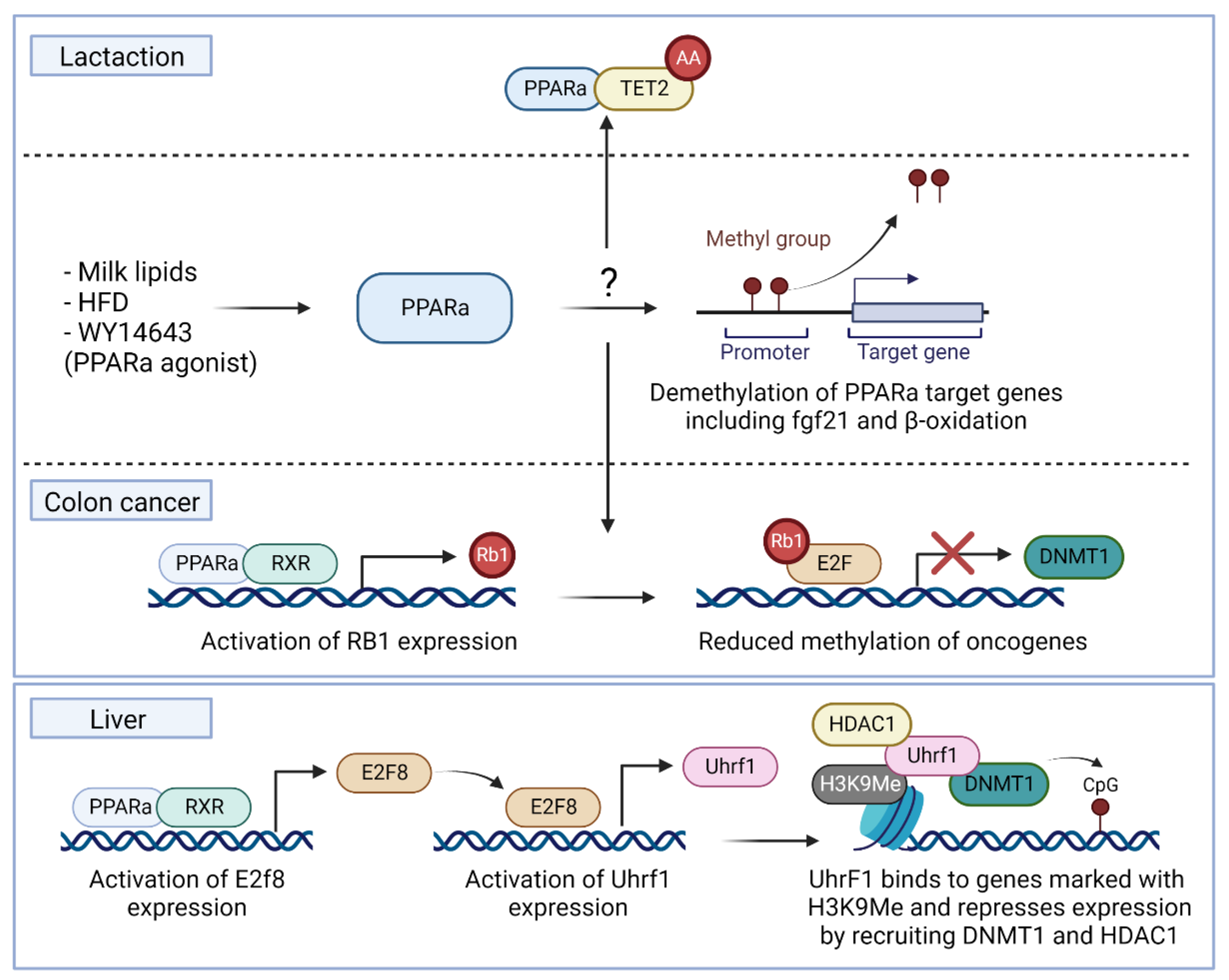
4.2.1. TET Enzymes
4.2.2. DNMT Enzymes
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Hyun, J.; Jung, Y. DNA Methylation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanova, M.; Younossi, Z.M. Independent association between nonalcoholic fatty liver disease and cardiovascular disease in the US population. Clin. Gastroenterol. Hepatol. 2012, 10, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Romero-Gomez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e365; quiz e314–e365. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.J.; Lee, K.E.; Kim, D.J.; Kim, S.K.; Ahn, C.W.; Lim, S.K.; Kim, K.R.; Lee, H.C.; Huh, K.B.; et al. Metabolic significance of nonalcoholic fatty liver disease in nonobese, nondiabetic adults. Arch. Intern. Med. 2004, 164, 2169–2175. [Google Scholar] [CrossRef] [Green Version]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Z.; Chen, J.; Ding, C.; Wong, K.; Chen, X.; Pu, L.; Huang, Q.; Chen, X.; Cheng, Z.; Liu, Y.; et al. Association of Hepatic Global DNA Methylation and Serum One-Carbon Metabolites with Histological Severity in Patients with NAFLD. Obesity 2020, 28, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Y.; Tang, D.; Du, Y.L.; Cao, C.Y.; Nie, Y.Q.; Cao, J.; Zhou, Y.J. Fatty liver mediated by peroxisome proliferator-activated receptor-alpha DNA methylation can be reversed by a methylation inhibitor and curcumin. J. Dig. Dis. 2018, 19, 421–430. [Google Scholar] [CrossRef]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef]
- Bugge, A.; Mandrup, S. Molecular Mechanisms and Genome-Wide Aspects of PPAR Subtype Specific Transactivation. PPAR Res. 2010, 2010, 169506. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, L.; Siersbaek, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.F. PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients. Int. J. Mol. Sci. 2022, 23, 4305. [Google Scholar] [CrossRef] [PubMed]
- Tahri-Joutey, M.; Andreoletti, P.; Surapureddi, S.; Nasser, B.; Cherkaoui-Malki, M.; Latruffe, N. Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARalpha. Int. J. Mol. Sci. 2021, 22, 8969. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef]
- Rimando, A.M.; Khan, S.I.; Mizuno, C.S.; Ren, G.; Mathews, S.T.; Kim, H.; Yokoyama, W. Evaluation of PPARalpha activation by known blueberry constituents. J. Sci. Food. Agric. 2016, 96, 1666–1671. [Google Scholar] [CrossRef]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARalpha in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Mann, J. Epigenetics and Liver Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claveria-Cabello, A.; Colyn, L.; Arechederra, M.; Urman, J.M.; Berasain, C.; Avila, M.A.; Fernandez-Barrena, M.G. Epigenetics in Liver Fibrosis: Could HDACs be a Therapeutic Target? Cells 2020, 9, 2321. [Google Scholar] [CrossRef]
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. Tet Enzymes, Variants, and Differential Effects on Function. Front. Cell Dev. Biol. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, K.P.; Yabuuchi, A.; Rao, S.; Huang, Y.; Cunniff, K.; Nardone, J.; Laiho, A.; Tahiliani, M.; Sommer, C.A.; Mostoslavsky, G.; et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem. Cell 2011, 8, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.P.; Guo, F.; Yang, H.; Wu, H.P.; Xu, G.F.; Liu, W.; Xie, Z.G.; Shi, L.; He, X.; Jin, S.G.; et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef]
- Iqbal, K.; Jin, S.G.; Pfeifer, G.P.; Szabo, P.E. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. USA 2011, 108, 3642–3647. [Google Scholar] [CrossRef] [Green Version]
- Good, C.R.; Madzo, J.; Patel, B.; Maegawa, S.; Engel, N.; Jelinek, J.; Issa, J.J. A novel isoform of TET1 that lacks a CXXC domain is overexpressed in cancer. Nucleic Acids Res. 2017, 45, 8269–8281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. Rev. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Ramarao-Milne, P.; Kondrashova, O.; Barry, S.; Hooper, J.D.; Lee, J.S.; Waddell, N. Histone Modifying Enzymes in Gynaecological Cancers. Cancers 2021, 13, 816. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenetics 2015, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirola, C.J.; Scian, R.; Gianotti, T.F.; Dopazo, H.; Rohr, C.; Martino, J.S.; Castano, G.O.; Sookoian, S. Epigenetic Modifications in the Biology of Nonalcoholic Fatty Liver Disease: The Role of DNA Hydroxymethylation and TET Proteins. Medicine 2015, 94, e1480. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueno, A.L.; Fernandez Gianotti, T.; Castano, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef]
- Lombardi, R.; Iuculano, F.; Pallini, G.; Fargion, S.; Fracanzani, A.L. Nutrients, Genetic Factors, and Their Interaction in Non-Alcoholic Fatty Liver Disease and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 8761. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, L.; Tan, Y.; Wei, J.; Chang, Y.; Jin, T.; Zhu, H. Betaine supplement alleviates hepatic triglyceride accumulation of apolipoprotein E deficient mice via reducing methylation of peroxisomal proliferator-activated receptor alpha promoter. Lipids Health Dis. 2013, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.J.; Zhang, H.W.; Zhou, J.Y.; Liu, Y.; Yang, Y.; Chen, X.L.; Zhu, C.H.; Zheng, R.D.; Ling, W.H.; Zhu, H.L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef]
- Wang, C.; Ma, C.; Gong, L.; Dai, S.; Li, Y. Preventive and therapeutic role of betaine in liver disease: A review on molecular mechanisms. Eur. J. Pharmacol. 2021, 912, 174604. [Google Scholar] [CrossRef]
- Xu, L.; Huang, D.; Hu, Q.; Wu, J.; Wang, Y.; Feng, J. Betaine alleviates hepatic lipid accumulation via enhancing hepatic lipid export and fatty acid oxidation in rats fed with a high-fat diet--CORRIGENDUM. Br. J. Nutr. 2015, 114, 995–996. [Google Scholar] [CrossRef] [Green Version]
- Cordero, P.; Gomez-Uriz, A.M.; Campion, J.; Milagro, F.I.; Martinez, J.A. Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 2013, 8, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pooya, S.; Blaise, S.; Moreno Garcia, M.; Giudicelli, J.; Alberto, J.M.; Gueant-Rodriguez, R.M.; Jeannesson, E.; Gueguen, N.; Bressenot, A.; Nicolas, B.; et al. Methyl donor deficiency impairs fatty acid oxidation through PGC-1alpha hypomethylation and decreased ER-alpha, ERR-alpha, and HNF-4alpha in the rat liver. J. Hepatol. 2012, 57, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Pruis, M.G.; Lendvai, A.; Bloks, V.W.; Zwier, M.V.; Baller, J.F.; de Bruin, A.; Groen, A.K.; PLoSch, T. Maternal western diet primes non-alcoholic fatty liver disease in adult mouse offspring. Acta Physiol. 2014, 210, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Yamada, H.; Munetsuna, E.; Yamazaki, M.; Kageyama, I.; Teshigawara, A.; Nouchi, Y.; Fujii, R.; Mizuno, G.; Sadamoto, N.; et al. Maternal high-fructose corn syrup consumption causes insulin resistance and hyperlipidemia in offspring via DNA methylation of the Pparalpha promoter region. J. Nutr. Biochem. 2022, 103, 108951. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Zhuo, Q.; Tseng, Y.; Wang, J.; Ma, Y.; Zhang, J.; Liu, J. TET1 promotes fatty acid oxidation and inhibits NAFLD progression by hydroxymethylation of PPARalpha promoter. Nutr. Metab. 2020, 17, 46. [Google Scholar] [CrossRef]
- Lestari, M.L.; Indrayanto, G. Curcumin. Profiles Drug Subst. Excip. Relat. Methodol. 2014, 39, 113–204. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K.; Aggarwal, B.B. Curcumin, a component of golden spice: From bedside to bench and back. Biotechnol. Adv. 2014, 32, 1053–1064. [Google Scholar] [CrossRef]
- Inagaki, T.; Tachibana, M.; Magoori, K.; Kudo, H.; Tanaka, T.; Okamura, M.; Naito, M.; Kodama, T.; Shinkai, Y.; Sakai, J. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 2009, 14, 991–1001. [Google Scholar] [CrossRef]
- Tateishi, K.; Okada, Y.; Kallin, E.M.; Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009, 458, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.J.; Kim, J.; Hoang, M.H.; Lee, S.J. Hepatic lipid accumulation alters global histone h3 lysine 9 and 4 trimethylation in the peroxisome proliferator-activated receptor alpha network. PLoS ONE 2012, 7, e44345. [Google Scholar] [CrossRef]
- Huang, L.; Liu, J.; Zhang, X.O.; Sibley, K.; Najjar, S.M.; Lee, M.M.; Wu, Q. Inhibition of protein arginine methyltransferase 5 enhances hepatic mitochondrial biogenesis. J. Biol. Chem. 2018, 293, 10884–10894. [Google Scholar] [CrossRef] [Green Version]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tee, W.W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrizio, E.; El Messaoudi, S.; Polanowska, J.; Paul, C.; Cook, J.R.; Lee, J.H.; Negre, V.; Rousset, M.; Pestka, S.; Le Cam, A.; et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 2002, 3, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–9645. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhao, X.; Zhao, L.; Li, J.; Yang, H.; Zhu, Z.; Liu, J.; Huang, G. Arginine Methylation of SREBP1a via PRMT5 Promotes De Novo Lipogenesis and Tumor Growth. Cancer Res. 2016, 76, 1260–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.; Hwang, J.T.; Park, J.H.; Choi, H.K. Free fatty acid-induced histone acetyltransferase activity accelerates lipid accumulation in HepG2 cells. Nutr. Res. Pract. 2019, 13, 196–204. [Google Scholar] [CrossRef]
- Sun, B.; Jia, Y.; Hong, J.; Sun, Q.; Gao, S.; Hu, Y.; Zhao, N.; Zhao, R. Sodium Butyrate Ameliorates High-Fat-Diet-Induced Non-alcoholic Fatty Liver Disease through Peroxisome Proliferator-Activated Receptor alpha-Mediated Activation of beta Oxidation and Suppression of Inflammation. J. Agric. Food Chem. 2018, 66, 7633–7642. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, G.N.; Dominguez-Perez, M.; Uribe, M.; Chavez-Tapia, N.C.; Nuno-Lambarri, N. Non-alcoholic fatty liver disease and microRNAs expression, how it affects the development and progression of the disease. Ann. Hepatol. 2021, 21, 100212. [Google Scholar] [CrossRef]
- Huang, R.; Duan, X.; Liu, X.; Cao, H.; Wang, Y.; Fan, J.; Wang, B. Upregulation of miR-181a impairs lipid metabolism by targeting PPARalpha expression in nonalcoholic fatty liver disease. Biochem. Biophys. Res. Commun. 2019, 508, 1252–1258. [Google Scholar] [CrossRef]
- Wang, D.R.; Wang, B.; Yang, M.; Liu, Z.L.; Sun, J.; Wang, Y.; Sun, H.; Xie, L.J. Suppression of miR-30a-3p Attenuates Hepatic Steatosis in Non-alcoholic Fatty Liver Disease. Biochem. Genet. 2020, 58, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, R.; Nardelli, C.; Pilone, V.; Buonomo, T.; Liguori, R.; Castano, I.; Buono, P.; Masone, S.; Persico, G.; Forestieri, P.; et al. miR-519d overexpression is associated with human obesity. Obesity 2010, 18, 2170–2176. [Google Scholar] [CrossRef]
- Yang, Z.; Qin, W.; Huo, J.; Zhuo, Q.; Wang, J.; Wang, L. MiR-22 modulates the expression of lipogenesis-related genes and promotes hepatic steatosis in vitro. FEBS Open Bio 2021, 11, 322–332. [Google Scholar] [CrossRef]
- Lee, Y.H.; Jang, H.J.; Kim, S.; Choi, S.S.; Khim, K.W.; Eom, H.J.; Hyun, J.; Shin, K.J.; Chae, Y.C.; Kim, H.; et al. Hepatic MIR20B promotes nonalcoholic fatty liver disease by suppressing PPARA. Elife 2021, 10, e70472. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.; Rodrigues, C.M.P.; Castro, R.E. Modulation of liver steatosis by miR-21/PPARalpha. Cell Death Discov. 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.M.; Qiu, Y.; Yang, Z.; Kim, H.; Qian, Q.; Sun, Q.; Zhang, C.; Yin, L.; Fang, D.; Back, S.H.; et al. IRE1alpha prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal 2018, 11, eaao4617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, H.; Sun, L.; Shen, S.; Zhou, Q.; Yuan, Y.; Xing, C. Epigenetic Alternations of MicroRNAs and DNA Methylation Contribute to Liver Metastasis of Colorectal Cancer. Dig. Dis. Sci. 2019, 64, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, M.; Wan, X.; Jin, X.; Chen, S.; Yu, C.; Li, Y. Effect of miR-34a in regulating steatosis by targeting PPARalpha expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.Y.; Sun, F.; Chen, J.N.; Wang, Y.Q.; Pan, Q.; Fan, J.G. circRNA_0046366 inhibits hepatocellular steatosis by normalization of PPAR signaling. World J. Gastroenterol. 2018, 24, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.F.; Cogliati, B.; Otton, R. Green Tea Prevents NAFLD by Modulation of miR-34a and miR-194 Expression in a High-Fat Diet Mouse Model. Oxid. Med. Cell. Longev. 2019, 2019, 4168380. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrin. Met. 2014, 25, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; de Boer, V.C.; Finley, L.W.; Sweeney, M.; Lu, H.; Schug, T.T.; Cen, Y.; Jeong, S.M.; Li, X.; Sauve, A.A.; et al. SIRT4 represses peroxisome proliferator-activated receptor alpha activity to suppress hepatic fat oxidation. Mol. Cell Biol. 2013, 33, 4552–4561. [Google Scholar] [CrossRef] [Green Version]
- Watroba, M.; Szukiewicz, D. Sirtuins at the Service of Healthy Longevity. Front. Physiol. 2021, 12, 724506. [Google Scholar] [CrossRef]
- McCarty, M.F. Practical prospects for boosting hepatic production of the “pro-longevity” hormone FGF21. Horm. Mol. Biol. Clin. Investig. 2015, 30, 20150057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, S.Y.; Qi-Li, F.R.; Liu, X.X.; Zhang, W.T.; Peng, C.; Wu, P.; Li, P.; Li, P.; Xu, X. Administration of isoliquiritigenin prevents nonalcoholic fatty liver disease through a novel IQGAP2-CREB-SIRT1 axis. Phytother. Res. 2021, 35, 3898–3915. [Google Scholar] [CrossRef]
- Hua, Y.Q.; Zeng, Y.; Xu, J.; Xu, X.L. Naringenin alleviates nonalcoholic steatohepatitis in middle-aged Apoe(-/-)mice: Role of SIRT1. Phytomedicine 2021, 81, 153412. [Google Scholar] [CrossRef] [PubMed]
- Suga, T.; Sato, K.; Ohyama, T.; Matsui, S.; Kobayashi, T.; Tojima, H.; Horiguchi, N.; Yamazaki, Y.; Kakizaki, S.; Nishikido, A.; et al. Ipragliflozin-induced improvement of liver steatosis in obese mice may involve sirtuin signaling. World J. Hepatol. 2020, 12, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Majeed, Y.; Halabi, N.; Madani, A.Y.; Engelke, R.; Bhagwat, A.M.; Abdesselem, H.; Agha, M.V.; Vakayil, M.; Courjaret, R.; Goswami, N.; et al. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Sci. Rep. 2021, 11, 8177. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Lin, Q.; Lin, R.; Zhang, J.; Ren, F.; Zhang, J.; Ji, M.; Li, Y. PPARalpha agonist fenofibrate attenuates TNF-alpha-induced CD40 expression in 3T3-L1 adipocytes via the SIRT1-dependent signaling pathway. Exp. Cell Res. 2013, 319, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Pantazi, E.; Folch-Puy, E.; Bejaoui, M.; Panisello, A.; Varela, A.T.; Rolo, A.P.; Palmeira, C.M.; Rosello-Catafau, J. PPARalpha Agonist WY-14643 Induces SIRT1 Activity in Rat Fatty Liver Ischemia-Reperfusion Injury. Biomed. Res. Int. 2015, 2015, 894679. [Google Scholar] [CrossRef] [Green Version]
- Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Meza-Rios, A.; Garcia-Banuelos, J.; Vera-Cruz, J.; Gutierrez-Cuevas, J.; Silva-Gomez, J.; Staels, B.; Dominguez-Rosales, J.; Galicia-Moreno, M.; et al. Pirfenidone Is an Agonistic Ligand for PPARalpha and Improves NASH by Activation of SIRT1/LKB1/pAMPK. Hepatol. Commun. 2020, 4, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Hayashida, S.; Arimoto, A.; Kuramoto, Y.; Kozako, T.; Honda, S.; Shimeno, H.; Soeda, S. Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Mol. Cell Biochem. 2010, 339, 285–292. [Google Scholar] [CrossRef]
- Gong, K.; Qu, B.; Wang, C.; Zhou, J.; Liao, D.; Zheng, W.; Pan, X. Peroxisome Proliferator-Activated Receptor alpha Facilitates Osteogenic Differentiation in MC3T3-E1 Cells via the Sirtuin 1-Dependent Signaling Pathway. Mol. Cells 2017, 40, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroya, J.; Redondo-Angulo, I.; Iglesias, R.; Giralt, M.; Villarroya, F.; Planavila, A. Sirt1 mediates the effects of a short-term high-fat diet on the heart. J. Nutr. Biochem. 2015, 26, 1328–1337. [Google Scholar] [CrossRef]
- Oka, S.; Alcendor, R.; Zhai, P.; Park, J.Y.; Shao, D.; Cho, J.; Yamamoto, T.; Tian, B.; Sadoshima, J. PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 2011, 14, 598–611. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Zhai, P.; Yamamoto, T.; Ikeda, Y.; Byun, J.; Hsu, C.P.; Sadoshima, J. Peroxisome Proliferator Activated Receptor-alpha Association With Silent Information Regulator 1 Suppresses Cardiac Fatty Acid Metabolism in the Failing Heart. Circ. Heart. Fail. 2015, 8, 1123–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2011, 90, 276–284. [Google Scholar] [CrossRef]
- Huang, K.; Du, M.; Tan, X.; Yang, L.; Li, X.; Jiang, Y.; Wang, C.; Zhang, F.; Zhu, F.; Cheng, M.; et al. PARP1-mediated PPARalpha poly(ADP-ribosyl)ation suppresses fatty acid oxidation in non-alcoholic fatty liver disease. J. Hepatol. 2017, 66, 962–977. [Google Scholar] [CrossRef]
- Wu, T.; Liu, Y.H.; Fu, Y.C.; Liu, X.M.; Zhou, X.H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar] [PubMed]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Chen, D.; Gong, Q.; Xu, Q.; Pan, D.; Lu, F.; Tang, Q. Elucidation of SIRT-1/PGC-1alpha-associated mitochondrial dysfunction and autophagy in nonalcoholic fatty liver disease. Lipids Health Dis. 2021, 20, 40. [Google Scholar] [CrossRef]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Yao, W.; Wang, K.; Wang, X.; Li, X.; Dong, J.; Zhang, Y.; Ding, X. Icariin ameliorates endothelial dysfunction in type 1 diabetic rats by suppressing ER stress via the PPARalpha/Sirt1/AMPKalpha pathway. J. Cell. Physiol. 2021, 236, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Cai, C.Z.; Yu, M.L.; Feng, Z.M.; Zhang, Y.W.; Liu, P.H.; Zeng, H.; Yu, C.H. LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway. World J. Gastroenterol. 2019, 25, 6607–6618. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Q.; Li, Y.; Tang, Q.; Wu, T.; Chen, L.; Pu, S.; Zhao, Y.; Zhang, G.; Huang, C.; et al. The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK-Sirt1-Pgc-1alpha signalling pathway. Adipocyte 2020, 9, 484–494. [Google Scholar] [CrossRef]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [Green Version]
- Byun, S.; Seok, S.; Kim, Y.C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seok, S.; Kim, Y.C.; Byun, S.; Choi, S.; Xiao, Z.; Iwamori, N.; Zhang, Y.; Wang, C.; Ma, J.; Ge, K.; et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid beta-oxidation. J. Clin. Investig. 2018, 128, 3144–3159. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Czaja, M.J. Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Pang, H.; Ling, D.; Cheng, Y.; Akbar, R.; Jin, L.; Ren, J.; Wu, H.; Chen, B.; Zhou, Y.; Zhu, H.; et al. Gestational high-fat diet impaired demethylation of Pparalpha and induced obesity of offspring. J. Cell. Mol. Med. 2021, 25, 5404–5416. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Ogawa, Y. Epigenetic Switching and Neonatal Nutritional Environment. Adv. Exp. Med. Biol. 2018, 1012, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Tsujimoto, K.; Hashimoto, K.; Kawahori, K.; Hanzawa, N.; Hamaguchi, M.; Seki, T.; Nawa, M.; Ehara, T.; Kitamura, Y.; et al. Epigenetic modulation of Fgf21 in the perinatal mouse liver ameliorates diet-induced obesity in adulthood. Nat. Commun. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehara, T.; Kamei, Y.; Yuan, X.; Takahashi, M.; Kanai, S.; Tamura, E.; Tsujimoto, K.; Tamiya, T.; Nakagawa, Y.; Shimano, H.; et al. Ligand-activated PPARalpha-dependent DNA demethylation regulates the fatty acid beta-oxidation genes in the postnatal liver. Diabetes 2015, 64, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta. Gene Regul. Mech. 2019, 1862, 141–152. [Google Scholar] [CrossRef]
- Kim, J.H.; Wahyudi, L.D.; Kim, K.K.; Gonzalez, F.J. PPARalpha activation drives demethylation of the CpG islands of the Gadd45b promoter in the mouse liver. Biochem. Biophys. Res. Commun. 2016, 476, 293–298. [Google Scholar] [CrossRef]
- Kawahori, K.; Kondo, Y.; Yuan, X.; Kawasaki, Y.; Hanzawa, N.; Tsujimoto, K.; Wada, F.; Kohda, T.; Ishigami, A.; Yamada, T.; et al. Ascorbic acid during the suckling period is required for proper DNA demethylation in the liver. Sci. Rep. 2020, 10, 21228. [Google Scholar] [CrossRef]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARalpha Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. Fenofibrate Exerts Antitumor Effects in Colon Cancer via Regulation of DNMT1 and CDKN2A. PPAR Res. 2021, 2021, 6663782. [Google Scholar] [CrossRef]
- McCabe, M.T.; Davis, J.N.; Day, M.L. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res. 2005, 65, 3624–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aibara, D.; Takahashi, S.; Yagai, T.; Kim, D.; Brocker, C.N.; Levi, M.; Matsusue, K.; Gonzalez, F.J. Gene repression through epigenetic modulation by PPARA enhances hepatocellular proliferation. iScience 2022, 25, 104196. [Google Scholar] [CrossRef]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Dnmt1/Transcription factor interactions: An alternative mechanism of DNA methylation inheritance. Genes Cancer 2010, 1, 434–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlhoff, C.; Desmarchelier, C.; Sailer, M.; Furst, R.W.; Haag, A.; Ulbrich, S.E.; Hummel, B.; Obeid, R.; Geisel, J.; Bader, B.L.; et al. Hepatic methionine homeostasis is conserved in C57BL/6N mice on high-fat diet despite major changes in hepatic one-carbon metabolism. PLoS ONE 2013, 8, e57387. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Nikam, A.; Patankar, J.V.; Somlapura, M.; Lahiri, P.; Sachdev, V.; Kratky, D.; Denk, H.; Zatloukal, K.; Abuja, P.M. The PPARalpha Agonist Fenofibrate Prevents Formation of Protein Aggregates (Mallory-Denk bodies) in a Murine Model of Steatohepatitis-like Hepatotoxicity. Sci. Rep. 2018, 8, 12964. [Google Scholar] [CrossRef]
- Rodriguez-Vilarrupla, A.; Lavina, B.; Garcia-Caldero, H.; Russo, L.; Rosado, E.; Roglans, N.; Bosch, J.; Garcia-Pagan, J.C. PPARalpha activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J. Hepatol. 2012, 56, 1033–1039. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Fernandez-Miranda, C.; Perez-Carreras, M.; Colina, F.; Lopez-Alonso, G.; Vargas, C.; Solis-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver. Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef]
- Nikolic, D.; Castellino, G.; Banach, M.; Toth, P.P.; Ivanova, E.; Orekhov, A.N.; Montalto, G.; Rizzo, M. PPAR Agonists, Atherogenic Dyslipidemia and Cardiovascular Risk. Curr. Pharm. Des. 2017, 23, 894–902. [Google Scholar] [CrossRef]
- Ida, S.; Kaneko, R.; Murata, K. Efficacy and safety of pemafibrate administration in patients with dyslipidemia: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Yamashita, S.; Arai, H.; Araki, E.; Yokote, K.; Suganami, H.; Fruchart, J.C.; Kodama, T.; Group, K.S. Effects of K-877, a novel selective PPARalpha modulator (SPPARMalpha), in dyslipidaemic patients: A randomized, double blind, active- and placebo-controlled, phase 2 trial. Atherosclerosis 2016, 249, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) alpha/gamma/delta Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef] [PubMed]
- Giglio, R.V.; Papanas, N.; Rizvi, A.A.; Ciaccio, M.; Patti, A.M.; Ilias, I.; Pantea Stoian, A.; Sahebkar, A.; Janez, A.; Rizzo, M. An Update on the Current and Emerging Use of Thiazolidinediones for Type 2 Diabetes. Medicina 2022, 58, 1475. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients with Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Corey, K.E.; Wilson, L.A.; Altinbas, A.; Yates, K.P.; Kleiner, D.E.; Chung, R.T.; Krauss, R.M.; Chalasani, N.; Network, N.C.R. Relationship between resolution of non-alcoholic steatohepatitis and changes in lipoprotein sub-fractions: A post-hoc analysis of the PIVENS trial. Aliment. Pharmacol. Ther. 2019, 49, 1205–1213. [Google Scholar] [CrossRef]
- Remely, M.; Ferk, F.; Sterneder, S.; Setayesh, T.; Kepcija, T.; Roth, S.; Noorizadeh, R.; Greunz, M.; Rebhan, I.; Wagner, K.H.; et al. Vitamin E Modifies High-Fat Diet-Induced Increase of DNA Strand Breaks, and Changes in Expression and DNA Methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice. Nutrients 2017, 9, 607. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yi, L.; Ye, X.; Zeng, X.; Liu, K.; Qin, Y.; Zhang, Q.; Mi, M. Resveratrol Ameliorates Lipid Droplet Accumulation in Liver through a SIRT1/ATF6-Dependent Mechanism. Cell Physiol. Biochem. 2018, 51, 2397–2420. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theys, C.; Lauwers, D.; Perez-Novo, C.; Vanden Berghe, W. PPARα in the Epigenetic Driver Seat of NAFLD: New Therapeutic Opportunities for Epigenetic Drugs? Biomedicines 2022, 10, 3041. https://doi.org/10.3390/biomedicines10123041
Theys C, Lauwers D, Perez-Novo C, Vanden Berghe W. PPARα in the Epigenetic Driver Seat of NAFLD: New Therapeutic Opportunities for Epigenetic Drugs? Biomedicines. 2022; 10(12):3041. https://doi.org/10.3390/biomedicines10123041
Chicago/Turabian StyleTheys, Claudia, Dorien Lauwers, Claudina Perez-Novo, and Wim Vanden Berghe. 2022. "PPARα in the Epigenetic Driver Seat of NAFLD: New Therapeutic Opportunities for Epigenetic Drugs?" Biomedicines 10, no. 12: 3041. https://doi.org/10.3390/biomedicines10123041