

The Interplay between Finasteride-Induced Androgen Imbalance, Endoplasmic Reticulum Stress, Oxidative Stress, and Liver Disorders in Paternal and Filial Generation
 , , , and
, , , and
Abstract
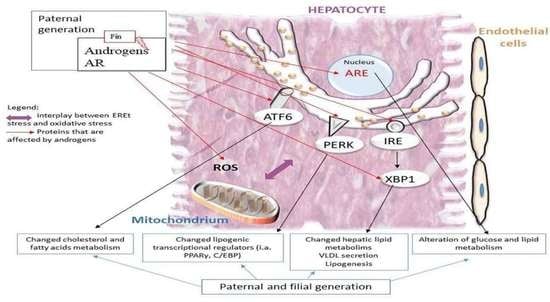
:
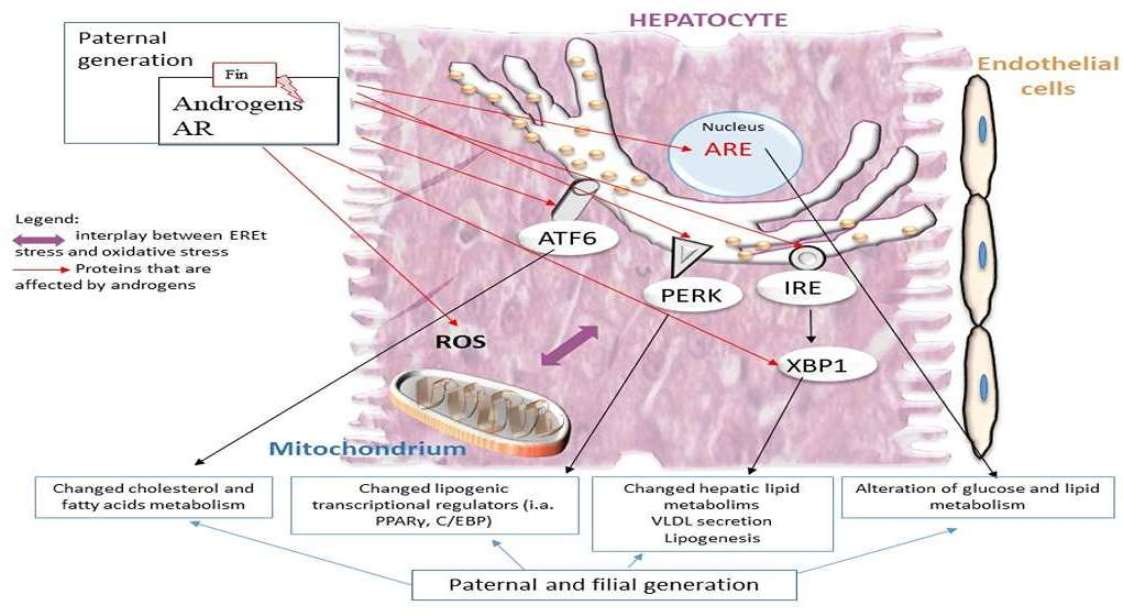{kind=link}
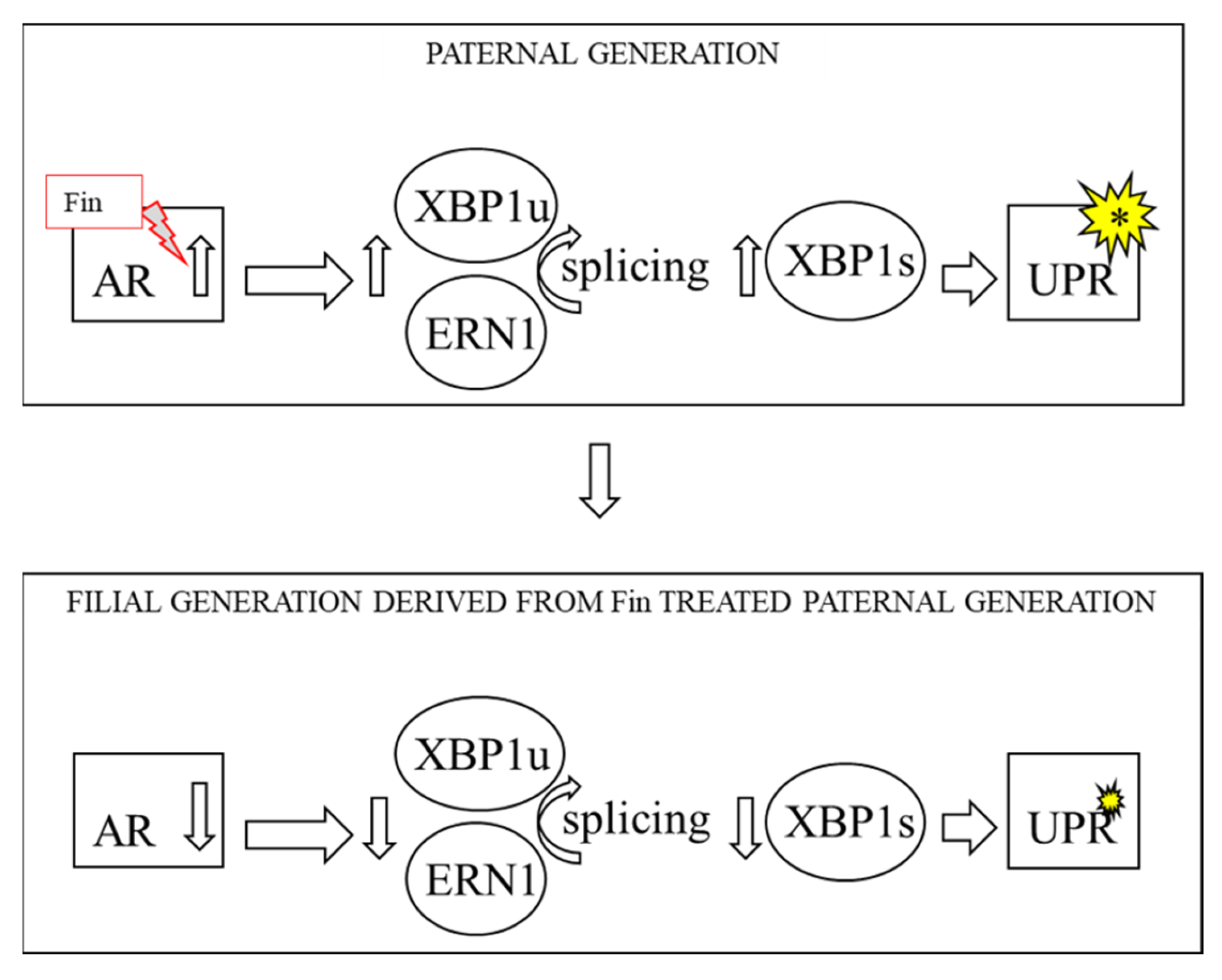{kind=link}
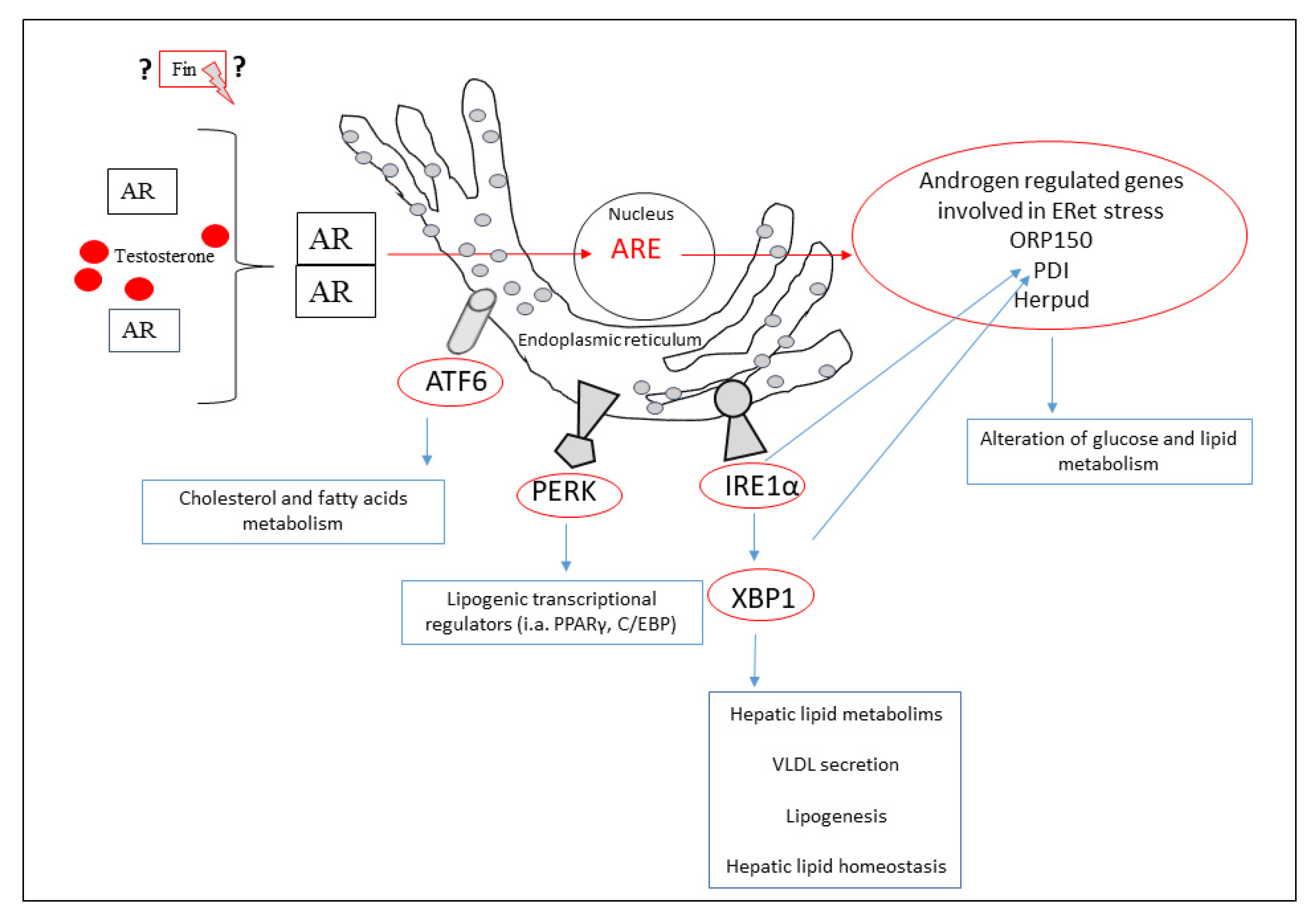{kind=link}
1. Basic Components of the Endoplasmic Reticulum Stress Machinery
2. Meaning of Unfolded Protein Response
3. Finasteride, Testosterone and Androgen Receptor
4. Androgens, Endoplasmic Reticulum Stress, and Liver
5. Oxidative Stress and Endoplasmic Reticulum Stress
6. Relationship between Androgen Imbalance and Endoplasmic Reticulum Stress
7. Androgens, Endoplasmic Reticulum Stress, and Liver Disorders
8. Autophagy, Unfolded Protein Response, and Liver
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banhegyi, G.; Baumeister, P.; Benedetti, A.; Dong, D.; Fu, Y.; Lee, A.S.; Li, J.; Mao, C.; Margittai, E.; Ni, M.; et al. Endoplasmic Reticulum Stress. Ann. N. Y. Acad. Sci. 2007, 1113, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef] [Green Version]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the Endoplasmic Reticulum: Atypical GRP78 in Cell Viability, Signalling and Therapeutic Targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Sidrauski, C.; Walter, P. The Transmembrane Kinase Ire1p Is a Site-Specific Endonuclease That Initiates MRNA Splicing in the Unfolded Protein Response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Chlebowska, J. Stres siateczki śródplazmatycznej i stres oksydacyjny w ostrych białaczkach szpikowych. Acta Haematol. Pol. 2016, 47, 197–204. [Google Scholar] [CrossRef]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A Stress Response Pathway from the Endoplasmic Reticulum to the Nucleus Requires a Novel Bifunctional Protein Kinase/Endoribonuclease (Ire1p) in Mammalian Cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef] [Green Version]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signalling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Unfolded Protein Response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The Unfolded Protein Response in Immunity and Inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuckin, M.A.; Eri, R.D.; Das, I.; Lourie, R.; Florin, T.H. ER Stress and the Unfolded Protein Response in Intestinal Inflammation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2010, 298, G820–G832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikawa, D.; Kimata, Y.; Kohno, K. Self-Association and BiP Dissociation Are Not Sufficient for Activation of the ER Stress Sensor Ire1. J. Cell Sci. 2007, 120, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The Luminal Domain of the ER Stress Sensor Protein PERK Binds Misfolded Proteins and Thereby Triggers PERK Oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [Green Version]
- Gardner, B.M.; Walter, P. Unfolded Proteins Are Ire1-Activating Ligands That Directly Induce the Unfolded Protein Response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [Green Version]
- Maiers, J.; Malhi, H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin. Liver Dis. 2019, 39, 235–248. [Google Scholar] [CrossRef]
- Blais, J.D.; Filipenko, V.; Bi, M.; Harding, H.P.; Ron, D.; Koumenis, C.; Wouters, B.G.; Bell, J.C. Activating Transcription Factor 4 Is Translationally Regulated by Hypoxic Stress. Mol. Cell. Biol. 2004, 24, 7469–7482. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Haeri, M.; Knox, B.E. Endoplasmic Reticulum Stress and Unfolded Protein Response Pathways: Potential for Treating Age-Related Retinal Degeneration. J. Ophthalmic Vis. Res. 2012, 7, 45–59. [Google Scholar]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the Mechanisms of Apoptosis Induced by Endoplasmic Reticulum Stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-L.; Ha, D.P.; Zhao, H.; Carlos, A.J.; Wei, S.; Pun, T.K.; Wu, K.; Zandi, E.; Kelly, K.; Lee, A.S. Endoplasmic Reticulum Stress Activates SRC, Relocating Chaperones to the Cell Surface Where GRP78/CD109 Blocks TGF-β Signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E4245–E4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, A.; Cheng, S.-B.; Kusabiraki, T.; Motomura, K.; Aoki, A.; Ushijima, A.; Ono, Y.; Tsuda, S.; Shima, T.; Yoshino, O.; et al. Endoplasmic Reticulum Stress Disrupts Lysosomal Homeostasis and Induces Blockade of Autophagic Flux in Human Trophoblasts. Sci. Rep. 2019, 9, 11466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharmacol. 2019, 10, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Green, R.M. Endoplasmic Reticulum Stress and Liver Diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef]
- Smith, L.B.; Walker, W.H. The Regulation of Spermatogenesis by Androgens. Semin. Cell Dev. Biol. 2014, 30, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Burger, H.G. Androgen Production in Women. Fertil. Steril. 2002, 77, 3–5. [Google Scholar] [CrossRef]
- Diver, M. Analytical and Physiological Factors Affecting the Interpretation of Serum Testosterone Concentration in Men. Ann. Clin. Biochem. Int. J. Lab. Med. 2006, 43, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Stukenborg, J.-B.; Mitchell, R.T.; Söder, O. Endocrine Disruptors and the Male Reproductive System. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101567. [Google Scholar] [CrossRef]
- Eggert, A.; Cisneros-Montalvo, S.; Anandan, S.; Musilli, S.; Stukenborg, J.-B.; Adamsson, A.; Nurmio, M.; Toppari, J. The Effects of Perfluorooctanoic Acid (PFOA) on Fetal and Adult Rat Testis. Reprod. Toxicol. 2019, 90, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kolasa-Wolosiuk, A.; Misiakiewicz-Has, K.; Baranowska-Bosiacka, I.; Gutowska, I.; Wiszniewska, B. Androgen Levels and Apoptosis in the Testis during Postnatal Development of Finasteride-Treated Male Rat Offspring. Folia Histochem. Cytobiol. 2015, 53, 236–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K.; Yamaguchi, K.; Li, F.; Ando, M.; Fujisawa, M. Finasteride-Associated Male Infertility. Fertil. Steril. 2011, 95, 1786.e9–1786.e11. [Google Scholar] [CrossRef] [PubMed]
- Lucas-Herald, A.K.; Alves-Lopes, R.; Montezano, A.C.; Ahmed, S.F.; Touyz, R.M. Genomic and Non-Genomic Effects of Androgens in the Cardiovascular System: Clinical Implications. Clin. Sci. Lond. Engl. 1979 2017, 131, 1405–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, K.D.; Olsen, E.A.; Whiting, D.; Savin, R.; DeVillez, R.; Bergfeld, W.; Price, V.H.; Van Neste, D.; Roberts, J.L.; Hordinsky, M.; et al. Finasteride in the Treatment of Men with Androgenetic Alopecia. J. Am. Acad. Dermatol. 1998, 39, 578–589. [Google Scholar] [CrossRef]
- Traish, A.M. The Post-Finasteride Syndrome: Clinical Manifestation of Drug-Induced Epigenetics Due to Endocrine Disruption. Curr. Sex. Health Rep. 2018, 10, 88–103. [Google Scholar] [CrossRef]
- Traish, A.M. Post-Finasteride Syndrome: A Surmountable Challenge for Clinicians. Fertil. Steril. 2020, 113, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Fertig, R.M.; Gamret, A.C.; Darwin, E.; Gaudi, S. Sexual Side Effects of 5-α-Reductase Inhibitors Finasteride and Dutasteride: A Comprehensive Review. Dermatol. Online J. 2017, 23, 3. [Google Scholar] [CrossRef]
- Ganzer, C.A.; Jacobs, A.R. Emotional Consequences of Finasteride: Fool’s Gold. Am. J. Mens Health 2018, 12, 90–95. [Google Scholar] [CrossRef]
- Thompson, I.M.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Miller, G.J.; Ford, L.G.; Lieber, M.M.; Cespedes, R.D.; Atkins, J.N.; Lippman, S.M.; et al. The Influence of Finasteride on the Development of Prostate Cancer. N. Engl. J. Med. 2003, 349, 215–224. [Google Scholar] [CrossRef]
- Hsieh, J.-T.; Chen, S.-C.; Yu, H.-J.; Chang, H.-C. Finasteride Upregulates Expression of Androgen Receptor in Hyperplastic Prostate and LNCaP Cells: Implications for Chemoprevention of Prostate Cancer: Finasteride Upregulates AR in Prostate Cell. Prostate 2011, 71, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.; Song, W.; Pastuszak, A.; Khera, M. Differential Gene Expression in Post-Finasteride Syndrome Patients. J. Sex. Med. 2021, 18, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Shi, H. Sex Hormones and Their Receptors Regulate Liver Energy Homeostasis. Int. J. Endocrinol. 2015, 2015, 294278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhou, D.; Cai, Y.; Yu, X.; Zheng, X.; Chen, B.; Li, W.; Zeng, H.; Hassan, M.; Zhao, Y.; et al. Endoplasmic Reticulum Stress Inhibits AR Expression via the PERK/EIF2α/ATF4 Pathway in Luminal Androgen Receptor Triple-Negative Breast Cancer and Prostate Cancer. NPJ Breast Cancer 2022, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Hinchliffe, S.A.; Woods, S.; Gray, S.; Burt, A.D. Cellular Distribution of Androgen Receptors in the Liver. J. Clin. Pathol. 1996, 49, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The Cell Biology of the Hepatocyte: A Membrane Trafficking Machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR Pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Stelloo, S.; Linder, S.; Nevedomskaya, E.; Valle-Encinas, E.; de Rink, I.; Wessels, L.F.A.; van der Poel, H.; Bergman, A.M.; Zwart, W. Androgen Modulation of XBP1 Is Functionally Driving Part of the AR Transcriptional Program. Endocr. Relat. Cancer 2020, 27, 67–79. [Google Scholar] [CrossRef]
- Erzurumlu, Y.; Ballar, P. Androgen Mediated Regulation of Endoplasmic Reticulum-Associated Degradation and Its Effects on Prostate Cancer. Sci. Rep. 2017, 7, 40719. [Google Scholar] [CrossRef] [Green Version]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Kolasa, A.; Rogińska, D.; Rzeszotek, S.; Machaliński, B.; Wiszniewska, B. Paternal Finasteride Treatment Can Influence the Testicular Transcriptome Profile of Male Offspring-Preliminary Study. Curr. Issues Mol. Biol. 2021, 43, 868–886. [Google Scholar] [CrossRef] [PubMed]
- Segawa, T.; Nau, M.E.; Xu, L.L.; Chilukuri, R.N.; Makarem, M.; Zhang, W.; Petrovics, G.; Sesterhenn, I.A.; McLeod, D.G.; Moul, J.W.; et al. Androgen-Induced Expression of Endoplasmic Reticulum (ER) Stress Response Genes in Prostate Cancer Cells. Oncogene 2002, 21, 8749–8758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusaczuk, M.; Cechowska-Pasko, M. Molecular Chaperone ORP150 in ER Stress–Related Diseases. Curr. Pharm. Des. 2013, 19, 2807–2818. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The Endoplasmic Reticulum and the Unfolded Protein Response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlot, A.M.; Porter, G.M.; Sahni, S.; Lim, E.G.; Peres, P.; Richardson, D.R. The Metastasis Suppressor, NDRG1, Differentially Modulates the Endoplasmic Reticulum Stress Response. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2019, 1865, 2094–2110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yu, C.; Yang, X.; Hong, H.; Lu, J.; Hu, W.; Hao, X.; Li, S.; Aikemu, B.; Yang, G.; et al. N-Myc Downstream-Regulated Gene 1 Inhibits the Proliferation of Colorectal Cancer through Emulative Antagonizing NEDD4-Mediated Ubiquitylation of P21. J. Exp. Clin. Cancer Res. 2019, 38, 490. [Google Scholar] [CrossRef] [Green Version]
- Ulrix, W.; Swinnen, J.V.; Heyns, W.; Verhoeven, G. Androgens Down-Regulate the Expression of the Human Homologue of Paternally Expressed Gene-3 in the Prostatic Adenocarcinoma Cell Line LNCaP. Mol. Cell. Endocrinol. 1999, 155, 69–76. [Google Scholar] [CrossRef]
- Jung, T.W.; Kyung, E.J.; Kim, H.-C.; Shin, Y.K.; Lee, S.H.; Park, E.S.; Hacımüftüoğlu, A.; Abd El-Aty, A.M.; Jeong, J.H. Protectin DX Ameliorates Hepatic Steatosis by Suppression of Endoplasmic Reticulum Stress via AMPK-Induced ORP150 Expression. J. Pharmacol. Exp. Ther. 2018, 365, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Arnoldussen, Y.J.; Storm, M.; Tesikova, M.; Nenseth, H.Z.; Zhao, S.; Fazli, L.; Rennie, P.; Risberg, B.; Wæhre, H.; et al. Divergent Androgen Regulation of Unfolded Protein Response Pathways Drives Prostate Cancer. EMBO Mol. Med. 2015, 7, 788–801. [Google Scholar] [CrossRef]
- Lackner, L.L.; Voeltz, G.K. The Mechanisms and Functions of Interorganelle Interactions. Mol. Biol. Cell 2017, 28, 703–704. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System. Cells 2022, 11, 2607. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E. The Interplay of Mitochondria with Calcium: An Historical Appraisal. Cell Calcium 2012, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Diogo, C.V.; Yambire, K.F.; Fernández Mosquera, L.; Branco, F.T.; Raimundo, N. Mitochondrial Adventures at the Organelle Society. Biochem. Biophys. Res. Commun. 2018, 500, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Filiponi, M.; Gougoura, S.; Befani, C.; Bargiota, A.; Liakos, P.; Koukoulis, G. Androgen Modulate the Pro-Oxidant and Antioxidant Activity of Macroendothelial Cells. Endocr. Abstr. 2020, 70, EP374. [Google Scholar] [CrossRef]
- Lu, J.P.; Monardo, L.; Bryskin, I.; Hou, Z.F.; Trachtenberg, J.; Wilson, B.C.; Pinthus, J.H. Androgens Induce Oxidative Stress and Radiation Resistance in Prostate Cancer Cells Though NADPH Oxidase. Prostate Cancer Prostatic Dis. 2010, 13, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Pinthus, J.H.; Bryskin, I.; Trachtenberg, J.; Luz, J.-P.; Singh, G.; Fridman, E.; Wilson, B.C. Androgen Induces Adaptation to Oxidative Stress in Prostate Cancer: Implications for Treatment with Radiation Therapy. Neoplasia 2007, 9, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Muthu, S.J.; Seppan, P. Apoptosis in Hippocampal Tissue Induced by Oxidative Stress in Testosterone Deprived Male Rats. Aging Male Off. J. Int. Soc. Study Aging Male 2020, 23, 1598–1610. [Google Scholar] [CrossRef]
- Hu, K.; Li, S.-Y.; Xiao, B.; Bi, F.; Lu, X.-Q.; Wu, X.-M. Protective Effects of Quercetin against Status Epilepticus Induced Hippocampal Neuronal Injury in Rats: Involvement of X-Linked Inhibitor of Apoptosis Protein. Acta Neurol. Belg. 2011, 111, 205–212. [Google Scholar]
- Seo, E.; Kang, H.; Choi, H.; Choi, W.; Jun, H.-S. Reactive Oxygen Species-Induced Changes in Glucose and Lipid Metabolism Contribute to the Accumulation of Cholesterol in the Liver during Aging. Aging Cell 2019, 18, e12895. [Google Scholar] [CrossRef] [Green Version]
- Mc Auley, M.T.; Mooney, K.M. Computationally Modeling Lipid Metabolism and Aging: A Mini-Review. Comput. Struct. Biotechnol. J. 2015, 13, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Salmon, A.B. Oxidative Stress in the Etiology of Age-Associated Decline in Glucose Metabolism. Longev. Heal. 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, L.; Feng, H.; Gong, D.; Zhao, X.; Cai, L.; Wu, Q.; Yuan, B.; Yang, M.; Zhao, J.; Zou, Y. Genipin Ameliorates Age-Related Insulin Resistance through Inhibiting Hepatic Oxidative Stress and Mitochondrial Dysfunction. Exp. Gerontol. 2013, 48, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Wang, X.; Lu, Y.; Wang, E.; Zhang, Z.; Yang, J.; Zhang, H.; Li, X. Hepatic Steatosis Exacerbated by Endoplasmic Reticulum Stress-Mediated Downregulation of FXR in Aging Mice. J. Hepatol. 2014, 60, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.K.; Shin, Y.S.; Choi, B.R.; Karna, K.K.; Kim, H.K.; Lee, S.W.; Kim, C.Y.; Park, J.K. Protective Effect of DA-9401 in Finasteride-Induced Apoptosis in Rat Testis: Inositol Requiring Kinase 1 and c-Jun N-Terminal Kinase Pathway. Drug Des. Devel. Ther. 2017, 11, 2969–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, W.; Shastri, M.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef] [PubMed]
- Serra, L.; Estienne, A.; Vasseur, C.; Froment, P.; Dupont, J. Review: Mechanisms of Glyphosate and Glyphosate-Based Herbicides Action in Female and Male Fertility in Humans and Animal Models. Cells 2021, 10, 3079. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, J.; Yang, L.; Zhang, H.; Zhang, Y.; Gao, D.; Jiang, H.; Li, Y.; Dong, H.; Ma, T.; et al. Glyphosate Exposure Attenuates Testosterone Synthesis via NR1D1 Inhibition of StAR Expression in Mouse Leydig Cells. Sci. Total Environ. 2021, 785, 147323. [Google Scholar] [CrossRef]
- Lytridou, A.A.; Demetriadou, A.; Christou, M.; Potamiti, L.; Mastroyiannopoulos, N.P.; Kyriacou, K.; Phylactou, L.A.; Drousiotou, A.; Petrou, P.P. Stbd1 Promotes Glycogen Clustering during Endoplasmic Reticulum Stress and Supports Survival of Mouse Myoblasts. J. Cell Sci. 2020, 133, jcs.244855. [Google Scholar] [CrossRef]
- Li, Z.; Wu, F.; Zhang, X.; Chai, Y.; Chen, D.; Yang, Y.; Xu, K.; Yin, J.; Li, R.; Shi, H.; et al. Valproate Attenuates Endoplasmic Reticulum Stress-Induced Apoptosis in SH-SY5Y Cells via the AKT/GSK3β Signaling Pathway. Int. J. Mol. Sci. 2017, 18, 315. [Google Scholar] [CrossRef] [Green Version]
- Park, E.C.; Kim, S.I.; Hong, Y.; Hwang, J.W.; Cho, G.-S.; Cha, H.-N.; Han, J.-K.; Yun, C.-H.; Park, S.-Y.; Jang, I.-S.; et al. Inhibition of CYP4A Reduces Hepatic Endoplasmic Reticulum Stress and Features of Diabetes in Mice. Gastroenterology 2014, 147, 860–869. [Google Scholar] [CrossRef]
- Kur, P.; Kolasa-Wołosiuk, A.; Grabowska, M.; Kram, A.; Tarnowski, M.; Baranowska-Bosiacka, I.; Rzeszotek, S.; Piasecka, M.; Wiszniewska, B. The Postnatal Offspring of Finasteride-Treated Male Rats Shows Hyperglycaemia, Elevated Hepatic Glycogen Storage and Altered GLUT2, IR, and AR Expression in the Liver. Int. J. Mol. Sci. 2021, 22, 1242. [Google Scholar] [CrossRef]
- Maseroli, E.; Comeglio, P.; Corno, C.; Cellai, I.; Filippi, S.; Mello, T.; Galli, A.; Rapizzi, E.; Presenti, L.; Truglia, M.C.; et al. Testosterone Treatment Is Associated with Reduced Adipose Tissue Dysfunction and Nonalcoholic Fatty Liver Disease in Obese Hypogonadal Men. J. Endocrinol. Invest. 2021, 44, 819–842. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Yu, I.-C.; Wang, R.-S.; Chen, Y.-T.; Liu, N.-C.; Altuwaijri, S.; Hsu, C.-L.; Ma, W.-L.; Jokinen, J.; Sparks, J.D.; et al. Increased Hepatic Steatosis and Insulin Resistance in Mice Lacking Hepatic Androgen Receptor. Hepatology 2008, 47, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Völzke, H.; Aumann, N.; Krebs, A.; Nauck, M.; Steveling, A.; Lerch, M.M.; Rosskopf, D.; Wallaschofski, H. Hepatic Steatosis Is Associated with Low Serum Testosterone and High Serum DHEAS Levels in Men. Int. J. Androl. 2010, 33, 45–53. [Google Scholar] [CrossRef]
- Dowman, J.K.; Hopkins, L.J.; Reynolds, G.M.; Armstrong, M.J.; Nasiri, M.; Nikolaou, N.; van Houten, E.L.A.F.; Visser, J.A.; Morgan, S.A.; Lavery, G.G.; et al. Loss of 5α-Reductase Type 1 Accelerates the Development of Hepatic Steatosis but Protects Against Hepatocellular Carcinoma in Male Mice. Endocrinology 2013, 154, 4536–4547. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Jiang, X.; Yokosuka, O. Androgen Receptor Signaling in Hepatocellular Carcinoma and Pancreatic Cancers. World J. Gastroenterol. 2014, 20, 9229–9236. [Google Scholar] [CrossRef]
- Panzhinskiy, E.; Hua, Y.; Culver, B.; Ren, J.; Nair, S. Endoplasmic Reticulum Stress Upregulates Protein Tyrosine Phosphatase 1B and Impairs Glucose Uptake in Cultured Myotubes. Diabetologia 2013, 56, 598–607. [Google Scholar] [CrossRef]
- Tang, X.; Shen, H.; Chen, J.; Wang, X.; Zhang, Y.; Chen, L.; Rukachaisirikul, V.; Jiang, H.; Shen, X. Activating Transcription Factor 6 Protects Insulin Receptor from ER Stress-Stimulated Desensitization via P42/44 ERK Pathway. Acta Pharmacol. Sin. 2011, 32, 1138–1147. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.Y.; Chalasani, U.; Pan, N.; Friedline, R.H.; Prosdocimo, D.A.; Nam, M.; Azuma, Y.; Maganti, R.; Yu, K.; Velagapudi, A.; et al. KLF15 Is a Molecular Link between Endoplasmic Reticulum Stress and Insulin Resistance. PLoS ONE 2013, 8, e77851. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Rosenfield, R.L.; Qin, K. KLF15 Is a Transcriptional Regulator of the Human 17β-Hydroxysteroid Dehydrogenase Type 5 Gene. A Potential Link between Regulation of Testosterone Production and Fat Stores in Women. J. Clin. Endocrinol. Metab. 2009, 94, 2594–2601. [Google Scholar] [CrossRef] [Green Version]
- Luu-The, V.; Dufort, I.; Pelletier, G.; Labrie, F. Type 5 17β-Hydroxysteroid Dehydrogenase: Its Role in the Formation of Androgens in Women. Mol. Cell. Endocrinol. 2001, 171, 77–82. [Google Scholar] [CrossRef]
- Feng, M.W.; Hanley, K.L.; Feng, G.-S. Androgen Receptor, Neovascularization and Liver Cancer Metastasis. J. Hepatol. 2021, 75, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Yan, D.; Li, J.; Chen, S.; Liu, Y.; Chen, R.; Duan, C.; Wei, M.; Li, H.; He, T. Activation of PKR/EIF2α Signaling Cascade Is Associated with Dihydrotestosterone-Induced Cell Cycle Arrest and Apoptosis in Human Liver Cells. J. Cell. Biochem. 2012, 113, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Ray, G.; Bhargav, P.M. A Study of Hormonal Abnormalities in Chronic Liver Disease. J. Assoc. Physicians India 2019, 67, 47–52. [Google Scholar]
- Okamura, T.; Hamaguchi, M.; Bamba, R.; Nakajima, H.; Yoshimura, Y.; Kimura, T.; Nishida, K.; Hashimoto, Y.; Fukuda, T.; Senmaru, T.; et al. Immune Modulating Effects of Additional Supplementation of Estradiol Combined with Testosterone in Murine Testosterone-Deficient NAFLD Model. Am. J. Physiol.-Gastrointest. Liver Physiol. 2020, 318, G989–G999. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, X.; Xu, W.; Cai, J.; Zhang, Y.; Wu, C.; Li, S.; Sun, Y.; Liu, W.; Tao, T. Role of Androgen in Liver Fat Content in Women: Metabolically Advantageous or Disadvantageous? Endocr. Pract. 2020, 26, 1003–1016. [Google Scholar] [CrossRef]
- Song, M.J.; Malhi, H. The Unfolded Protein Response and Hepatic Lipid Metabolism in Non Alcoholic Fatty Liver Disease. Pharmacol. Ther. 2019, 203, 107401. [Google Scholar] [CrossRef]
- Kur, P.; Kolasa-Wołosiuk, A.; Misiakiewicz-Has, K.; Wiszniewska, B. Sex Hormone-Dependent Physiology and Diseases of Liver. Int. J. Environ. Res. Public. Health 2020, 17, 2620. [Google Scholar] [CrossRef] [Green Version]
- Czaja, M.J.; Ding, W.-X.; Donohue, T.M.; Friedman, S.L.; Kim, J.-S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.J.; et al. Functions of Autophagy in Normal and Diseased Liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (3rd Edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Allaire, M.; Rautou, P.-E.; Codogno, P.; Lotersztajn, S. Autophagy in Liver Diseases: Time for Translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaipitinca, L.; Mandatori, S.; Mancinelli, R.; Giulitti, F. Petrungaro, S.; Moresi, V.; Giampietri, C. The Role of Autophagy in Liver Epithelial Cells and Its Impact on Systemic Homeostasis. Nutrients 2019, 11, 827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.-M.; Ni, J.-D.; Song, D.; Ding, M.; Huang, J. Interplay between Unfolded Protein Response and Autophagy Promotes Tumor Drug Resistance. Oncol. Lett. 2015, 10, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Kwanten, W.J.; Vandewynckel, Y.-P.; Martinet, W.; De Winter, B.Y.; Michielsen, P.P.; Van Hoof, V.O.; Driessen, A.; Timmermans, J.-P.; Bedossa, P.; Van Vlierberghe, H.; et al. Hepatocellular Autophagy Modulates the Unfolded Protein Response and Fasting-Induced Steatosis in Mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 311, G599–G609. [Google Scholar] [CrossRef] [Green Version]
- Blessing, A.M.; Rajapakshe, K.; Reddy Bollu, L.; Shi, Y.; White, M.A.; Pham, A.H.; Lin, C.; Jonsson, P.; Cortes, C.J.; Cheung, E.; et al. Transcriptional Regulation of Core Autophagy and Lysosomal Genes by the Androgen Receptor Promotes Prostate Cancer Progression. Autophagy 2017, 13, 506–521. [Google Scholar] [CrossRef]
- Pootrakul, L.; Datar, R.H.; Shi, S.-R.; Cai, J.; Hawes, D.; Groshen, S.G.; Lee, A.S.; Cote, R.J. Expression of Stress Response Protein Grp78 Is Associated with the Development of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2006, 12, 5987–5993. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Han, J.J.; Tennakoon, J.B.; Mehta, F.F.; Merchant, F.A.; Burns, A.R.; Howe, M.K.; McDonnell, D.P.; Frigo, D.E. Androgens Promote Prostate Cancer Cell Growth through Induction of Autophagy. Mol. Endocrinol. 2013, 27, 280–295. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Blomenkamp, K.S.; Fickert, P.; Trauner, M.; Teckman, J.H. NorUDCA Promotes Degradation of A1-Antitrypsin Mutant Z Protein by Inducing Autophagy through AMPK/ULK1 Pathway. PLoS ONE 2018, 13, e0200897. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy Modulation as a Potential Therapeutic Target for Diverse Diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzeszotek, S.; Kolasa, A.; Pilutin, A.; Misiakiewicz-Has, K.; Sielatycka, K.; Wiszniewska, B. The Interplay between Finasteride-Induced Androgen Imbalance, Endoplasmic Reticulum Stress, Oxidative Stress, and Liver Disorders in Paternal and Filial Generation. Biomedicines 2022, 10, 2725. https://doi.org/10.3390/biomedicines10112725
Rzeszotek S, Kolasa A, Pilutin A, Misiakiewicz-Has K, Sielatycka K, Wiszniewska B. The Interplay between Finasteride-Induced Androgen Imbalance, Endoplasmic Reticulum Stress, Oxidative Stress, and Liver Disorders in Paternal and Filial Generation. Biomedicines. 2022; 10(11):2725. https://doi.org/10.3390/biomedicines10112725
Chicago/Turabian StyleRzeszotek, Sylwia, Agnieszka Kolasa, Anna Pilutin, Kamila Misiakiewicz-Has, Katarzyna Sielatycka, and Barbara Wiszniewska. 2022. "The Interplay between Finasteride-Induced Androgen Imbalance, Endoplasmic Reticulum Stress, Oxidative Stress, and Liver Disorders in Paternal and Filial Generation" Biomedicines 10, no. 11: 2725. https://doi.org/10.3390/biomedicines10112725