
New Insights into Molecular Mechanisms of Chronic Kidney Disease
 , , , ,
, , , ,
Abstract
:1. Introduction
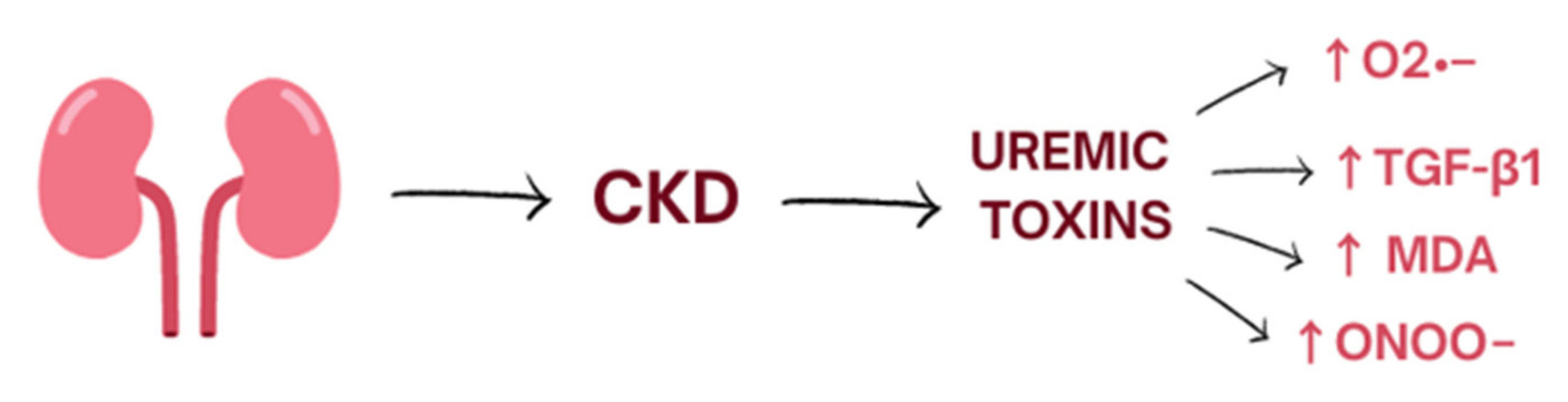
2. Oxidative Stress
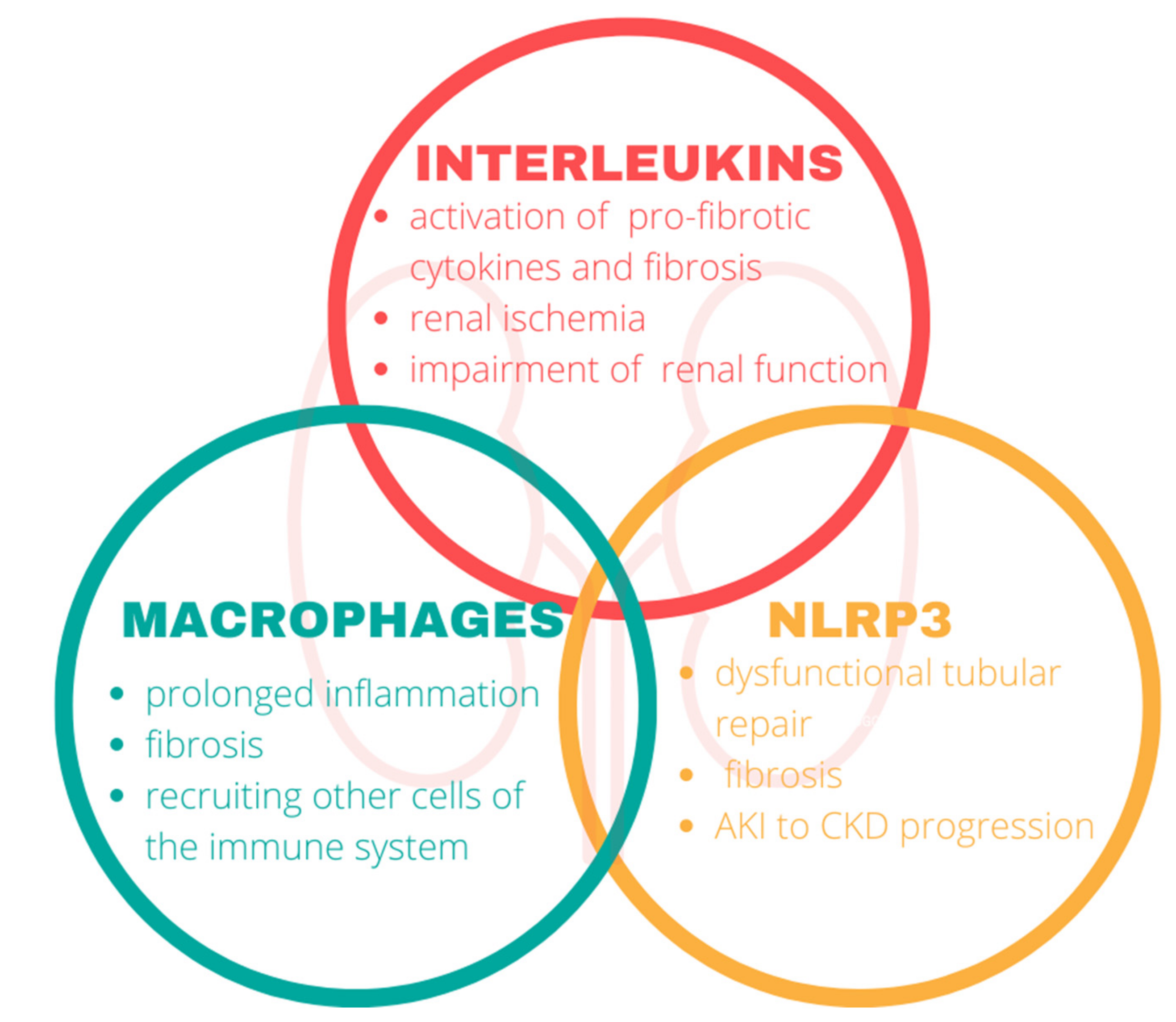
3. Inflammation
3.1. Interleukins
3.2. Macrophages
3.3. Nod-Like Receptor Protein 3
4. Neutrophil Gelatinase-Associated Lipocalin
5. Matrix Metalloproteinases
6. Gut–Kidney Axis
7. New Targets of Treatment
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADMA | Asymmetric dimethylarginine |
| AKI | Acute kidney injury |
| AOPPs | Advanced oxidation protein products |
| CKD | Chronic kidney disease |
| CVD | Cardiovascular diseases |
| DFO | Desferrioxamine |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptors |
| EPCs | Endothelial progenitor cells |
| ESRD | End-Stage Renal Disease |
| GFR | Glomerular filtration rate |
| MAD | Malonyldialdehyde |
| MMPs | Metalloproteinases |
| NGAL | Neutrophil gelatinase-associated lipocalin |
| NO | Nitric oxide |
| PCO | Protein carbonyls |
| ROS | Reactive oxygen species |
| TGF | Transforming Growth Factor |
| TNF | Tumor Necrosis Factor |
References
- Ammirati, A.L. Chronic Kidney Disease. Rev. Assoc. Med. Bras. 2020, 66 (Suppl. S1), s03–s09. [Google Scholar] [CrossRef] [PubMed]
- Ene-Iordache, B.; Perico, N.; Bikbov, B.; Carminati, S.; Remuzzi, A.; Perna, A.; Islam, N.; Bravo, R.F.; Aleckovic-Halilovic, M.; Zou, H.; et al. Chronic Kidney Disease and Cardiovascular Risk in Six Regions of the World (ISN-KDDC): A Cross-Sectional Study. Lancet Glob. Health 2016, 4, e307–e319. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Oh, H.J.; Choi, H.Y.; Lee, H.; Ryu, D.-R. Impact of Chronic Kidney Disease on Mortality: A Nationwide Cohort Study. Kidney Res. Clin. Pract. 2019, 38, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- Chapter 1: Definition and Classification of CKD. Kidney Int. Suppl. 2013, 3, 19–62. [CrossRef] [Green Version]
- Charles, C.; Ferris, A.H. Chronic Kidney Disease. Prim. Care 2020, 47, 585–595. [Google Scholar] [CrossRef]
- Vaidya, S.R.; Aeddula, N.R. Chronic Renal Failure. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic Kidney Disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef]
- Kućmierz, J.; Frąk, W.; Młynarska, E.; Franczyk, B.; Rysz, J. Molecular Interactions of Arterial Hypertension in Its Target Organs. Int. J. Mol. Sci. 2021, 22, 9669. [Google Scholar] [CrossRef]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef] [Green Version]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T. Indoxyl Sulfate Is a Nephro-Vascular Toxin. J. Ren. Nutr. 2010, 20 (Suppl. S5), S2–S6. [Google Scholar] [CrossRef]
- Schei, J.; Fuskevåg, O.-M.; Stefansson, V.T.N.; Solbu, M.D.; Jenssen, T.G.; Eriksen, B.O.; Melsom, T. Urinary Markers of Oxidative Stress Are Associated With Albuminuria But Not GFR Decline. Kidney Int. Rep. 2018, 3, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.; Morrow, J.D. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic. Biol. Med. 2000, 28, 505–513. [Google Scholar] [CrossRef]
- Colombo, G.; Reggiani, F.; Angelini, C.; Finazzi, S.; Astori, E.; Garavaglia, M.L.; Landoni, L.; Portinaro, N.M.; Giustarini, D.; Rossi, R.; et al. Plasma Protein Carbonyls as Biomarkers of Oxidative Stress in Chronic Kidney Disease, Dialysis, and Transplantation. Oxidative Med. Cell. Longev. 2020, 2020, 2975256. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative Stress in Chronic Kidney Disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Metodiewa, D.; Kośka, C. Reactive oxygen species and reactive nitrogen species: Relevance to cyto(neuro)toxic events and neurologic disorders. An overview. Neurotox. Res. 2000, 1, 197–233. [Google Scholar] [CrossRef]
- Nagata, M. Podocyte Injury and Its Consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.; Su, Y.; Paueksakon, P.; Hu, W.; Zhang, M.-Z.; Harris, R.C.; Blackwell, T.S.; Zent, R.; Pozzi, A. P47(Phox) Contributes to Albuminuria and Kidney Fibrosis in Mice. Kidney Int. 2015, 87, 948–962. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [Green Version]
- Mack, M.; Yanagita, M. Origin of Myofibroblasts and Cellular Events Triggering Fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-beta1 induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Modlinger, P.S.; Wilcox, C.S.; Aslam, S. Nitric Oxide, Oxidative Stress, and Progression of Chronic Renal Failure. Semin. Nephrol. 2004, 24, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Justiz Vaillant, A.A.; Qurie, A. Interleukin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mertowska, P.; Mertowski, S.; Smarz-Widelska, I.; Grywalska, E. Biological Role, Mechanism of Action and the Importance of Interleukins in Kidney Diseases. Int. J. Mol. Sci. 2022, 23, 647. [Google Scholar] [CrossRef] [PubMed]
- Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Temmar, M.; Lemke, H.-D.; Tribouilloy, C.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). Plasma Interleukin-6 Is Independently Associated with Mortality in Both Hemodialysis and Pre-Dialysis Patients with Chronic Kidney Disease. Kidney Int. 2010, 77, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Mima, T.; Nishimoto, N. Clinical Value of Blocking IL-6 Receptor. Curr. Opin. Rheumatol. 2009, 21, 224–230. [Google Scholar] [CrossRef]
- Kreiner, F.F.; Kraaijenhof, J.M.; von Herrath, M.; Hovingh, G.K.K.; von Scholten, B.J. Interleukin 6 in Diabetes, Chronic Kidney Disease, and Cardiovascular Disease: Mechanisms and Therapeutic Perspectives. Expert Rev. Clin. Immunol. 2022, 18, 377–389. [Google Scholar] [CrossRef]
- Jones, S.A.; Fraser, D.J.; Fielding, C.A.; Jones, G.W. Interleukin-6 in Renal Disease and Therapy. Nephrol. Dial. Transplant. 2015, 30, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in Innate and Adaptive Immunity in Health and Disease. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Bandach, I.; Segev, Y.; Landau, D. Experimental Modulation of Interleukin 1 Shows Its Key Role in Chronic Kidney Disease Progression and Anemia. Sci. Rep. 2021, 11, 6288. [Google Scholar] [CrossRef]
- Schunk, S.J.; Triem, S.; Schmit, D.; Zewinger, S.; Sarakpi, T.; Becker, E.; Hütter, G.; Wrublewsky, S.; Küting, F.; Hohl, M.; et al. Interleukin-1α Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation 2021, 144, 893–908. [Google Scholar] [CrossRef]
- Lemos, D.R.; McMurdo, M.; Karaca, G.; Wilflingseder, J.; Leaf, I.A.; Gupta, N.; Miyoshi, T.; Susa, K.; Johnson, B.G.; Soliman, K.; et al. Interleukin-1β Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 1690–1705. [Google Scholar] [CrossRef]
- Romanova, Y.; Laikov, A.; Markelova, M.; Khadiullina, R.; Makseev, A.; Hasanova, M.; Rizvanov, A.; Khaiboullina, S.; Salafutdinov, I. Proteomic Analysis of Human Serum from Patients with Chronic Kidney Disease. Biomolecules 2020, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.-C.; Li, H.-H.; Hsu, Y.-H.; Hsing, C.-H.; Sung, J.-M.; Chang, M.-S. Interleukin-20 Targets Renal Cells and Is Associated with Chronic Kidney Disease. Biochem. Biophys. Res. Commun. 2008, 374, 448–453. [Google Scholar] [CrossRef]
- Chang, M.S.; Hsu, Y.H. The Role of IL-20 in Chronic Kidney Disease and Diabetic Nephropathy: Pathogenic and Therapeutic Implications. J. Leukoc. Biol. 2018, 104, 919–923. [Google Scholar] [CrossRef]
- Hsu, Y.; Wu, C.; Chiu, C.; Chen, W.; Chang, Y.; Wabitsch, M.; Chang, M. IL-20 Is Involved in Obesity by Modulation of Adipogenesis and Macrophage Dysregulation. Immunology 2021, 164, 817–833. [Google Scholar] [CrossRef]
- Chiu, S.; Bharat, A. Role of Monocytes and Macrophages in Regulating Immune Response Following Lung Transplantation. Curr. Opin. Organ Transplant. 2016, 21, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Nair, M.G. Macrophages in Wound Healing: Activation and Plasticity. Immunol. Cell. Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef]
- Stout, R.D.; Suttles, J. Immunosenescence and Macrophage Functional Plasticity: Dysregulation of Macrophage Function by Age-Associated Microenvironmental Changes. Immunol. Rev. 2005, 205, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Caillon, A.; Paradis, P.; Schiffrin, E.L. Role of Immune Cells in Hypertension. Br. J. Pharmacol. 2019, 176, 1818–1828. [Google Scholar] [CrossRef]
- Duffield, J.S. Macrophages and Immunologic Inflammation of the Kidney. Semin. Nephrol. 2010, 30, 234–254. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Li, G.; Chen, Y.; Conley, S.M.; Gehr, T.W.; Boini, K.M.; Li, P.-L. Nod-like Receptor Protein 3 (NLRP3) Inflammasome Activation and Podocyte Injury via Thioredoxin-Interacting Protein (TXNIP) during Hyperhomocysteinemia. J. Biol. Chem. 2014, 289, 27159–27168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E.; Xiao, T.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantero-Navarro, E.; Rayego-Mateos, S.; Orejudo, M.; Tejedor-Santamaria, L.; Tejera-Muñoz, A.; Sanz, A.B.; Marquez-Exposito, L.; Marchant, V.; Santos-Sanchez, L.; Egido, J.; et al. Role of Macrophages and Related Cytokines in Kidney Disease. Front. Med. 2021, 8, 688060. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Nazir, S.; Gadi, I.; Al-Dabet, M.M.; Elwakiel, A.; Kohli, S.; Ghosh, S.; Manoharan, J.; Ranjan, S.; Bock, F.; Braun-Dullaeus, R.C.; et al. Cytoprotective Activated Protein C Averts Nlrp3 Inflammasome-Induced Ischemia-Reperfusion Injury via MTORC1 Inhibition. Blood 2017, 130, 2664–2677. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Xu, K.; Li, C.; Qi, C.; Fang, Y.; Zhu, N.; Bao, J.; Zhao, Z.; Yu, Q.; Wu, H.; et al. NLRP3 Associated with Chronic Kidney Disease Progression after Ischemia/Reperfusion-Induced Acute Kidney Injury. Cell Death Discov. 2021, 7, 324. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, D.W.; Ravichandran, K.; Keys, D.O.; Akcay, A.; Nguyen, Q.; He, Z.; Jani, A.; Ljubanovic, D.; Edelstein, C.L. NLRP3 Inflammasome Knockout Mice Are Protected against Ischemic but Not Cisplatin-Induced Acute Kidney Injury. J. Pharmacol. Exp. Ther. 2013, 346, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Faubel, S.; Edelstein, C.L. Caspases as Drug Targets in Ischemic Organ Injury. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 269–287. [Google Scholar] [CrossRef]
- Jang, H.N.; Kim, J.H.; Jung, M.H.; Tak, T.; Jung, J.H.; Lee, S.; Jung, S.; Chang, S.-H.; Kim, H.-J. Human Endothelial Progenitor Cells Protect the Kidney against Ischemia-Reperfusion Injury via the NLRP3 Inflammasome in Mice. Int. J. Mol. Sci. 2022, 23, 1546. [Google Scholar] [CrossRef]
- Anders, H.-J.; Suarez-Alvarez, B.; Grigorescu, M.; Foresto-Neto, O.; Steiger, S.; Desai, J.; Marschner, J.A.; Honarpisheh, M.; Shi, C.; Jordan, J.; et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1–mediated tissue injury. Kidney Int. 2017, 93, 656–669. [Google Scholar] [CrossRef]
- Liu, D.; Xu, M.; Ding, L.-H.; Lv, L.-L.; Liu, H.; Ma, K.-L.; Zhang, A.-H.; Crowley, S.D.; Liu, B.-C. Activation of the Nlrp3 Inflammasome by Mitochondrial Reactive Oxygen Species: A Novel Mechanism of Albumin-Induced Tubulointerstitial Inflammation. Int. J. Biochem. Cell Biol. 2014, 57, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Glassford, N.; Schneider, A.; Eastwood, G.; Peck, L.; Young, H.; Bellomo, R. Neutrophil gelatinase-associated lipocalin as a marker of tubular damage appears to be unrelated to fractional excretion of sodium as a marker of tubular function in septic patients, with or without AKI. Crit. Care 2011, 15 (Suppl. S3), P10. [Google Scholar] [CrossRef] [Green Version]
- Devarajan, P. Neutrophil gelatinase-associated lipocalin—An emerging troponin for kidney injury. Nephrol. Dial. Transplant. 2008, 23, 3737–3743. [Google Scholar] [CrossRef] [Green Version]
- Banai, A.; Rozenfeld, K.-L.; Levit, D.; Merdler, I.; Loewenstein, I.; Banai, S.; Shacham, Y. Neutrophil gelatinase-associated lipocalin (NGAL) for the prediction of acute kidney injury in chronic kidney disease patients treated with primary percutaneous coronary intervention. Int. J. Cardiol. Heart Vasc. 2020, 32, 100695. [Google Scholar] [CrossRef]
- Sise, M.E.; Barasch, J.; Devarajan, P.; Nickolas, T.L. Elevated urine neutrophil gelatinase-associated lipocalin can diagnose acute kidney injury in patients with chronic kidney diseases. Kidney Int. 2009, 75, 115–116. [Google Scholar] [CrossRef] [Green Version]
- Viau, A.; El Karoui, K.; Laouari, D.; Burtin, M.; Nguyen, C.; Mori, K.; Pillebout, E.; Berger, T.; Mak, T.W.; Knebelmann, B.; et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J. Clin. Investig. 2010, 120, 4065–4076. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Luo, Q.; Wang, L.; Han, L. Diagnostic value of neutrophil gelatinase-associated lipocalin for early diagnosis of cardiac surgery-associated acute kidney injury: A meta-analysis. Eur. J. Cardiothorac. Surg. 2016, 49, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Cai, Y.; Wang, P.-F.; Qu, J.-N.; Luo, Z.-C.; Chen, X.-D.; Huang, B.; Liu, Y.; Huang, W.-Q.; Wu, J.; et al. Diagnosis and prognosis of neutrophil gelatinase-associated lipocalin for acute kidney injury with sepsis: A systematic review and meta-analysis. Crit. Care 2016, 20, 41. [Google Scholar] [CrossRef] [Green Version]
- Tecson, K.M.; Erhardtsen, E.; Eriksen, P.M.; Gaber, A.O.; Germain, M.; Golestaneh, L.; Lavoria, M.D.L.A.; Moore, L.W.; McCullough, P.A. Optimal cut points of plasma and urine neutrophil gelatinase-associated lipocalin for the prediction of acute kidney injury among critically ill adults: Retrospective determination and clinical validation of a prospective multicentre study. BMJ Open 2017, 7, e016028. [Google Scholar] [CrossRef] [Green Version]
- Albert, C.; Zapf, A.; Haase, M.; Röver, C.; Pickering, J.W.; Albert, A.; Bellomo, R.; Breidthardt, T.; Camou, F.; Chen, Z.; et al. Neutrophil Gelatinase-Associated Lipocalin Measured on Clinical Laboratory Platforms for the Prediction of Acute Kidney Injury and the Associated Need for Dialysis Therapy: A Systematic Review and Meta-analysis. Am. J. Kidney Dis. 2020, 76, 826–841.e1. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ott, K.; Mori, K.; Kalandadze, A.; Li, J.-Y.; Paragas, N.; Nicholas, T.; Devarajan, P.; Barasch, J. Neutrophil gelatinase-associated lipocalin-mediated iron traffic in kidney epithelia. Curr. Opin. Nephrol. Hypertens. 2006, 15, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Marouf, R.; Adekile, A.D.; El-Muzaini, H.; Abdulla, R.; Mojiminiyi, O.A. Neutrophil gelatinase-associated lipocalin as a biomarker of nephropathy in sickle cell disease. Ann. Hematol. 2021, 100, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.H.; Xie, D.; Wang, X.; Baudier, R.L.; Orlandi, P.; Appel, L.J.; Dember, L.M.; He, J.; Kusek, J.W.; Lash, J.P.; et al. Novel Risk Factors for Progression of Diabetic and Nondiabetic CKD: Findings From the Chronic Renal Insufficiency Cohort (CRIC) Study. Am. J. Kidney Dis. 2020, 77, 56–73.e1. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Singh, A.B.; Harris, R.C. The Role of the EGF Family of Ligands and Receptors in Renal Development, Physiology and Pathophysiology. Exp. Cell Res. 2009, 315, 602–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, M.; Andreucci, M.; Garofalo, C.; Faga, T.; Michael, A.; Ielapi, N.; Grande, R.; Sapienza, P.; de Franciscis, S.; Mastroroberto, P.; et al. The Association of Matrix Metalloproteinases with Chronic Kidney Disease and Peripheral Vascular Disease: A Light at the End of the Tunnel? Biomolecules 2020, 10, 154. [Google Scholar] [CrossRef] [Green Version]
- Thrailkill, K.M.; Clay Bunn, R.; Fowlkes, J.L. Matrix Metalloproteinases: Their Potential Role in the Pathogenesis of Diabetic Nephropathy. Endocrine 2009, 35, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.M.; Lu, K.C.; Chen, Y.J.; Li, C.Y.; Lee, Y.H.; Chiu, H.W. Matrix metalloproteinase-7 promotes chronic kidney disease progression via the induction of inflammasomes and the suppression of autophagy. Biomed. Pharmacother. 2022, 154, 113565. [Google Scholar] [CrossRef]
- Tan, R.J.; Li, Y.; Rush, B.M.; Cerqueira, D.M.; Zhou, D.; Fu, H.; Ho, J.; Beer Stolz, Y.D.; Liu, Y. Tubular injury triggers podocyte dysfunction by β-catenin-driven release of MMP-7. JCI Insight 2019, 4, e122399. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Tan, R.J.; Liu, Y. The Many Faces of Matrix Metalloproteinase-7 in Kidney Diseases. Biomolecules 2020, 10, 960. [Google Scholar] [CrossRef]
- Rysz, J.; Gluba-Brzózka, A.; Franczyk, B.; Jabłonowski, Z.; Ciałkowska-Rysz, A. Novel Biomarkers in the Diagnosis of Chronic Kidney Disease and the Prediction of Its Outcome. Int. J. Mol. Sci. 2017, 18, 1702. [Google Scholar] [CrossRef]
- Yang, X.; Chen, C.; Teng, S.; Fu, X.; Zha, Y.; Liu, H.; Wang, L.; Tian, J.; Zhang, X.; Liu, Y.; et al. Urinary matrix metalloproteinase-7 predicts severe AKI and poor outcomes after cardiac surgery. J. Am. Soc. Nephrol. 2017, 28, 3373–3382. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Tian, Y.; Sun, L.; Zhou, L.; Xiao, L.; Tan, R.J.; Tian, J.; Fu, F.; Hou, F.F.; Liu, Y. Matrix metalloproteinase-7 is a urinary biomarker and pathogenic mediator of kidney fibrosis. J. Am. Soc. Nephrol. 2017, 28, 598–611. [Google Scholar] [CrossRef] [Green Version]
- Andreucci, M.; Provenzano, M.; Faga, T.; Michael, A.; Patella, G.; Mastroroberto, P.; Serraino, G.F.; Bracale, U.M.; Ielapi, N.; Serra, R. Aortic Aneurysms, Chronic Kidney Disease and Metalloproteinases. Biomolecules 2021, 11, 194. [Google Scholar] [CrossRef]
- Sampieri, C.L.; Orozco-Ortega, R.A. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Chronic Kidney Disease and Acute Kidney Injury: A Systematic Review of the Literature. Hippokratia 2018, 22, 99–104. [Google Scholar]
- Peiskerová, M.; Kalousová, M.; Kratochvílová, M.; Dusilová-Sulková, S.; Uhrová, J.; Bandúr, S.; Malbohan, I.M.; Zima, T.; Tesar, V. Fibroblast Growth Factor 23 and Matrix-Metalloproteinases in Patients with Chronic Kidney Disease: Are They Associated with Cardiovascular Disease? Kidney Blood Press. Res. 2009, 32, 276–283. [Google Scholar] [CrossRef]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix Metalloproteinase 2 and Basement Membrane Integrity: A Unifying Mechanism for Progressive Renal Injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Ivashkiv, L.B. Costimulation of Chemokine Receptor Signaling by Matrix Metalloproteinase-9 Mediates Enhanced Migration of IFN-α Dendritic Cells. J. Immunol. 2006, 176, 6022–6033. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-X.; Wang, Y.-P. Gut Microbiota-brain Axis. Chin. Med. J. 2016, 129, 2373–2380. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Tokarek, J.; Gadzinowska, J.; Młynarska, E.; Franczyk, B.; Rysz, J. What Is the Role of Gut Microbiota in Obesity Prevalence? A Few Words about Gut Microbiota and Its Association with Obesity and Related Diseases. Microorganisms 2021, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Rysz, J.; Franczyk, B.; Ławiński, J.; Olszewski, R.; Ciałkowska-Rysz, A.; Gluba-Brzózka, A. The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins 2021, 13, 252. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, S.; Pecoits-Filho, R.; Perreto, S.; Barberato, S.H.; Stinghen, A.E.; Lima, E.G.; Fuerbringer, R.; Sauthier, S.M.; Riella, M.C. Associations between renal function, volume status and endotoxaemia in chronic kidney disease patients. Nephrol. Dial. Transplant. 2006, 21, 2788–2794. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Jiang, H.; Shi, K.; Ren, Y.; Zhang, P.; Cheng, S. Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology 2012, 17, 733–738. [Google Scholar] [CrossRef]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Dure-Smith, B.; Miller, R.; Mirahmadi, M.K. Pathology of gastrointestinal tract in chronic hemodialysis patients: An autopsy study of 78 cases. Am. J. Gastroenterol. 1985, 80, 608–611. [Google Scholar]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Sumida, K.; Kovesdy, C.P. The gut-kidney-heart axis in chronic kidney disease. Physiol. Int. 2019, 106, 195–206. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Li, H.H.; Sung, J.M.; Chen, W.Y.; Hou, Y.C.; Weng, Y.H.; Lai, W.T.; Wu, C.H.; Chang, M.S. Interleukin-20 targets podocytes and is upregulated in experimental murine diabetic nephropathy. Exp. Mol. Med. 2017, 49, e310. [Google Scholar] [CrossRef] [Green Version]
- Headley, S.A.; Chapman, D.J.; Germain, M.J.; Evans, E.E.; Hutchinson, J.; Madsen, K.L.; Ikizler, T.A.; Miele, E.M.; Kirton, K.; O’Neill, E.; et al. The effects of 16-weeks of prebiotic supplementation and aerobic exercise training on inflammatory markers, oxidative stress, uremic toxins, and the microbiota in pre-dialysis kidney patients: A randomized controlled trial-protocol paper. BMC Nephrol. 2020, 21, 517. [Google Scholar] [CrossRef]
- Lee, S.B.; Kalluri, R. Mechanistic connection between inflammation and fibrosis. Kidney Int. 2010, 78 (Suppl. S119), S22–S26. [Google Scholar] [CrossRef]
- Moon, J.A.; Kim, H.T.; Cho, I.S.; Sheen, Y.Y.; Kim, D.K. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006, 70, 1234–1243. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Modifiable | Not Modifiable |
|---|---|
| Smoking | Old age |
| Nephrotoxins (e.g., alcohol, drugs) | Male gender |
| Obesity | Family history |
| Other comorbidities (e.g., hypertension, diabetes mellitus, CVD) | Low birth weight |
| Metabolic factors (insulin resistance, dyslipidemia, and hyperuricemia) | A non-Caucasian ethnicity |
| MMP | Group of MMP | Pathophysiological Mechanisms in CKD |
|---|---|---|
| MMP-2, MMP-9 | Gelatinases |
|
| MMP-7 | Matrilysins |
|
| MMP-3 | Stromelysins |
|
| MMP-14 | Membrane-type MMPs |
|
| Medications | Mechanism |
|---|---|
| Sirukumab and siltuximab | Blockage if the classical signaling and trans-signaling by targeting IL-6 |
| Tocilizumab and sarilumab | Blockage of all 3 types of IL-6 signaling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frąk, W.; Kućmierz, J.; Szlagor, M.; Młynarska, E.; Rysz, J.; Franczyk, B. New Insights into Molecular Mechanisms of Chronic Kidney Disease. Biomedicines 2022, 10, 2846. https://doi.org/10.3390/biomedicines10112846
Frąk W, Kućmierz J, Szlagor M, Młynarska E, Rysz J, Franczyk B. New Insights into Molecular Mechanisms of Chronic Kidney Disease. Biomedicines. 2022; 10(11):2846. https://doi.org/10.3390/biomedicines10112846
Chicago/Turabian StyleFrąk, Weronika, Joanna Kućmierz, Magdalena Szlagor, Ewelina Młynarska, Jacek Rysz, and Beata Franczyk. 2022. "New Insights into Molecular Mechanisms of Chronic Kidney Disease" Biomedicines 10, no. 11: 2846. https://doi.org/10.3390/biomedicines10112846