
Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Characteristics of KMT2A
3. KMT2A—Clinical Presentation
4. Risk Stratification
5. Clinical Outcome and Interfant Protocol
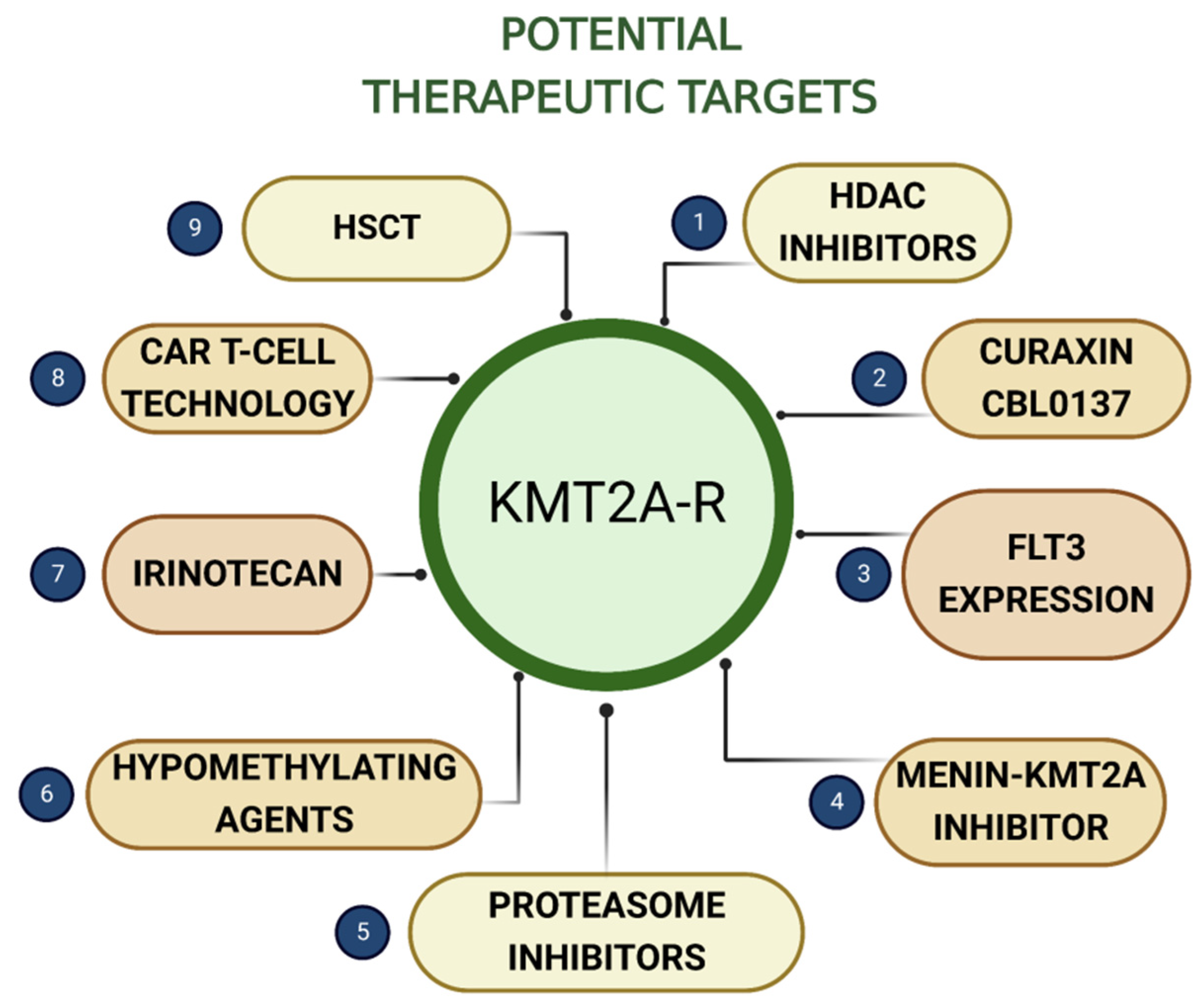
6. Potential Therapeutic Targets
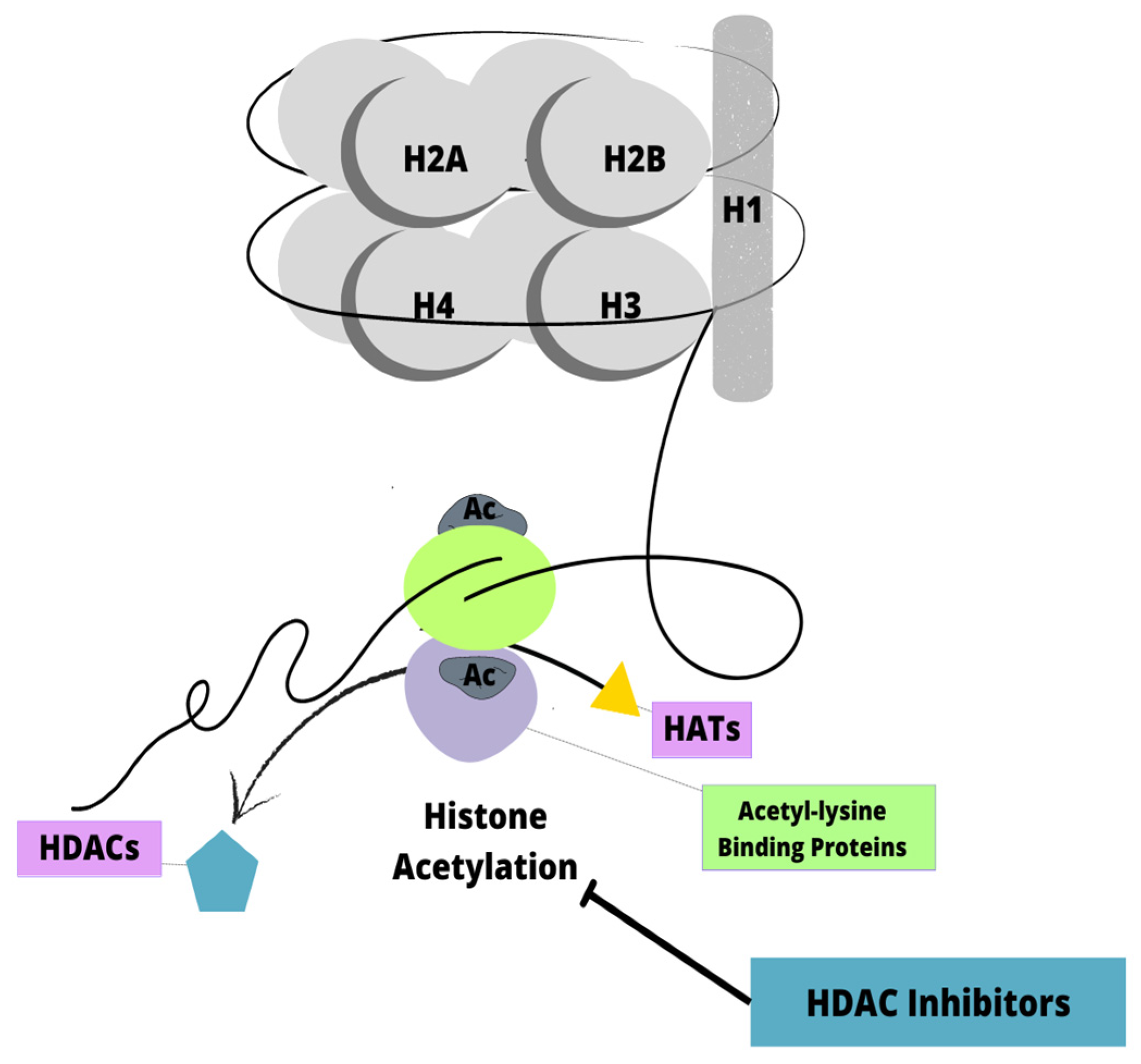
6.1. Histone Deacetylase Inhibitors
6.2. Curaxin CBL0137
6.3. FLT3 Expression
6.4. Menin-KMT2A Inhibitor
6.5. Proteasome Inhibitors
6.6. Hypomethylating Agents
6.7. Irinotecan
6.8. Chimeric Antigen Receptor T-Cell Technology
6.9. Hematopoietic Stem Cell Transplantation
7. Future Prospects
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dores, G.M.; Devesa, S.S.; Curtis, R.E.; Linet, M.S.; Morton, L.M. Acute leukemia incidence and patient survival among children and adults in the United States, 2001–2007. Blood 2012, 119, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Pulte, D.; Jansen, L.; Gondos, A.; Katalinic, A.; Barnes, B.; Ressing, M.; Holleczek, B.; Eberle, A.; Brenner, H.; the GEKID Cancer Survival Working Group. Survival of adults with acute lymphoblastic leukemia in Germany and the United States. PLoS ONE 2014, 9, e85554. [Google Scholar] [CrossRef] [PubMed]
- Gökbuget, N.; Hoelzer, D. Treatment of adult acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2006, 1, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef]
- Entry—*159555—Lysine-Specific Methyltransferase 2A; KMT2A—OMIM. (n.d.). Available online: https://www.omim.org/entry/159555 (accessed on 22 January 2023).
- Harris, N.L.; Jaffe, E.S.; Diebold, J.; Flandrin, G.; Muller-Hermelink, H.K.; Vardiman, J.; Lister, T.A.; Bloomfield, C.D. The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. Report of the Clinical Advisory Committee meeting, Airlie House, Virginia, November, 1997. Ann. Oncol. 1999, 10, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.D.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Duffield, A.S.; Mullighan, C.G.; Borowitz, M.J. International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Archiv. 2023, 482, 11–26. [Google Scholar] [CrossRef]
- Tkachuk, D.C.; Kohler, S.; Cleary, M.L. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992, 71, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Nakamura, T.; Alder, H.; Prasad, R.; Canaani, O.; Cimino, G.; Croce, C.M.; Canaani, E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 1992, 71, 701–708. [Google Scholar] [CrossRef]
- Ziemin-van der Poel, S.; McCabe, N.R.; Gill, H.J.; Espinosa, R.; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; LeBeau, M.M.; Rowley, J.D. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, S.; Di Fede, E.; Bernardelli, C.; Lettieri, A.; Parodi, C.; Grazioli, P.; Colombo, E.A.; Ancona, S.; Milani, D.; Ottaviano, E.; et al. KMT2A: Umbrella Gene for Multiple Diseases. Genes 2022, 13, 514. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Tipping the Balance of Chromatin States. Annu. Rev. Genom. Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornsson, H.T. The Mendelian Disorders of the Epigenetic Machinery. Genome Res. 2015, 25, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Postnatal Malleability and Therapeutic Prospects. Hum. Mol. Genet. 2019, 28, R254–R264. [Google Scholar] [CrossRef] [PubMed]
- WSS Foundation. Available online: http://www.wssfoundation.org/wiedemann-steiner-syndrome/ (accessed on 23 January 2023).
- Husmann, D.; Gozani, O. Histone Lysine Methyltransferases in Biology and Disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Paredes, M.; Esteller, M. Cancer Epigenetics Reaches Mainstream Oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef]
- Steinhilber, D.; Marschalek, R. How to effectively treat acute leukemia patients bearing MLL-rearrangements? Biochem. Pharmacol. 2018, 147, 183–190. [Google Scholar] [CrossRef]
- Marschalek, R. Systematic classification of mixed-lineage leukemia fusion partners predicts additional cancer pathways. Ann. Lab. Med. 2016, 36, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Ono, R.; Kumagai, H.; Nakajima, H.; Hishiya, A.; Taki, T.; Horikawa, K.; Takatsu, K.; Satoh, T.; Hayashi, Y.; Kitamura, T.; et al. Mixed-lineage-leukemia (MLL) fusion protein collaborates with Ras to induce acute leukemia through aberrant Hox expression and Raf activation. Leukemia 2009, 23, 2197–2209. [Google Scholar] [CrossRef]
- Chen, C.W.; Armstrong, S.A. Targeting DOT1L and HOX gene expression in MLL- rearranged leukemia and beyond. Exp. Hematol. 2015, 43, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muntean, A.G.; Hess, J.L. The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. 2012, 7, 283–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Matlawska-Wasowska, K.; Kang, H.; Devidas, M.; Wen, J.; Harvey, R.C.; Nickl, C.K.; Ness, S.A.; Rusch, M.; Li, Y.; Onozawa, M.; et al. MLL rearrangements impact outcome in HOXA-deregulated T-lineage acute lympho- blastic leukemia: A Children’s Oncology Group Study. Leukemia 2016, 30, 1909–1912. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- t(11;19)(q23;p13.3) KMT2A/MLLT1. Available online: https://atlasgeneticsoncology.org/haematological/1071/t(11;19)(q23;p13-3) (accessed on 26 February 2023).
- Wen, J.; Zhou, M.; Shen, Y.; Long, Y.; Guo, Y.; Song, L.; Xiao, J. Poor treatment responses were related to poor outcomes in pediatric B cell acute lymphoblastic leukemia with KMT2A rearrangements. BMC Cancer 2022, 22, 859. [Google Scholar] [CrossRef]
- Ries, R.E.; Leonti, A.R.; Triche, T.J.; Gerbing, R.B.; Hirsch, B.A.; Raimondi, S.C.; Smith, J.L.; Cooper, T.M.; Farrar, J.E.; Deshpande, A.J.; et al. Structural Variants Involving MLLT10/AF10 Are Associated with Adverse Outcome in AML Regardless of the Partner Gene—A COG/Tpaml Study. Blood 2019, 134, 461. [Google Scholar] [CrossRef]
- t(1;11)(p32;q23) KMT2A/EPS15. Available online: https://atlasgeneticsoncology.org/haematological/1046/t(1;11)(p32;q23) (accessed on 26 February 2023).
- t(6;11)(q27;q23) KMT2A/AFDN. Available online: https://atlasgeneticsoncology.org/haematological/1015/t(6;11)(q27;q23) (accessed on 26 February 2023).
- t(9;11)(p21;q23) KMT2A/MLLT3. Available online: https://atlasgeneticsoncology.org/haematological/1001/t(9;11)(p21;q23) (accessed on 23 January 2023).
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 2019, 104, 1176–1188. [Google Scholar] [CrossRef]
- Liang, D.C.; Chen, S.H.; Liu, H.C.; Yang, C.P.; Yeh, T.C.; Jaing, T.H.; Hung, I.J.; Hou, J.Y.; Lin, T.H.; Lin, C.H.; et al. Mutational status of NRAS, KRAS, and PTPN11 genes is associated with genetic/cytogenetic features in children with B-precursor acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e26786. [Google Scholar] [CrossRef] [PubMed]
- Fedders, H.; Alsadeq, A.; Schmäh, J.; Vogiatzi, F.; Zimmermann, M.; Möricke, A.; Lenk, L.; Stadt, U.Z.; Horstmann, M.A.; Pieters, R.; et al. The role of constitutive activation of FMS-related tyrosine kinase-3 and NRas/KRas mutational status in infants with KMT2A-rearranged acute lymphoblastic leukemia. Haematologica 2017, 102, e438–e442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trentin, L.; Bresolin, S.; Giarin, E.; Bardini, M.; Serafin, V.; Accordi, B.; Fais, F.; Tenca, C.; De Lorenzo, P.; Valsecchi, M.G.; et al. Deciphering KRAS and NRAS mutated clone dynamics in MLL-AF4 paediatric leukaemia by ultra deep sequencing analysis. Sci. Rep. 2016, 6, 34449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerenciano, M.; Barbosa Tda, C.; de Almeida Lopes, B.; Meyer, C.; Marschalek, R.; Pombo-de-Oliveira, M.S. Subclonality and prenatal origin of RAS mutations in KMT2A (MLL)-rearranged infant acute lymphoblastic leukaemia. Br. J. Haematol. 2015, 170, 268–271. [Google Scholar] [CrossRef]
- Driessen, E.M.; van Roon, E.H.; Spijkers-Hagelstein, J.A.; Schneider, P.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R.; Stam, R.W. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica 2013, 98, 937–944. [Google Scholar] [CrossRef]
- Prelle, C.; Bursen, A.; Dingermann, T.; Marschalek, R. Secondary mutations in t(4;11) leukemia patients. Leukemia 2013, 27, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Khalidi, H.S.; Chang, K.L.; Medeiros, L.J.; Brynes, R.K.; Slovak, M.L.; Murata-Collins, J.L.; Arber, D.A. Acute lymphoblastic leukemia. Survey of immunophenotype, French-American-British classification, frequency of myeloid antigen expression, and karyotypic abnormalities in 210 pediatric and adult cases. Am. J. Clin. Pathol. 1999, 111, 467–476. [Google Scholar] [CrossRef]
- Tamai, H.; Miyake, K.; Takatori, M.; Miyake, N.; Yamaguchi, H.; Dan, K.; Shimada, T.; Inokuchi, K. Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia 2011, 25, 888–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, C.; Gaidano, G.; Cimino, G.; Pastore, C.; Nomdedeu, J.; Volpe, G.; Vivenza, C.; Parvis, G.; Mazza, U.; Basso, G.; et al. Distribution of TP53 mutations among acute leukemias with MLL rearrangements. Genes Chromosom. Cancer 1996, 15, 48–53. [Google Scholar] [CrossRef]
- Stengel, A.; Schnittger, S.; Weissmann, S.; Kuznia, S.; Kern, W.; Kohlmann, A.; Haferlach, T.; Haferlach, C. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood 2014, 124, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Pieters, R.; Biondi, A. How I treat infant leukemia. Blood 2019, 133, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhou, M.; Zou, P.; Liao, X.; Xiao, J. Mature B cell acute lymphoblastic leukaemia with KMT2A-MLLT3 transcripts in children: Three case reports and literature reviews. Orphanet J. Rare Dis. 2021, 16, 331. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.A.; Pei, D.; Choi, J.; Cheng, C.; Coustan-Smith, E.; Campana, D.; Swanson, H.D.; Pauley, J.L.; Inaba, H.; Metzger, M.L.; et al. Clofarabine treatment of KMT2Ar infantile patients with acute lymphoblastic leukemia in St. Jude Total Therapy Study 16. Blood Adv. 2022, 6, 6131–6134. [Google Scholar] [CrossRef]
- Zhu, Y.; He, X.; Lin, Y.C.; Dong, H.; Zhang, L.; Chen, X.; Wang, Z.; Shen, Y.; Li, M.; Wang, H.; et al. Targeting PRMT1-mediated FLT3 methylation disrupts maintenance of MLL-rearranged acute lymphoblastic leukemia. Blood 2019, 134, 1257–1268. [Google Scholar] [CrossRef]
- Pieters, R.; De Lorenzo, P.; Ancliffe, P.; Aversa, L.A.; Brethon, B.; Biondi, A.; Campbell, M.; Escherich, G.; Ferster, A.; Gardner, R.A.; et al. Outcome of Infants Younger Than 1 Year With Acute Lymphoblastic Leukemia Treated With the Interfant-06 Protocol: Results From an International Phase III Randomized Study. J. Clin. Oncol. 2019, 37, 2246–2256. [Google Scholar] [CrossRef]
- Stutterheim, J.; de Lorenzo, P.; van der Sluis, I.M.; Alten, J.; Ancliffe, P.; Attarbaschi, A.; Aversa, L.; Boer, J.M.; Biondi, A.; Brethon, B.; et al. Minimal residual disease and outcome characteristics in infant KMT2A-germline acute lymphoblastic leukaemia treated on the Interfant-06 protocol. Eur. J. Cancer 2022, 160, 72–79. [Google Scholar] [CrossRef]
- Stutterheim, J.; van der Sluis, I.M.; de Lorenzo, P.; Alten, J.; Ancliffe, P.; Attarbaschi, A.; Brethon, B.; Biondi, A.; Campbell, M.; Cazzaniga, G.; et al. Clinical Implications of Minimal Residual Disease Detection in Infants With KMT2A-Rearranged Acute Lymphoblastic Leukemia Treated on the Interfant-06 Protocol. J. Clin. Oncol. 2021, 39, 652–662. [Google Scholar] [CrossRef]
- Pui, C.H.; Rubnitz, J.E.; Hancock, M.L.; Downing, J.R.; Raimondi, S.C.; Rivera, G.K.; Sandlund, J.T.; Ribeiro, R.C.; Head, D.R.; Relling, M.V.; et al. Reappraisal of the clinical and biologic significance of myeloid-associated antigen expression in childhood acute lymphoblastic leukemia. J. Clin. Oncol. 1998, 16, 3768–3773. [Google Scholar] [CrossRef]
- Sanjuan-Pla, A.; Bueno, C.; Prieto, C.; Acha, P.; Stam, R.W.; Marschalek, R.; Menéndez, P. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 2015, 126, 2676–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motlló, C.; Ribera, J.M.; Morgades, M.; Granada, I.; Montesinos, P.; Brunet, S.; Bergua, J.; Tormo, M.; García-Boyero, R.; Sarrà, J.; et al. Frequency and prognostic significance of t(v;11q23)/KMT2A rearrangements in adult patients with acute lymphoblastic leukemia treated with risk-adapted protocols. Leuk Lymphoma. 2017, 58, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Zini, G.; Bassan, R. Diagnosis and subclassification of acute lymphoblastic leukemia. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, C.; Montes, R.; Martín, L.; Prat, I.; Hernandez, M.C.; Orfao, A.; Menendez, P. NG2 antigen is expressed in CD34+ HPCs and plasmacytoid dendritic cell precursors: Is NG2 expression in leukemia dependent on the target cell where leukemogenesis is triggered? Leukemia 2008, 22, 1475–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, P.; Bueno, C. Expression of NG2 antigen in MLL-rearranged acute leukemias: How complex does it get? Leuk. Res. 2011, 35, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Prieto, C.; López-Millán, B.; Roca-Ho, H.; Stam, R.W.; Romero-Moya, D.; Rodríguez-Baena, F.J.; Sanjuan-Pla, A.; Ayllón, V.; Ramírez, M.; Bardini, M.; et al. NG2 antigen is involved in leukemia invasiveness and central nervous system infiltration in MLL-rearranged infant B-ALL. Leukemia 2018, 32, 633–644. [Google Scholar] [CrossRef]
- Smith, F.O.; Rauch, C.; Williams, D.E.; March, C.J.; Arthur, D.; Hilden, J.; Lampkin, B.C.; Buckley, J.D.; Buckley, C.V.; Woods, W.G.; et al. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996, 87, 1123–1133. [Google Scholar]
- Behm, F.G.; Smith, F.O.; Raimondi, S.C.; Pui, C.H.; Bernstein, I.D. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood 1996, 1, 1134–1139. [Google Scholar] [CrossRef]
- Wuchter, C.; Harbott, J.; Schoch, C.; Schnittger, S.; Borkhardt, A.; Karawajew, L.; Ratei, R.; Ruppert, V.; Haferlach, T.; Creutzig, U.; et al. Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000, 14, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Millan, B.; Sanchéz-Martínez, D.; Roca-Ho, H.; Gutiérrez-Agüera, F.; Molina, O.; Diaz de la Guardia, R.; Torres-Ruiz, R.; Fuster, J.L.; Ballerini, P.; Suessbier, U.; et al. NG2 antigen is a therapeutic target for MLL-rearranged B-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 1557–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilden, J.M.; Dinndorf, P.A.; Meerbaum, S.O.; Sather, H.; Villaluna, D.; Heerema, N.A.; McGlennen, R.; Smith, F.O.; Woods, W.G.; Salzer, W.L.; et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: Report on CCG 1953 from the Children’s Oncology Group. Blood 2006, 108, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Guest, E.M.; Stam, R.W. Updates in the biology and therapy for infant acute lymphoblastic leukemia. Curr. Opin. Pediatr. 2017, 29, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Van der Linden, M.H.; Valsecchi, M.G.; De Lorenzo, P.; Möricke, A.; Janka, G.; Leblanc, T.M.; Felice, M.; Biondi, A.; Campbell, M.; Hann, I.; et al. Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood 2009, 114, 3764–3768. [Google Scholar] [CrossRef] [PubMed]
- Takachi, T.; Watanabe, T.; Miyamura, T.; Moriya Saito, A.; Deguchi, T.; Hori, T.; Yamada, T.; Ohmori, S.; Haba, M.; Aoki, Y.; et al. Hematopoietic stem cell transplantation for infants with high-risk KMT2A gene-rearranged acute lymphoblastic leukemia. Blood Adv. 2021, 5, 3891–3899. [Google Scholar] [CrossRef]
- Tomizawa, D.; Miyamura, T.; Imamura, T.; Watanabe, T.; Moriya Saito, A.; Ogawa, A.; Takahashi, Y.; Hirayama, M.; Taki, T.; Deguchi, T.; et al. A risk-stratified therapy for infants with acute lymphoblastic leukemia: A report from the JPLSG MLL-10 trial. Blood 2020, 136, 1813–1823. [Google Scholar] [CrossRef]
- Montaño, A.; Forero-Castro, M.; Marchena-Mendoza, D.; Benito, R.; Hernández-Rivas, J.M. New Challenges in Targeting Signaling Pathways in Acute Lymphoblastic Leukemia by NGS Approaches: An Update. Cancers 2018, 10, 110. [Google Scholar] [CrossRef] [Green Version]
- Montaño, A.; Hernández-Sánchez, J.; Forero-Castro, M.; Matorra-Miguel, M.; Lumbreras, E.; Miguel, C.; Santos, S.; Ramírez-Maldonado, V.; Fuster, J.L.; de Las Heras, N.; et al. Comprehensive Custom NGS Panel Validation for the Improvement of the Stratification of B-Acute Lymphoblastic Leukemia Patients. J. Pers. Med. 2020, 10, 137. [Google Scholar] [CrossRef]
- Qiu, K.Y.; Zhou, D.H.; Liao, X.Y.; Huang, K.; Li, Y.; Xu, H.G.; Weng, W.J.; Xu, L.H.; Fang, J. Prognostic value and outcome for acute lymphocytic leukemia in children with MLL rearrangement: A case-control study. BMC Cancer 2022, 22, 1257. [Google Scholar] [CrossRef]
- Popov, A.; Tsaur, G.; Permikin, Z.; Fominikh, V.; Verzhbitskaya, T.; Riger, T.; Demina, A.; Shorikov, E.; Kustanovich, A.; Movchan, L.; et al. Incidence and prognostic value of central nervous system involvement in infants with B-cell precursor acute lymphoblastic leukemia treated according to the MLL-Baby protocol. Pediatr. Blood Cancer 2022, 69, e29860. [Google Scholar] [CrossRef]
- Zhao, M.; Duan, Y.; Wang, J.; Liu, Y.; Zhao, Y.; Wang, H.; Zhang, L.; Chen, Z.S.; Hu, Z.; Wei, L. Histone Deacetylase Inhibitor I3 Induces the Differentiation of Acute Myeloid Leukemia Cells with t (8; 21) or MLL Gene Translocation and Leukemic Stem-Like Cells. J. Oncol. 2022, 2022, 3345536. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Li, G.; Cui, Z.; Chen, P.; Wang, J.; Hu, Z.; Zhang, L.; Wei, L. The Histone Deacetylase Inhibitor I1 Induces Differentiation of Acute Leukemia Cells With MLL Gene Rearrangements via Epigenetic Modification. Front. Pharmacol. 2022, 13, 876076. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jiang, M.; Lau, C.W.; Luo, J.; Chan, A.M.; Wang, L.; Huang, Y. Curaxin CBL0137 inhibits endothelial inflammation and atherogenesis via suppression of the Src-YAP signalling axis. Br. J. Pharmacol. 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Leonova, K.; Safina, A.; Nesher, E.; Sandlesh, P.; Pratt, R.; Burkhart, C.; Lipchick, B.; Gitlin, I.; Frangou, C.; Koman, I.; et al. TRAIN (Transcription of Repeats Activates INterferon) in response to chromatin destabilization induced by small molecules in mammalian cells. eLife 2018, 7, e30842. [Google Scholar] [CrossRef] [PubMed]
- Somers, K.; Kosciolek, A.; Bongers, A.; El-Ayoubi, A.; Karsa, M.; Mayoh, C.; Wadham, C.; Middlemiss, S.; Neznanov, N.; Kees, U.R.; et al. Potent antileukemic activity of curaxin CBL0137 against MLL-rearranged leukemia. Int. J. Cancer 2020, 1, 1902–1916. [Google Scholar] [CrossRef]
- Garrido Castro, P.; van Roon, E.H.J.; Pinhanços, S.S.; Trentin, L.; Schneider, P.; Kerstjens, M.; Te Kronnie, G.; Heidenreich, O.; Pieters, R.; Stam, R.W. The HDAC inhibitor panobinostat (LBH589) exerts in vivo anti-leukaemic activity against MLL-rearranged acute lymphoblastic leukaemia and involves the RNF20/RNF40/WAC-H2B ubiquitination axis. Leukemia 2018, 32, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Karsa, M.; Ronca, E.; Bongers, A.; Kosciolek, A.; El-Ayoubi, A.; Revalde, J.L.; Seneviratne, J.A.; Cheung, B.B.; Cheung, L.C.; et al. The Combination of Curaxin CBL0137 and Histone Deacetylase Inhibitor Panobinostat Delays KMT2A-Rearranged Leukemia Progression. Front. Oncol. 2022, 12, 863329. [Google Scholar] [CrossRef]
- Gurova, K.V. Chromatin Stability as a Target for Cancer Treatment. Bioessays 2019, 41, e1800141. [Google Scholar] [CrossRef]
- Frikha, R.; Abdellaoui, N.; Kassar, O.; Rebai, T. Lack of FLT3-ITD in Tunisian childhood acute lymphoblastic leukemia. Afr. Health Sci. 2022, 22, 318–322. [Google Scholar] [CrossRef]
- El Chaer, F.; Keng, M.; Ballen, K.K. MLL-Rearranged Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2020, 15, 83–89. [Google Scholar] [CrossRef]
- Cooper, T.M.; Cassar, J.; Eckroth, E.; Malvar, J.; Sposto, R.; Gaynon, P.; Chang, B.H.; Gore, L.; August, K.; Pollard, J.A.; et al. A Phase I Study of Quizartinib Combined with Chemotherapy in Relapsed Childhood Leukemia: A Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Clin. Cancer Res. 2016, 22, 4014–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uckun, F.M.; Qazi, S. Tyrosine kinases in KMT2A/MLL-rearranged acute leukemias as potential therapeutic targets to overcome cancer drug resistance. Cancer Drug Resist. 2022, 9, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Guest, E.M.; Kairalla, J.A.; Hilden, J.M.; Dreyer, Z.E.; Carroll, A.J.; Heerema, N.A.; Wang, C.Y.; Devidas, M.; Gore, L.; Salzer, W.L.; et al. Outstanding outcomes in infants with KMT2A-germline acute lymphoblastic leukemia treated with chemotherapy alone: Results of the Children’s Oncology Group AALL0631 trial. Haematologica 2022, 1, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Heikamp, E.B.; Henrich, J.A.; Perner, F.; Wong, E.M.; Hatton, C.; Wen, Y.; Barwe, S.P.; Gopalakrishnapillai, A.; Xu, H.; Uckelmann, H.J.; et al. The menin-MLL1 interaction is a molecular dependency in NUP98-rearranged AML. Blood 2022, 139, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Brzezinka, K.; Nevedomskaya, E.; Lesche, R.; Steckel, M.; Eheim, A.L.; Haegebarth, A.; Stresemann, C. Functional diversity of inhibitors tackling the differentiation blockage of MLL-rearranged leukemia. J. Hematol. Oncol. 2019, 12, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Nissan, G.; Katzir, N.; Füzesi-Levi, M.G.; Sharon, M. Biology of the Extracellular Proteasome. Biomolecules 2022, 12, 619. [Google Scholar] [CrossRef] [PubMed]
- Mousavian, Z.; Nowzari-Dalini, A.; Rahmatallah, Y.; Masoudi-Nejad, A. Differential network analysis and protein-protein interaction study reveals active protein modules in glucocorticoid resistance for infant acute lymphoblastic leukemia. Mol. Med. 2019, 25, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, L.C.; de Kraa, R.; Oommen, J.; Chua, G.A.; Singh, S.; Hughes, A.M.; Ferrari, E.; Ford, J.; Chiu, S.K.; Stam, R.W.; et al. Preclinical Evaluation of Carfilzomib for Infant KMT2A-Rearranged Acute Lymphoblastic Leukemia. Front. Oncol. 2021, 11, 631594. [Google Scholar] [CrossRef]
- Jenkins, T.W.; Downey-Kopyscinski, S.L.; Fields, J.L.; Rahme, G.J.; Colley, W.C.; Israel, M.A.; Maksimenko, A.V.; Fiering, S.N.; Kisselev, A.F. Activity of immunoproteasome inhibitor ONX-0914 in acute lymphoblastic leukemia expressing MLL-AF4 fusion protein. Sci. Rep. 2021, 11, 10883. [Google Scholar] [CrossRef]
- Roolf, C.; Richter, A.; Konkolefski, C.; Knuebel, G.; Sekora, A.; Krohn, S.; Stenzel, J.; Krause, B.J.; Vollmar, B.; Murua Escobar, H.; et al. Decitabine demonstrates antileukemic activity in B cell precursor acute lymphoblastic leukemia with MLL rearrangements. J. Hematol. Oncol. 2018, 11, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Gao, X.; Zhao, X.; Wu, H.; Yan, M.; Li, Y.; Zeng, H.; Ji, Z.; Guo, X. Decitabine inhibits the proliferation of human T-cell acute lymphoblastic leukemia molt4 cells and promotes apoptosis partly by regulating the PI3K/AKT/mTOR pathway. Oncol. Lett. 2021, 21, 340. [Google Scholar] [CrossRef]
- Schneider, P.; Castro, P.G.; Pinhanços, S.M.; Kerstjens, M.; van Roon, E.H.; Essing, A.H.W.; Dolman, M.E.M.; Molenaar, J.J.; Pieters, R.; Stam, R.W. Decitabine mildly attenuates MLL-rearranged acute lymphoblastic leukemia in vivo, and represents a poor chemo-sensitizer. EJHaem 2020, 1, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan-Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef] [PubMed]
- Kerstjens, M.; Garrido Castro, P.; Pinhanços, S.S.; Schneider, P.; Wander, P.; Pieters, R.; Stam, R.W. Irinotecan Induces Disease Remission in Xenograft Mouse Models of Pediatric MLL-Rearranged Acute Lymphoblastic Leukemia. Biomedicines 2021, 9, 711. [Google Scholar] [CrossRef]
- Hayden, P.J.; Roddie, C.; Bader, P.; Basak, G.W.; Bonig, H.; Bonini, C.; Chabannon, C.; Ciceri, F.; Corbacioglu, S.; Ellard, R.; et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann. Oncol. 2022, 33, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Wölfl, M.; Rasche, M.; Eyrich, M.; Schmid, R.; Reinhardt, D.; Schlegel, P.G. Spontaneous reversion of a lineage switch following an initial blinatumomab-induced ALL-to-AML switch in MLL-rearranged infant ALL. Blood Adv. 2018, 2, 1382–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Kohler, M.E.; Fry, T.; Ernst, P. Does lineage plasticity enable escape from CAR-T cell therapy? Lessons from MLL-r leukemia. Exp. Hematol. 2021, 100, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.; Alati, C.; Canale, F.A.; Musuraca, G.; Martinelli, G.; Cerchione, C. A Review of Clinical Outcomes of CAR T-Cell Therapies for B-Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 2150. [Google Scholar] [CrossRef]
- Britten, O.; Ragusa, D.; Tosi, S.; Kamel, Y.M. MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)-Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells 2019, 8, 1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyer, Z.E.; Dinndorf, P.A.; Camitta, B.; Sather, H.; La, M.K.; Devidas, M.; Hilden, J.M.; Heerema, N.A.; Sanders, J.E.; McGlennen, R.; et al. Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: A report from the Children’s Oncology Group. J. Clin. Oncol. 2011, 29, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balduzzi, A.; Buechner, J.; Ifversen, M.; Dalle, J.H.; Colita, A.M.; Bierings, M. Acute Lymphoblastic Leukaemia in the Youngest: Haematopoietic Stem Cell Transplantation and Beyond. Front. Pediatr. 2022, 10, 807992. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
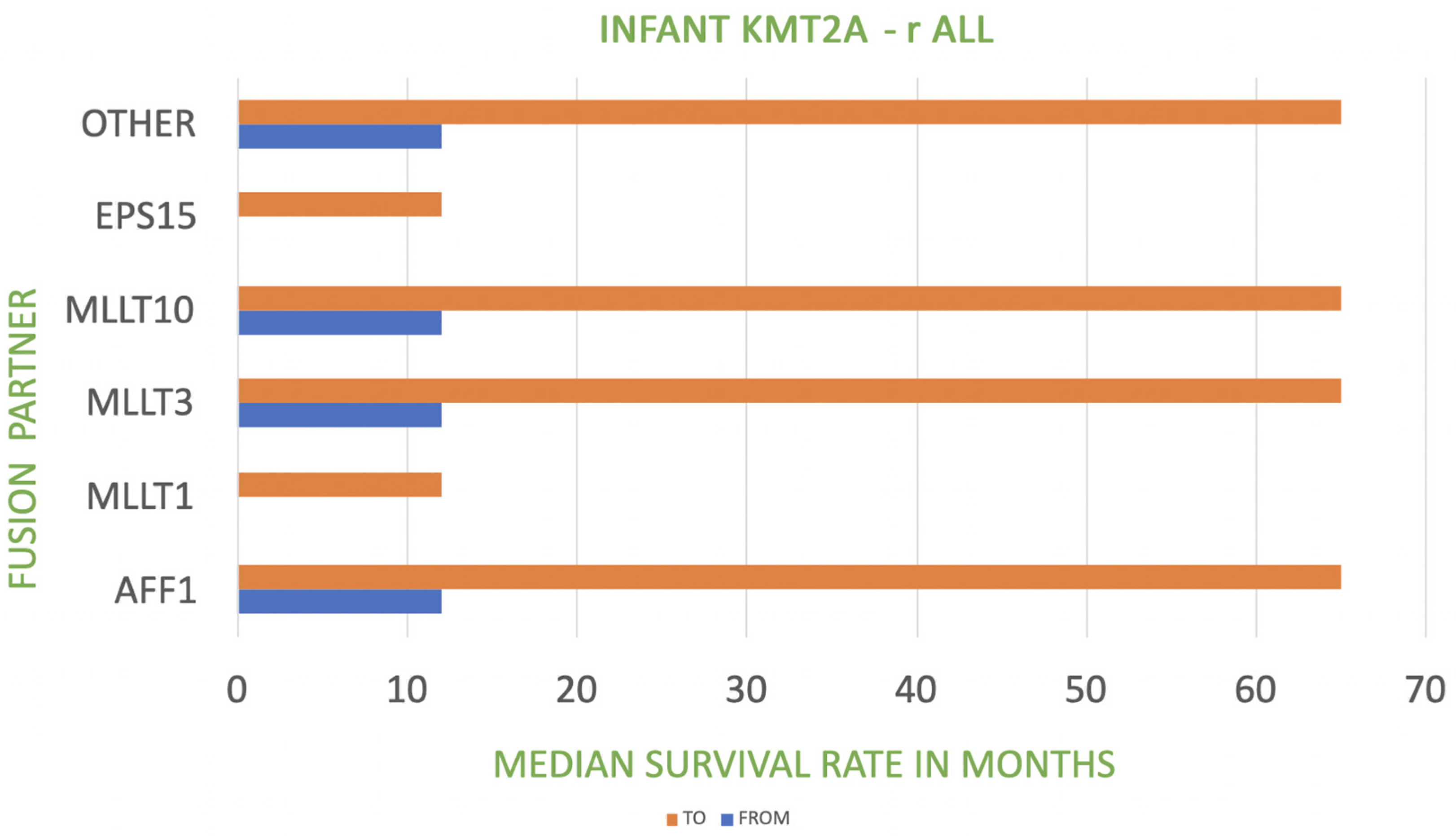
| KMT2A Fusion Partner in Infant KMT2A-r ALL | Frequency | Prognosis |
|---|---|---|
| AFF1-4q21 | 49% | poor |
| MLLT1-19p13 | 22% | very poor |
| MLLT3-9p21 | 16% | poor to intermediate |
| MLLT10-10p12 | 6% | poor |
| EPS15-1p32 | 2% | very poor |
| Other | 5% | poor |
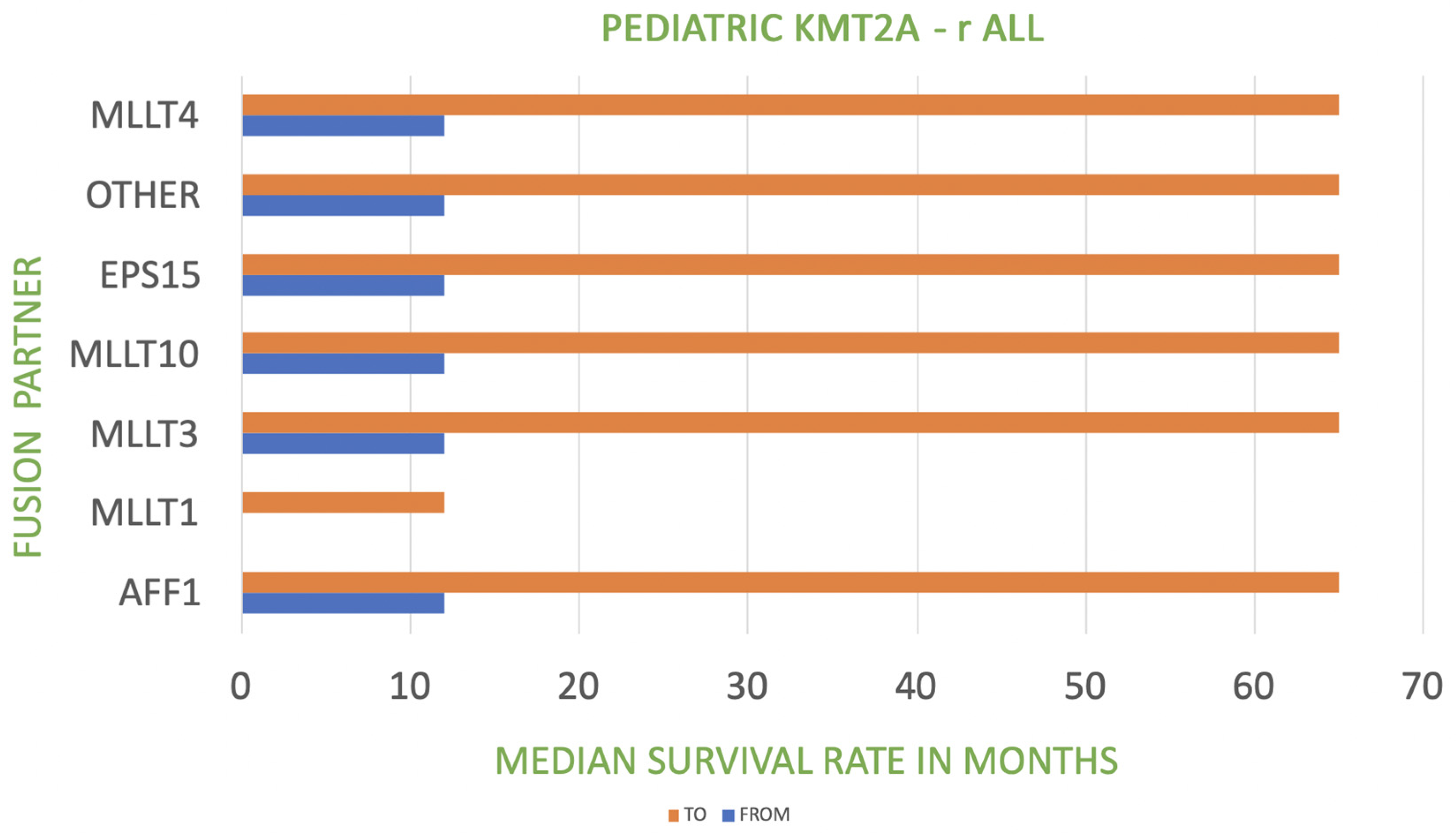
| KMT2A Fusion Partner in Pediatric KMT2A-r ALL | Frequency | Prognosis |
| AFF1-4q21 | 44% | poor |
| MLLT3-9p21 | 18% | poor to intermediate |
| MLLT1-19p13 | 18% | very poor |
| MLLT10-10p12 | 5% | poor |
| MLLT4-6q27 | 5% | poor |
| EPS15-1p32 | 2% | poor |
| other | 8% | poor |
| Risk | Interfant | COG | JPLSG | Approximate EFS, % |
|---|---|---|---|---|
| high | KMT2A-r and age <180 days and WBCs ≥ 300,000/μL | KMT2A-r and age <90 days | KMT2A-r and (age <180 days or CNS leukemia or poor prednisone response) | 20 |
| intermediate | other KMT2A-r | other KMT2A-r | other KMT2A-r | 50 |
| low | KMT2A-g | KMT2A-g | KMT2A-g | 75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Górecki, M.; Kozioł, I.; Kopystecka, A.; Budzyńska, J.; Zawitkowska, J.; Lejman, M. Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia. Biomedicines 2023, 11, 821. https://doi.org/10.3390/biomedicines11030821
Górecki M, Kozioł I, Kopystecka A, Budzyńska J, Zawitkowska J, Lejman M. Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia. Biomedicines. 2023; 11(3):821. https://doi.org/10.3390/biomedicines11030821
Chicago/Turabian StyleGórecki, Mateusz, Ilona Kozioł, Agnieszka Kopystecka, Julia Budzyńska, Joanna Zawitkowska, and Monika Lejman. 2023. "Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia" Biomedicines 11, no. 3: 821. https://doi.org/10.3390/biomedicines11030821