
Tissue Factor, Thrombosis, and Chronic Kidney Disease
{kind=link}
{kind=link}
Abstract
:1. Introduction
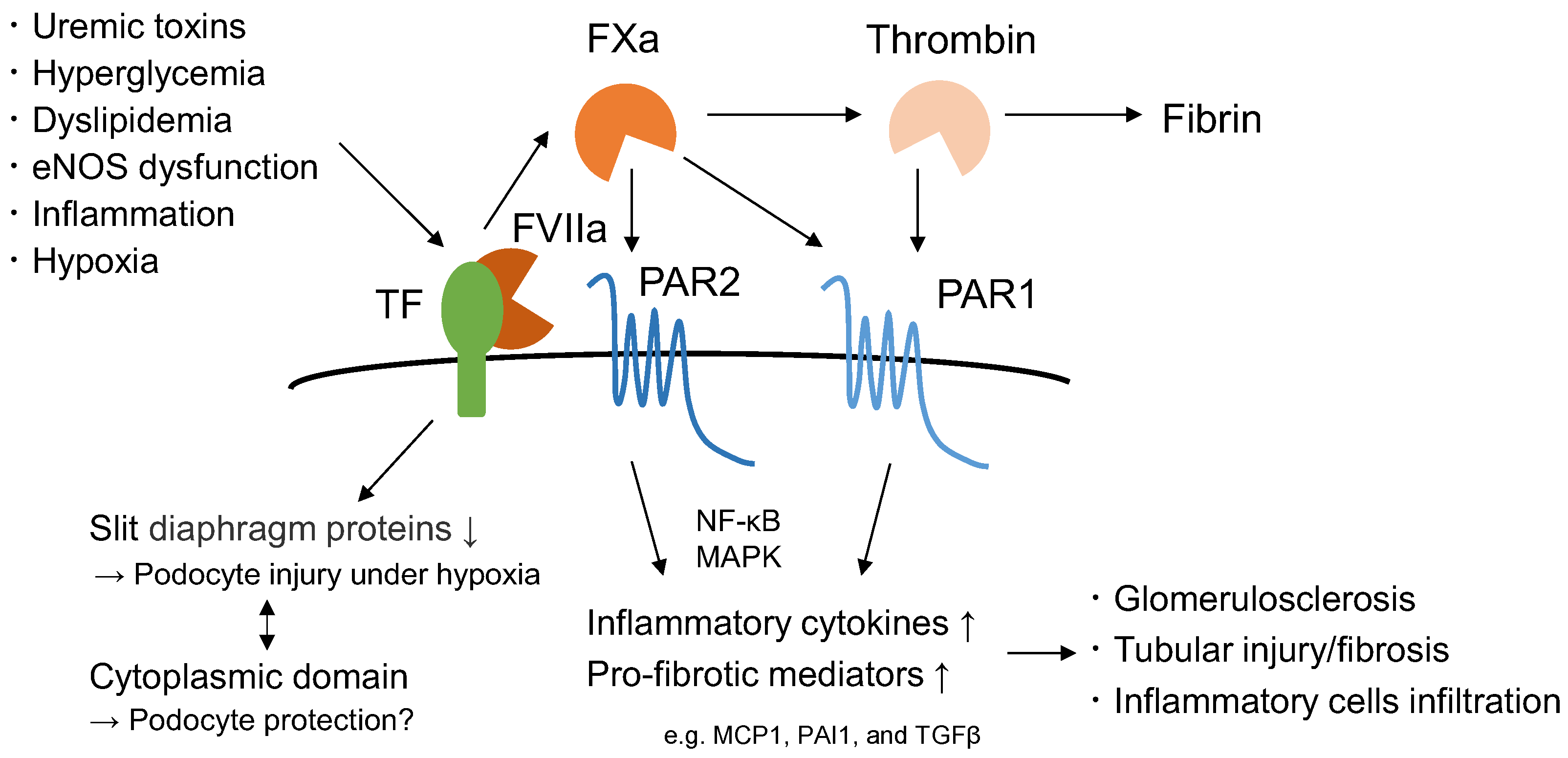
2. TF and the Extrinsic Coagulation System
3. Expression of TF in the Kidneys
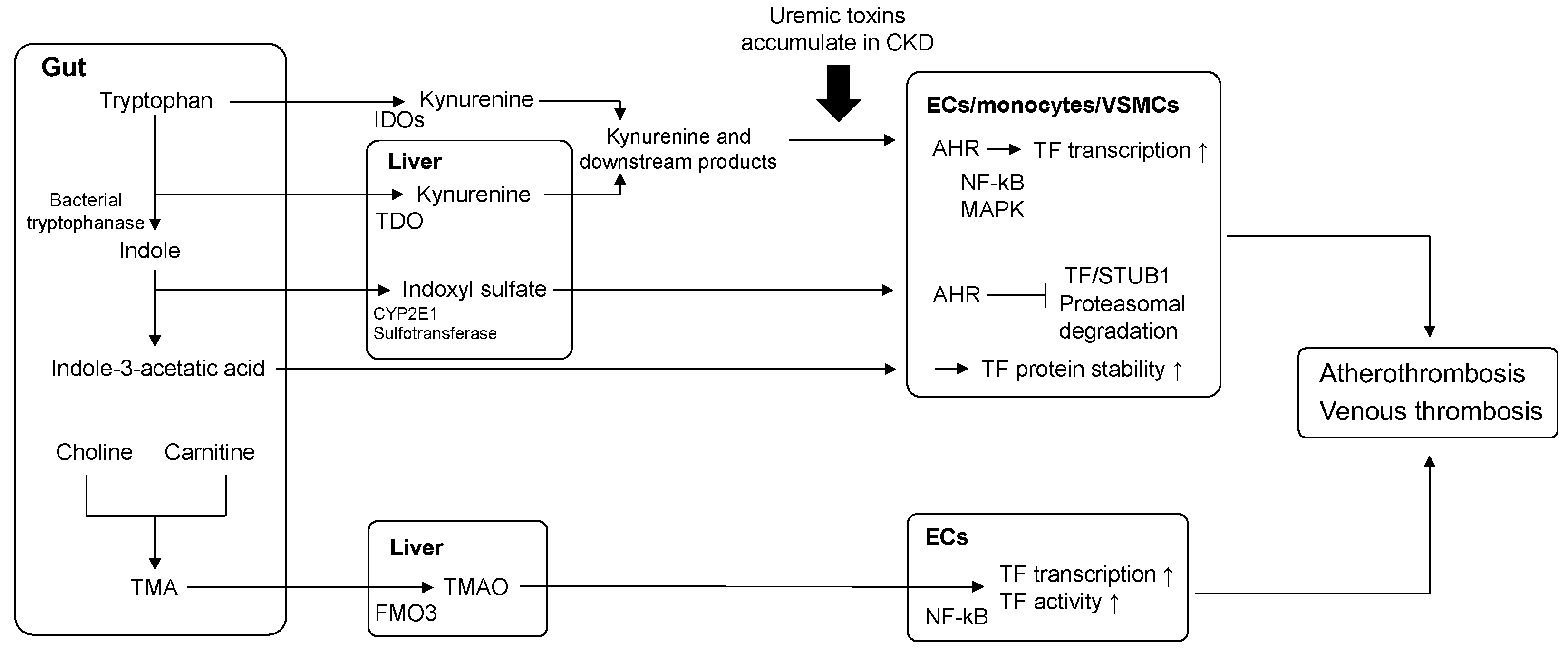
4. Relationship between Uremic Toxins and TF
4.1. Indoxyl Sulfate (IS) and Indole-3-Acetic Acid (IAA)
4.2. Kynurenine (Kyn)
4.3. p-Cresyl Sulfate (PCS)
4.4. Trimethylamine N-Oxide (TMAO)
5. Elevated Uremic Toxin-TF Axis and Risk of Thrombosis in CKD
6. Targeting Uremic Toxins for the Prevention of TF Activity and Thrombotic Risk
6.1. Role of Gut Microbiota in Reducing Uremic Toxins
6.2. AST-120: An Oral Adsorbent of Uremic Toxin
7. Hypoxia and TF in CKD
8. Targeting the Coagulation System Reduces Kidney Injury in CKD
8.1. Relationship among TF, eNOS, and Inflammation in DKD
8.2. Deletion of Myeloid TF Reduces Kidney Injury in Adenine-Induced CKD Model
8.3. Role of TF in Podocyte Injury
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Collaboration, G.C.K.D. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294–1304. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Lutz, J.; Menke, J.; Sollinger, D.; Schinzel, H.; Thürmel, K. Haemostasis in chronic kidney disease. Nephrol. Dial. Transpl. 2014, 29, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.J.; Wei, R.B.; Wang, Y.; Su, T.Y.; Di, P.; Li, Q.P.; Yang, X.; Li, P.; Chen, X.M. Blood coagulation system in patients with chronic kidney disease: A prospective observational study. BMJ Open 2017, 7, e014294. [Google Scholar] [CrossRef] [Green Version]
- Dubin, R.; Cushman, M.; Folsom, A.R.; Fried, L.F.; Palmas, W.; Peralta, C.A.; Wassel, C.; Shlipak, M.G. Kidney function and multiple hemostatic markers: Cross sectional associations in the multi-ethnic study of atherosclerosis. BMC Nephrol. 2011, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Tran, L.; Pannier, B.; Lacolley, P.; Serrato, T.; Benetos, A.; London, G.M.; Bézie, Y.; Regnault, V. A case-control study indicates that coagulation imbalance is associated with arteriosclerosis and markers of endothelial dysfunction in kidney failure. Kidney Int. 2021, 99, 1162–1172. [Google Scholar] [CrossRef]
- Kamiński, T.W.; Pawlak, K.; Karbowska, M.; Myśliwiec, M.; Pawlak, D. Indoxyl sulfate—The uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Ocak, G.; van Stralen, K.J.; Rosendaal, F.R.; Verduijn, M.; Ravani, P.; Palsson, R.; Leivestad, T.; Hoitsma, A.J.; Ferrer-Alamar, M.; Finne, P.; et al. Mortality due to pulmonary embolism, myocardial infarction, and stroke among incident dialysis patients. J. Thromb. Haemost. 2012, 10, 2484–2493. [Google Scholar] [CrossRef]
- Ocak, G.; Noordzij, M.; Rookmaaker, M.B.; Cases, A.; Couchoud, C.; Heaf, J.G.; Jarraya, F.; De Meester, J.; Groothoff, J.W.; Waldum-Grevbo, B.E.; et al. Mortality due to bleeding, myocardial infarction and stroke in dialysis patients. J. Thromb. Haemost. 2018, 16, 1953–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattanakit, K.; Cushman, M.; Stehman-Breen, C.; Heckbert, S.R.; Folsom, A.R. Chronic kidney disease increases risk for venous thromboembolism. J. Am. Soc. Nephrol. 2008, 19, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folsom, A.R.; Lutsey, P.L.; Astor, B.C.; Wattanakit, K.; Heckbert, S.R.; Cushman, M.; Study, A.R.i.C. Chronic kidney disease and venous thromboembolism: A prospective study. Nephrol. Dial. Transpl. 2010, 25, 3296–3301. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodi, B.K.; Gansevoort, R.T.; Næss, I.A.; Lutsey, P.L.; Brækkan, S.K.; Veeger, N.J.; Brodin, E.E.; Meijer, K.; Sang, Y.; Matsushita, K.; et al. Association of mild to moderate chronic kidney disease with venous thromboembolism: Pooled analysis of five prospective general population cohorts. Circulation 2012, 126, 1964–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.L.; Zakai, N.A.; Folsom, A.R.; Kurella Tamura, M.; Peralta, C.A.; Judd, S.E.; Callas, P.W.; Cushman, M. Measures of Kidney Disease and the Risk of Venous Thromboembolism in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) Study. Am. J. Kidney Dis. 2017, 70, 182–190. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [Green Version]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-Derived Uremic Toxins and Thrombosis in Chronic Kidney Disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef] [Green Version]
- Fryc, J.; Naumnik, B. Thrombolome and Its Emerging Role in Chronic Kidney Diseases. Toxins 2021, 13, 223. [Google Scholar] [CrossRef]
- Ravid, J.D.; Kamel, M.H.; Chitalia, V.C. Uraemic solutes as therapeutic targets in CKD-associated cardiovascular disease. Nat. Rev. Nephrol. 2021, 17, 402–416. [Google Scholar] [CrossRef]
- Shashar, M.; Francis, J.; Chitalia, V. Thrombosis in the uremic milieu--emerging role of “thrombolome”. Semin. Dial. 2015, 28, 198–205. [Google Scholar] [CrossRef]
- Posma, J.J.; Grover, S.P.; Hisada, Y.; Owens, A.P.; Antoniak, S.; Spronk, H.M.; Mackman, N. Roles of Coagulation Proteases and PARs (Protease-Activated Receptors) in Mouse Models of Inflammatory Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Palygin, O.; Ilatovskaya, D.V.; Staruschenko, A. Protease-activated receptors in kidney disease progression. Am. J. Physiol. Renal. Physiol. 2016, 311, F1140–F1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oe, Y.; Miyazaki, M.; Takahashi, N. Coagulation, Protease-Activated Receptors, and Diabetic Kidney Disease: Lessons from eNOS-Deficient Mice. Tohoku J. Exp. Med. 2021, 255, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Madhusudhan, T.; Kerlin, B.A.; Isermann, B. The emerging role of coagulation proteases in kidney disease. Nat. Rev. Nephrol. 2016, 12, 94–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Østerud, B.; Bjørklid, E. Sources of tissue factor. Semin. Thromb. Hemost. 2006, 32, 11–23. [Google Scholar] [CrossRef]
- Samad, F.; Ruf, W. Inflammation, obesity, and thrombosis. Blood 2013, 122, 3415–3422. [Google Scholar] [CrossRef]
- Owens, A.P.; Passam, F.H.; Antoniak, S.; Marshall, S.M.; McDaniel, A.L.; Rudel, L.; Williams, J.C.; Hubbard, B.K.; Dutton, J.A.; Wang, J.; et al. Monocyte tissue factor-dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J. Clin. Investig. 2012, 122, 558–568. [Google Scholar] [CrossRef] [Green Version]
- Flössel, C.; Luther, T.; Müller, M.; Albrecht, S.; Kasper, M. Immunohistochemical detection of tissue factor (TF) on paraffin sections of routinely fixed human tissue. Histochemistry 1994, 101, 449–453. [Google Scholar] [CrossRef]
- Osterholm, C.; Veress, B.; Simanaitis, M.; Hedner, U.; Ekberg, H. Differential expression of tissue factor (TF) in calcineurin inhibitor-induced nephrotoxicity and rejection--implications for development of a possible diagnostic marker. Transpl. Immunol. 2005, 15, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Narita, I.; Shimada, M.; Yamabe, H.; Kinjo, T.; Tanno, T.; Nishizaki, K.; Kawai, M.; Nakamura, M.; Murakami, R.; Nakamura, N.; et al. NF-κB-dependent increase in tissue factor expression is responsible for hypoxic podocyte injury. Clin. Exp. Nephrol. 2016, 20, 679–688. [Google Scholar] [CrossRef]
- Shimosawa, M.; Sakamoto, K.; Tomari, Y.; Kamikado, K.; Otsuka, H.; Liu, N.; Kitamura, H.; Uemura, K.; Nogaki, F.; Mori, N.; et al. Lipopolysaccharide-triggered acute aggravation of mesangioproliferative glomerulonephritis through activation of coagulation in a high IgA strain of ddY mice. Nephron Exp. Nephrol. 2009, 112, e81–e91. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Liu, N.; Nagai, K.; Hasegawa, T.; Kobayashi, I.; Nogaki, F.; Tanaka, M.; Arai, H.; Fukatsu, A.; Kita, T.; et al. Roles of coagulation pathway and factor Xa in rat mesangioproliferative glomerulonephritis. Lab. Investig. 2007, 87, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettelaie, C.; Su, S.; Li, C.; Collier, M.E. Tissue factor-containing microparticles released from mesangial cells in response to high glucose and AGE induce tube formation in microvascular cells. Microvasc. Res. 2008, 76, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Wendt, T.; Zhang, Y.M.; Bierhaus, A.; Kriegsmann, J.; Deng, Y.; Waldherr, R.; Teske, T.; Luther, T.; Fünfstück, R.; Nawroth, P.P. Tissue factor expression in an animal model of hydronephrosis. Nephrol. Dial. Transpl. 1995, 10, 1820–1828. [Google Scholar]
- Watanabe, M.; Oe, Y.; Sato, E.; Sekimoto, A.; Sato, H.; Ito, S.; Takahashi, N. Protease-activated receptor 2 exacerbates cisplatin-induced nephrotoxicity. Am. J. Physiol. Renal. Physiol. 2019, 316, F654–F659. [Google Scholar] [CrossRef] [PubMed]
- Lwaleed, B.A.; Vayro, S.; Racusen, L.C.; Cooper, A.J. Tissue factor expression by a human kidney proximal tubular cell line in vitro: A model relevant to urinary tissue factor secretion in disease? J. Clin. Pathol. 2007, 60, 762–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommeijer, D.W.; Hansen, H.R.; van Oerle, R.; Hamulyak, K.; van Zanten, A.P.; Meesters, E.; Spronk, H.M.; ten Cate, H. Soluble tissue factor is a candidate marker for progression of microvascular disease in patients with Type 2 diabetes. J. Thromb. Haemost. 2006, 4, 574–580. [Google Scholar] [CrossRef]
- Iyer, A.; Humphries, T.L.R.; Owens, E.P.; Zhao, K.N.; Masci, P.P.; Johnson, D.W.; Nikolic-Paterson, D.; Gobe, G.C.; Fairlie, D.P.; Vesey, D.A. PAR2 Activation on Human Kidney Tubular Epithelial Cells Induces Tissue Factor Synthesis, That Enhances Blood Clotting. Front. Physiol. 2021, 12, 615428. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; Group, E.U.T.W. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Liabeuf, S.; Pepin, M.; Franssen, C.F.M.; Viggiano, D.; Carriazo, S.; Gansevoort, R.T.; Gesualdo, L.; Hafez, G.; Malyszko, J.; Mayer, C.; et al. Chronic kidney disease and neurological disorders: Are uraemic toxins the missing piece of the puzzle? Nephrol. Dial. Transpl. 2021, 37, ii33–ii44. [Google Scholar] [CrossRef]
- Chalupsky, M.; Goodson, D.A.; Gamboa, J.L.; Roshanravan, B. New insights into muscle function in chronic kidney disease and metabolic acidosis. Curr. Opin. Nephrol. Hypertens. 2021, 30, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients 2017, 9, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; Nigam, S.K.; Burtey, S.; Glorieux, G. What If Not All Metabolites from the Uremic Toxin Generating Pathways Are Toxic? A Hypothesis. Toxins 2022, 14, 221. [Google Scholar] [CrossRef] [PubMed]
- Oe, Y.; Sato, E.; Sato, H.; Miyazaki, M.; Ito, S.; Takahashi, N. Uremic toxins alter coagulation and fibrinolysis-related genes expression in human endothelial cells. Thromb. Res. 2020, 186, 75–77. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Yamakage, S.; Oe, Y.; Sato, E.; Okamoto, K.; Sekimoto, A.; Kumakura, S.; Sato, H.; Yoshida, M.; Nagasawa, T.; Miyazaki, M.; et al. Myeloid cell-derived coagulation tissue factor is associated with renal tubular damage in mice fed an adenine diet. Sci. Rep. 2021, 11, 12159. [Google Scholar] [CrossRef]
- Shashar, M.; Belghasem, M.E.; Matsuura, S.; Walker, J.; Richards, S.; Alousi, F.; Rijal, K.; Kolachalama, V.B.; Balcells, M.; Odagi, M.; et al. Targeting STUB1-tissue factor axis normalizes hyperthrombotic uremic phenotype without increasing bleeding risk. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2019, 93, 121–136. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I.; Kolandaivelu, K.; Edelman, E.R. Uremic serum and solutes increase post-vascular interventional thrombotic risk through altered stability of smooth muscle cell tissue factor. Circulation 2013, 127, 365–376. [Google Scholar] [CrossRef]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.Y.; Bolati, W.; Lee, C.T.; Chien, Y.S.; Yisireyili, M.; Saito, S.; Pei, S.N.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Downregulates Mas Receptor via Aryl Hydrocarbon Receptor/Nuclear Factor-kappa B, and Induces Cell Proliferation and Tissue Factor Expression in Vascular Smooth Muscle Cells. Nephron 2016, 133, 205–212. [Google Scholar] [CrossRef]
- Yisireyili, M.; Saito, S.; Abudureyimu, S.; Adelibieke, Y.; Ng, H.Y.; Nishijima, F.; Takeshita, K.; Murohara, T.; Niwa, T. Indoxyl sulfate-induced activation of (pro)renin receptor promotes cell proliferation and tissue factor expression in vascular smooth muscle cells. PLoS ONE 2014, 9, e109268. [Google Scholar] [CrossRef] [Green Version]
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D. Indoxyl Sulfate Promotes Arterial Thrombosis in Rat Model via Increased Levels of Complex TF/VII, PAI-1, Platelet Activation as Well as Decreased Contents of SIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623. [Google Scholar] [CrossRef] [Green Version]
- Karbowska, M.; Kaminski, T.W.; Marcinczyk, N.; Misztal, T.; Rusak, T.; Smyk, L.; Pawlak, D. The Uremic Toxin Indoxyl Sulfate Accelerates Thrombotic Response after Vascular Injury in Animal Models. Toxins 2017, 9, 229. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, D.; Song, P.; Zou, M.H. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front. Biosci. (Landmark Ed.) 2015, 20, 1116–1143. [Google Scholar] [CrossRef] [Green Version]
- Zakrocka, I.; Załuska, W. Kynurenine pathway in kidney diseases. Pharmacol. Rep. 2022, 74, 27–39. [Google Scholar] [CrossRef]
- Pawlak, K.; Tankiewicz, J.; Mysliwiec, M.; Pawlak, D. Tissue factor/its pathway inhibitor system and kynurenines in chronic kidney disease patients on conservative treatment. Blood Coagul. Fibrinolysis. 2009, 20, 590–594. [Google Scholar] [CrossRef]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Hypercoagulability is independently associated with kynurenine pathway activation in dialysed uraemic patients. Thromb. Haemost. 2009, 102, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.A.; Richards, S.; Whelan, S.A.; Yoo, S.B.; Russell, T.L.; Arinze, N.; Lotfollahzadeh, S.; Napoleon, M.A.; Belghasem, M.; Lee, N.; et al. Indoleamine 2,3-dioxygenase-1, a Novel Therapeutic Target for Post-Vascular Injury Thrombosis in CKD. J. Am. Soc. Nephrol. 2021, 32, 2834–2850. [Google Scholar] [CrossRef]
- Belghasem, M.; Roth, D.; Richards, S.; Napolene, M.A.; Walker, J.; Yin, W.; Arinze, N.; Lyle, C.; Spencer, C.; Francis, J.M.; et al. Metabolites in a mouse cancer model enhance venous thrombogenicity through the aryl hydrocarbon receptor-tissue factor axis. Blood 2019, 134, 2399–2413. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.K.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [Green Version]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Maré, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef]
- Meijers, B.K.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Missailidis, C.; Hällqvist, J.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, C.C.; Croyal, M.; Ene, L.; Aguesse, A.; Billon-Crossouard, S.; Krempf, M.; Lemoine, S.; Guebre-Egziabher, F.; Juillard, L.; Soulage, C.O. Elevation of Trimethylamine-N-Oxide in Chronic Kidney Disease: Contribution of Decreased Glomerular Filtration Rate. Toxins 2019, 11, 635. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Friebel, J.; Buffa, J.A.; Li, X.S.; Wang, Z.; Sangwan, N.; Li, L.; DiDonato, J.A.; Tizian, C.; Haghikia, A.; et al. Vascular endothelial Tissue Factor contributes to trimethylamine N-oxide-enhanced arterial thrombosis. Cardiovasc. Res. 2021, 118, 2367–2384. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Qiu, X.; Liu, Y.; Yuan, C.; Yang, X. Trimethylamine N-oxide promotes tissue factor expression and activity in vascular endothelial cells: A new link between trimethylamine N-oxide and atherosclerotic thrombosis. Thromb. Res. 2019, 177, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Mercier, E.; Branger, B.; Vecina, F.; Al-Sabadani, B.; Berlan, J.; Dauzat, M.; Fourcade, J.; Gris, J.C. Tissue factor coagulation pathway and blood cells activation state in renal insufficiency. Hematol. J. 2001, 2, 18–25. [Google Scholar] [CrossRef]
- Wu, C.C.; Hsieh, M.Y.; Hung, S.C.; Kuo, K.L.; Tsai, T.H.; Lai, C.L.; Chen, J.W.; Lin, S.J.; Huang, P.H.; Tarng, D.C. Serum Indoxyl Sulfate Associates with Postangioplasty Thrombosis of Dialysis Grafts. J. Am. Soc. Nephrol. 2016, 27, 1254–1264. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Evenepoel, P.; Anders, H.J. Intestinal microbiome and fitness in kidney disease. Nat. Rev. Nephrol. 2019, 15, 531–545. [Google Scholar] [CrossRef]
- Markowiak, P.; Śliżewska, K. Effects of Probiotics, Prebiotics, and Synbiotics on Human Health. Nutrients 2017, 9, 1021. [Google Scholar] [CrossRef]
- Liu, S.; Liu, H.; Chen, L.; Liang, S.S.; Shi, K.; Meng, W.; Xue, J.; He, Q.; Jiang, H. Effect of probiotics on the intestinal microbiota of hemodialysis patients: A randomized trial. Eur. J. Nutr. 2020, 59, 3755–3766. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.S.; Wang, H.F.; Lee, M.C.; Chiu, L.S.; Wu, M.Y.; Chang, W.C.; Wu, T.K. The Efficacy of Lactobacillus-Containing Probiotic Supplementation in Hemodialysis Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Ren. Nutr. 2021, 31, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Kumakura, S.; Kikuchi, M. Review of the efficacy of AST-120 (KREMEZIN). Ren. Fail. 2019, 41, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Schulman, G.; Agarwal, R.; Acharya, M.; Berl, T.; Blumenthal, S.; Kopyt, N. A multicenter, randomized, double-blind, placebo-controlled, dose-ranging study of AST-120 (Kremezin) in patients with moderate to severe CKD. Am. J. Kidney Dis. 2006, 47, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Nomura, T.; Sugiyama, S.; Miyazaki, T.; Tsukushi, S.; Tsutsui, S. The protein metabolite hypothesis, a model for the progression of renal failure: An oral adsorbent lowers indoxyl sulfate levels in undialyzed uremic patients. Kidney Int. Suppl. 1997, 62, S23–S28. [Google Scholar]
- Kikuchi, K.; Itoh, Y.; Tateoka, R.; Ezawa, A.; Murakami, K.; Niwa, T. Metabolomic search for uremic toxins as indicators of the effect of an oral sorbent AST-120 by liquid chromatography/tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010, 878, 2997–3002. [Google Scholar] [CrossRef]
- Sato, E.; Saigusa, D.; Mishima, E.; Uchida, T.; Miura, D.; Morikawa-Ichinose, T.; Kisu, K.; Sekimoto, A.; Saito, R.; Oe, Y.; et al. Impact of the Oral Adsorbent AST-120 on Organ-Specific Accumulation of Uremic Toxins: LC-MS/MS and MS Imaging Techniques. Toxins 2017, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Wu, M.Y.; Hu, P.J.; Chen, T.T.; Shen, W.C.; Chang, W.C.; Wu, M.S. Effects and Safety of an Oral Adsorbent on Chronic Kidney Disease Progression: A Systematic Review and Meta-Analysis. J. Clin. Med. 2019, 8, 1718. [Google Scholar] [CrossRef] [Green Version]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [Green Version]
- Cha, R.H.; Kang, S.W.; Park, C.W.; Cha, D.R.; Na, K.Y.; Kim, S.G.; Yoon, S.A.; Han, S.Y.; Chang, J.H.; Park, S.K.; et al. A Randomized, Controlled Trial of Oral Intestinal Sorbent AST-120 on Renal Function Deterioration in Patients with Advanced Renal Dysfunction. Clin. J. Am. Soc. Nephrol. 2016, 11, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Nakada, Y.; Onoue, K.; Nakano, T.; Ishihara, S.; Kumazawa, T.; Nakagawa, H.; Ueda, T.; Nishida, T.; Soeda, T.; Okayama, S.; et al. AST-120, an Oral Carbon Absorbent, Protects against the Progression of Atherosclerosis in a Mouse Chronic Renal Failure Model by Preserving sFlt-1 Expression Levels. Sci. Rep. 2019, 9, 15571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Zuo, Y.; Ma, J.; Yancey, P.G.; Hunley, T.E.; Motojima, M.; Fogo, A.B.; Linton, M.F.; Fazio, S.; Ichikawa, I.; et al. Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice. Nephrol. Dial. Transpl. 2011, 26, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Pruijm, M.; Milani, B.; Pivin, E.; Podhajska, A.; Vogt, B.; Stuber, M.; Burnier, M. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int. 2018, 93, 932–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzén, S.; Pihl, L.; Khan, N.; Gustafsson, H.; Palm, F. Pronounced kidney hypoxia precedes albuminuria in type 1 diabetic mice. Am. J. Physiol. Renal. Physiol. 2016, 310, F807–F809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, M.; Tanaka, T.; Nangaku, M. Hypoxia-Inducible Factor and Oxygen Biology in the Kidney. Kidney360 2020, 1, 1021–1031. [Google Scholar] [CrossRef]
- Gupta, N.; Zhao, Y.Y.; Evans, C.E. The stimulation of thrombosis by hypoxia. Thromb. Res. 2019, 181, 77–83. [Google Scholar] [CrossRef]
- Sun, L.; Liu, Y.; Lin, S.; Shang, J.; Liu, J.; Li, J.; Yuan, S.; Zhang, L. Early growth response gene-1 and hypoxia-inducible factor-1α affect tumor metastasis via regulation of tissue factor. Acta Oncol. 2013, 52, 842–851. [Google Scholar] [CrossRef]
- Stavik, B.; Espada, S.; Cui, X.Y.; Iversen, N.; Holm, S.; Mowinkel, M.C.; Halvorsen, B.; Skretting, G.; Sandset, P.M. EPAS1/HIF-2 alpha-mediated downregulation of tissue factor pathway inhibitor leads to a pro-thrombotic potential in endothelial cells. Biochim. Biophys. Acta 2016, 1862, 670–678. [Google Scholar] [CrossRef]
- Ahn, Y.T.; Chua, M.S.; Whitlock, J.P.; Shin, Y.C.; Song, W.H.; Kim, Y.; Eom, C.Y.; An, W.G. Rodent-specific hypoxia response elements enhance PAI-1 expression through HIF-1 or HIF-2 in mouse hepatoma cells. Int. J. Oncol. 2010, 37, 1627–1638. [Google Scholar] [CrossRef]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, C.A.; Yan, S.D.; Yan, S.F.; Liao, H.; Zhou, Y.S.; Sobel, J.; Kisiel, W.; Stern, D.M.; Pinsky, D.J. Monocytes and tissue factor promote thrombosis in a murine model of oxygen deprivation. J. Clin. Investig. 1997, 99, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wish, J.B. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am. J. Kidney Dis. 2017, 69, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Kurata, Y.; Tanaka, T.; Nangaku, M. Hypoxia-inducible factor prolyl hydroxylase inhibitor in the treatment of anemia in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cheng, Q.; Wang, J.; Zhao, X.; Zhu, S. Long-term efficacy and safety of hypoxia-inducible factor prolyl hydroxylase inhibitors in anaemia of chronic kidney disease: A meta-analysis including 13,146 patients. J. Clin. Pharm. Ther. 2021, 46, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Landmesser, U.; Rauch, U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc. Med. 2016, 26, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Emekli-Alturfan, E.; Kasikci, E.; Yarat, A. Tissue factor activities of streptozotocin induced diabetic rat tissues and the effect of peanut consumption. Diabetes Metab. Res. Rev. 2007, 23, 653–658. [Google Scholar] [CrossRef]
- Sommeijer, D.W.; Florquin, S.; Hoedemaker, I.; Timmerman, J.J.; Reitsma, P.H.; Ten Cate, H. Renal tissue factor expression is increased in streptozotocin-induced diabetic mice. Nephron Exp. Nephrol. 2005, 101, e86–e94. [Google Scholar] [CrossRef]
- Wang, C.H.; Li, F.; Hiller, S.; Kim, H.S.; Maeda, N.; Smithies, O.; Takahashi, N. A modest decrease in endothelial NOS in mice comparable to that associated with human NOS3 variants exacerbates diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2011, 108, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Oe, Y.; Hayashi, S.; Fushima, T.; Sato, E.; Kisu, K.; Sato, H.; Ito, S.; Takahashi, N. Coagulation Factor Xa and Protease-Activated Receptor 2 as Novel Therapeutic Targets for Diabetic Nephropathy. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, C.H.; Wang, J.G.; Thai, T.; Boysen, G.; Xu, L.; Turner, A.L.; Wolberg, A.S.; Mackman, N.; Maeda, N.; et al. Elevated tissue factor expression contributes to exacerbated diabetic nephropathy in mice lacking eNOS fed a high fat diet. J. Thromb. Haemost. 2010, 8, 2122–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsui, S.; Oe, Y.; Sekimoto, A.; Sato, E.; Hashizume, Y.; Yamakage, S.; Kumakura, S.; Sato, H.; Ito, S.; Takahashi, N. Dual blockade of protease-activated receptor 1 and 2 additively ameliorates diabetic kidney disease. Am. J. Physiol. Renal. Physiol. 2020, 318, F1067–F1073. [Google Scholar] [CrossRef] [PubMed]
- Passauer, J.; Pistrosch, F.; Büssemaker, E. Nitric oxide in chronic renal failure. Kidney Int. 2005, 67, 1665–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oe, Y.; Mitsui, S.; Sato, E.; Shibata, N.; Kisu, K.; Sekimoto, A.; Miyazaki, M.; Sato, H.; Ito, S.; Takahashi, N. Lack of Endothelial Nitric Oxide Synthase Accelerates Ectopic Calcification in Uremic Mice Fed an Adenine and High Phosphorus Diet. Am. J. Pathol. 2021, 191, 283–293. [Google Scholar] [CrossRef]
- Dong, J.; Ping, Y.; Wang, Y.; Zhang, Y. The roles of endothelial nitric oxide synthase gene polymorphisms in diabetes mellitus and its associated vascular complications: A systematic review and meta-analysis. Endocrine 2018, 62, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Azushima, K.; Gurley, S.B.; Coffman, T.M. Modelling diabetic nephropathy in mice. Nat. Rev. Nephrol. 2018, 14, 48–56. [Google Scholar] [CrossRef]
- Jia, T.; Olauson, H.; Lindberg, K.; Amin, R.; Edvardsson, K.; Lindholm, B.; Andersson, G.; Wernerson, A.; Sabbagh, Y.; Schiavi, S.; et al. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol. 2013, 14, 116. [Google Scholar] [CrossRef] [Green Version]
- Makhloufi, C.; Crescence, L.; Darbousset, R.; McKay, N.; Massy, Z.A.; Dubois, C.; Panicot-Dubois, L.; Burtey, S.; Poitevin, S. Assessment of Thrombotic and Bleeding Tendency in Two Mouse Models of Chronic Kidney Disease: Adenine-Diet and 5/6th Nephrectomy. TH Open 2020, 4, e66–e76. [Google Scholar] [CrossRef] [Green Version]
- Horinouchi, Y.; Ikeda, Y.; Fukushima, K.; Imanishi, M.; Hamano, H.; Izawa-Ishizawa, Y.; Zamami, Y.; Takechi, K.; Miyamoto, L.; Fujino, H.; et al. Renoprotective effects of a factor Xa inhibitor: Fusion of basic research and a database analysis. Sci. Rep. 2018, 8, 10858. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Ohashi, K.; Ogawa, H.; Otaka, N.; Kawanishi, H.; Takikawa, T.; Ozaki, Y.; Takahara, K.; Tatsumi, M.; Takefuji, M.; et al. Factor Xa inhibitor, edoxaban ameliorates renal injury after subtotal nephrectomy by reducing epithelial-mesenchymal transition and inflammatory response. Physiol. Rep. 2022, 10, e15218. [Google Scholar] [CrossRef]
- Saifi, M.A.; Annaldas, S.; Godugu, C. A direct thrombin inhibitor, dabigatran etexilate protects from renal fibrosis by inhibiting protease activated receptor-1. Eur. J. Pharmacol. 2021, 893, 173838. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Oe, Y.; Fushima, T.; Sato, E.; Sato, H.; Ito, S.; Takahashi, N. Protease-activated receptor 2 exacerbates adenine-induced renal tubulointerstitial injury in mice. Biochem. Biophys. Res. Commun. 2017, 483, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Ramachandran, R.; Hollenberg, M.D.; Muruve, D.A. Proteinase-activated receptor-2 transactivation of epidermal growth factor receptor and transforming growth factor-β receptor signaling pathways contributes to renal fibrosis. J. Biol. Chem. 2013, 288, 37319–37331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Zhang, T.; Xiao, X.; Shi, Y.; Duan, H.; Ren, Y. Protease-activated receptor-2 promotes kidney tubular epithelial inflammation by inhibiting autophagy via the PI3K/Akt/mTOR signalling pathway. Biochem. J. 2017, 474, 2733–2747. [Google Scholar] [CrossRef]
- Lok, S.W.Y.; Yiu, W.H.; Li, H.; Xue, R.; Zou, Y.; Li, B.; Chan, K.W.; Chan, L.Y.Y.; Leung, J.C.K.; Lai, K.N.; et al. The PAR-1 antagonist vorapaxar ameliorates kidney injury and tubulointerstitial fibrosis. Clin. Sci. 2020, 134, 2873–2891. [Google Scholar] [CrossRef]
- Ha, S.; Chung, K.W.; Lee, J.; Chung, H.Y.; Moon, H.R. Renal tubular PAR2 promotes interstitial fibrosis by increasing inflammatory responses and EMT process. Arch. Pharm. Res. 2022, 45, 159–173. [Google Scholar] [CrossRef]
- Brinkkoetter, P.T.; Ising, C.; Benzing, T. The role of the podocyte in albumin filtration. Nat. Rev. Nephrol. 2013, 9, 328–336. [Google Scholar] [CrossRef]
- Leeuwis, J.W.; Nguyen, T.Q.; Dendooven, A.; Kok, R.J.; Goldschmeding, R. Targeting podocyte-associated diseases. Adv. Drug Deliv. Rev. 2010, 62, 1325–1336. [Google Scholar] [CrossRef]
- Trimarchi, H.; Coppo, R. Podocytopathy in the mesangial proliferative immunoglobulin A nephropathy: New insights into the mechanisms of damage and progression. Nephrol. Dial. Transpl. 2019, 34, 1280–1285. [Google Scholar] [CrossRef]
- Farquhar, A.; MacDonald, M.K.; Ireland, J.T. The role of fibrin deposition in diabetic glomerulosclerosis: A light, electron and immunofluorescence microscopy study. J. Clin. Pathol. 1972, 25, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulos, J.; Moussa, L.; Tipping, P.G. The cytoplasmic domain of tissue factor restricts physiological albuminuria and pathological proteinuria associated with glomerulonephritis in mice. Nephron Exp. Nephrol. 2010, 116, e72–e83. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oe, Y.; Takahashi, N. Tissue Factor, Thrombosis, and Chronic Kidney Disease. Biomedicines 2022, 10, 2737. https://doi.org/10.3390/biomedicines10112737
Oe Y, Takahashi N. Tissue Factor, Thrombosis, and Chronic Kidney Disease. Biomedicines. 2022; 10(11):2737. https://doi.org/10.3390/biomedicines10112737
Chicago/Turabian StyleOe, Yuji, and Nobuyuki Takahashi. 2022. "Tissue Factor, Thrombosis, and Chronic Kidney Disease" Biomedicines 10, no. 11: 2737. https://doi.org/10.3390/biomedicines10112737