
Interplay of Oxidative Stress and Necrosis-like Cell Death in Cardiac Ischemia/Reperfusion Injury: A Focus on Necroptosis

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Mechanisms of Necroptosis Induction and Execution
2.1. Canonical Signaling Pathway of Necroptosis
2.2. Non-Canonical Signaling Pathway of Necroptosis
3. General Role of OS in Necroptosis Signaling
4. Possible Involvement of OS in Necroptotic Injury in the Heart
5. Anti-Necroptotic Agents: Pharmacodynamic Features and Modulation of Oxidative Stress
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Timmis, A.; Townsend, N.; Gale, C.; Grobbee, R.; Maniadakis, N.; Flather, M.; Vardas, P. European Society of Cardiology: Cardiovascular Disease Statistics 2021. Eur. Heart J. 2021, 39, 508–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Ginks, W.R.; Sybers, H.D.; Maroko, P.R.; Covell, J.W.; Sobel, B.E.; Ross, J., Jr. Coronary artery reperfusion. II. Reduction of myocardial infarct size at 1 week after the coronary occlusion. J. Clin. Investig. 1972, 51, 2717–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellon, D.M.; Baxter, G.F. Protecting the ischaemic and reperfused myocardium in acute myocardial infarction: Distant dream or near reality? Heart 2000, 83, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, G.W.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Maehara, A.; Eitel, I.; Granger, C.B.; Jenkins, P.L.; et al. Relationship Between Infarct Size and Outcomes Following Primary PCI: Patient-Level Analysis From 10 Randomized Trials. J. Am. Coll. Cardiol. 2016, 67, 1674–1683. [Google Scholar] [CrossRef]
- Ma, W.; Wei, S.; Zhang, B.; Li, W. Molecular Mechanisms of Cardiomyocyte Death in Drug-Induced Cardiotoxicity. Front. Cell Dev. Biol. 2020, 8, 434. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Chen, Y.; Hua, Y.; Li, X.; Arslan, I.M.; Zhang, W.; Meng, G. Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front. Pharmacol. 2020, 11, 42. [Google Scholar] [CrossRef]
- Cai, L.; Kang, Y.J. Cell death and diabetic cardiomyopathy. Cardiovasc. Toxicol. 2003, 3, 219–228. [Google Scholar] [CrossRef]
- Szobi, A.; Goncalvesova, E.; Varga, Z.V.; Leszek, P.; Kusmierczyk, M.; Hulman, M.; Kyselovic, J.; Ferdinandy, P.; Adameova, A. Analysis of necroptotic proteins in failing human hearts. J. Transl. Med. 2017, 15, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ 2007, 14, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharmacol. 2000, 130, 197–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaoita, H.; Ogawa, K.; Maehara, K.; Maruyama, Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 1998, 97, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Holly, T.A.; Drincic, A.; Byun, Y.; Nakamura, S.; Harris, K.; Klocke, F.J.; Cryns, V.L. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J. Mol. Cell Cardiol. 1999, 31, 1709–1715. [Google Scholar] [CrossRef]
- Mishra, P.K.; Adameova, A.; Hill, J.A.; Baines, C.P.; Kang, P.M.; Downey, J.; Narula, J.; Takahashi, M.; Abbate, A.; Piristine, H.C.; et al. Guidelines for evaluating myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, 891–922. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Lichy, M.; Szobi, A.; Hrdlicka, J.; Horvath, C.; Kormanova, V.; Rajtik, T.; Neckar, J.; Kolar, F.; Adameova, A. Different signalling in infarcted and non-infarcted areas of rat failing hearts: A role of necroptosis and inflammation. J. Cell. Mol. Med. 2019, 23, 6429–6441. [Google Scholar] [CrossRef] [Green Version]
- Lichy, M.; Szobi, A.; Hrdlicka, J.; Neckar, J.; Kolar, F.; Adameova, A. Programmed Cell Death in the Left and Right Ventricle of the Late Phase of Post-Infarction Heart Failure. Int. J. Mol. Sci. 2020, 21, 7782. [Google Scholar] [CrossRef]
- Szobi, A.; Farkasova-Ledvenyiova, V.; Lichy, M.; Murarikova, M.; Carnicka, S.; Ravingerova, T.; Adameova, A. Cardioprotection of ischaemic preconditioning is associated with inhibition of translocation of MLKL within the plasma membrane. J. Cell. Mol. Med. 2018, 22, 4183–4196. [Google Scholar] [CrossRef] [PubMed]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef]
- Guerra, S.; Leri, A.; Wang, X.; Finato, N.; Di Loreto, C.; Beltrami, C.A.; Kajstura, J.; Anversa, P. Myocyte death in the failing human heart is gender dependent. Circ. Res. 1999, 85, 856–866. [Google Scholar] [CrossRef]
- Jose Corbalan, J.; Vatner, D.E.; Vatner, S.F. Myocardial apoptosis in heart disease: Does the emperor have clothes? Basic Res. Cardiol. 2016, 111, 31. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Shen, Y.T.; Gaussin, V.; Heyndrickx, G.R.; Bartunek, J.; Resuello, R.R.; Natividad, F.F.; Kitsis, R.N.; Vatner, D.E.; Vatner, S.F. Apoptosis predominates in nonmyocytes in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H785–H791. [Google Scholar] [CrossRef] [Green Version]
- Fliss, H. Accelerated apoptosis in reperfused myocardium: Friend of foe? Basic Res. Cardiol. 1998, 93, 90–93. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Burleson, K.O.; Kloner, R.A.; Babior, B.M.; Engler, R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Investig. 1994, 94, 1621–1628. [Google Scholar] [CrossRef] [Green Version]
- Bromme, H.J.; Holtz, J. Apoptosis in the heart: When and why? Mol. Cell. Biochem. 1996, 163–164, 261–275. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.e.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ. Res. 2021, 129, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Shen, J.; Li, Y.; Wu, J.; Luo, X.; Yu, Y.; Zhang, Y.; Gu, L.; Zhang, X.; Jiang, C.; et al. Pyroptosis inhibition improves the symptom of acute myocardial infarction. Cell Death Dis. 2021, 12, 852. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef]
- Horvath, C.; Young, M.; Jarabicova, I.; Kindernay, L.; Ferenczyova, K.; Ravingerova, T.; Lewis, M.; Suleiman, M.S.; Adameova, A. Inhibition of Cardiac RIP3 Mitigates Early Reperfusion Injury and Calcium-Induced Mitochondrial Swelling without Altering Necroptotic Signalling. Int. J. Mol. Sci. 2021, 22, 7893. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, C.; Zhang, C.; Li, J.; Guo, W.; Yan, D.; Yang, C.; Zhao, J.; Xia, T.; Wang, Y.; et al. Heat shock protein 70 inhibits cardiomyocyte necroptosis through repressing autophagy in myocardial ischemia/reperfusion injury. Vitr. Cell. Dev. Biol. Anim. 2016, 52, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, M.; Yano, T.; Tanno, M.; Abe, K.; Ishikawa, S.; Miki, T.; Kuno, A.; Tobisawa, T.; Muratsubaki, S.; Ohno, K.; et al. Suppression of autophagic flux contributes to cardiomyocyte death by activation of necroptotic pathways. J. Mol. Cell Cardiol. 2017, 108, 203–213. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, L.; Yuan, W.; Sun, L.; Xia, Z.; Zhang, Z.; Yao, W. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J. Cell. Mol. Med. 2020, 24, 6670–6679. [Google Scholar] [CrossRef]
- Bertrand, R.L. Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med. Hypotheses 2017, 101, 69–74. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Golfman, L.; Takeda, S.; Takeda, N.; Nagano, M. Evidence for the role of oxidative stress in acute ischemic heart disease: A brief review. Can. J. Cardiol. 1999, 15, 587–593. [Google Scholar] [PubMed]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens 2000, 18, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Shah, A.K.; Dhalla, N.S. Role of Oxidative Stress in the Genesis of Ventricular Arrhythmias. Int. J. Mol. Sci. 2020, 21, 4200. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M. Sarcoplasmic reticulum and cardiac oxidative stress: An emerging target for heart disease. Expert Opin. Ther. Targets 2001, 5, 205–217. [Google Scholar] [CrossRef]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1986–H1994. [Google Scholar] [CrossRef] [PubMed]
- Ostadal, P.; Elmoselhi, A.B.; Zdobnicka, I.; Lukas, A.; Elimban, V.; Dhalla, N.S. Role of oxidative stress in ischemia-reperfusion-induced changes in Na+,K(+)-ATPase isoform expression in rat heart. Antioxid. Redox Signal. 2004, 6, 914–923. [Google Scholar] [CrossRef]
- Temsah, R.M.; Netticadan, T.; Chapman, D.; Takeda, S.; Mochizuki, S.; Dhalla, N.S. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am. J. Physiol. 1999, 277, H584–H594. [Google Scholar] [CrossRef]
- Siwik, D.A.; Colucci, W.S. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail. Rev. 2004, 9, 43–51. [Google Scholar] [CrossRef]
- Singh, R.B.; Hryshko, L.; Freed, D.; Dhalla, N.S. Activation of proteolytic enzymes and depression of the sarcolemmal Na+/K+-ATPase in ischemia-reperfused heart may be mediated through oxidative stress. Can. J. Physiol. Pharmacol. 2012, 90, 249–260. [Google Scholar] [CrossRef]
- Kwon, S.H.; Pimentel, D.R.; Remondino, A.; Sawyer, D.B.; Colucci, W.S. H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J. Mol. Cell Cardiol. 2003, 35, 615–621. [Google Scholar] [CrossRef]
- Kumar, D.; Lou, H.; Singal, P.K. Oxidative stress and apoptosis in heart dysfunction. Herz 2002, 27, 662–668. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef]
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.J.; Han, B.S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 835. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.V.; Kupai, K.; Szucs, G.; Gaspar, R.; Paloczi, J.; Farago, N.; Zvara, A.; Puskas, L.G.; Razga, Z.; Tiszlavicz, L.; et al. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J. Mol. Cell Cardiol. 2013, 62, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schröder, K.; Streckfuß-Bömeke, K.; Doroshow, J.H.; et al. Nox4 regulates InsP(3) receptor-dependent Ca(2+) release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.V.; Pipicz, M.; Baán, J.A.; Baranyai, T.; Koncsos, G.; Leszek, P.; Kuśmierczyk, M.; Sánchez-Cabo, F.; García-Pavía, P.; Brenner, G.J.; et al. Alternative Splicing of NOX4 in the Failing Human Heart. Front. Physiol. 2017, 8, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Duan, F.; Hu, J.; Luo, B.; Huang, B.; Lou, X.; Sun, X.; Li, H.; Zhang, X.; Yin, S.; et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020, 34, 101523. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Goncalvesova, E.; Szobi, A.; Dhalla, N.S. Necroptotic cell death in failing heart: Relevance and proposed mechanisms. Heart Fail. Rev. 2016, 21, 213–221. [Google Scholar] [CrossRef]
- Adameova, A.; Hrdlicka, J.; Szobi, A.; Farkasova, V.; Kopaskova, K.; Murarikova, M.; Neckar, J.; Kolar, F.; Ravingerova, T.; Dhalla, N.S. Evidence of necroptosis in hearts subjected to various forms of ischemic insults. Can. J. Physiol. Pharmacol. 2017, 95, 1163–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, S.M.; Adameova, A.; Barile, L.; Cabrera-Fuentes, H.A.; Lazou, A.; Pagliaro, P.; Stenslokken, K.O.; Garcia-Dorado, D.; Action, E.-C.C. Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J. Cell. Mol. Med. 2020, 24, 3795–3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moquin, D.; Chan, F.K. The molecular regulation of programmed necrotic cell injury. Trends Biochem. Sci. 2010, 35, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Panayotova-Dimitrova, D.; Leverkus, M. Pick your poison: The Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 2012, 11, 460–467. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, K.; Chan, F.K. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013, 27, 1640–1649. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.F.; Huang, Z.; Ren, J.M.; Zhang, Z.R.; He, P.; Li, Y.X.; Ma, J.H.; Chen, W.Z.; Zhang, Y.Y.; Zhou, X.J.; et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013, 23, 994–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef]
- Cai, Z.Y.; Zhang, A.L.; Choksi, S.; Li, W.H.; Li, T.; Zhang, X.M.; Liu, Z.G. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016, 26, 886–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conos, S.A.; Chen, K.W.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.N.; Yang, Z.H.; Wang, X.K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.R.; Vila-Petroff, M.; Delbridge, L.M. CaMKII-dependent responses to ischemia and reperfusion challenges in the heart. Front. Pharmacol. 2014, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Weinreuter, M.; Kreusser, M.M.; Beckendorf, J.; Schreiter, F.C.; Leuschner, F.; Lehmann, L.H.; Hofmann, K.P.; Rostosky, J.S.; Diemert, N.; Xu, C.; et al. CaM Kinase II mediates maladaptive post-infarct remodeling and pro-inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol. Med. 2014, 6, 1231–1245. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, K.; Liu, X.; Qin, M.; Xiang, Y. CaMKII in Regulation of Cell Death During Myocardial Reperfusion Injury. Front. Mol. Biosci. 2021, 8, 478. [Google Scholar] [CrossRef]
- Szobi, A.; Rajtik, T.; Carnicka, S.; Ravingerova, T.; Adameova, A. Mitigation of postischemic cardiac contractile dysfunction by CaMKII inhibition: Effects on programmed necrotic and apoptotic cell death. Mol. Cell. Biochem. 2014, 388, 269–276. [Google Scholar] [CrossRef]
- Zhou, T.; DeRoo, E.; Yang, H.; Stranz, A.; Wang, Q.; Ginnan, R.; Singer, H.A.; Liu, B. MLKL and CaMKII Are Involved in RIPK3-Mediated Smooth Muscle Cell Necroptosis. Cells 2021, 10, 2397. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar] [PubMed]
- Hou, H.; Wang, Y.; Li, Q.; Li, Z.; Teng, Y.; Li, J.; Wang, X.; Chen, J.; Huang, N. The role of RIP3 in cardiomyocyte necrosis induced by mitochondrial damage of myocardial ischemia-reperfusion. Acta Biochim. Biophys. Sin. 2018, 50, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- She, L.; Tu, H.; Zhang, Y.Z.; Tang, L.J.; Li, N.S.; Ma, Q.L.; Liu, B.; Li, Q.; Luo, X.J.; Peng, J. Inhibition of Phosphoglycerate Mutase 5 Reduces Necroptosis in Rat Hearts Following Ischemia/Reperfusion Through Suppression of Dynamin-Related Protein 1. Cardiovasc. Drugs Ther. 2019, 33, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Farias Luz, N.; Balaji, S.; De Rosa, M.J.; O’Donnell, C.L.; Gough, P.J.; Bertin, J.; Welsh, R.M.; Chan, F.K. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. J. Immunol. 2016, 196, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moujalled, D.M.; Cook, W.D.; Murphy, J.M.; Vaux, D.L. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014, 5, e1086. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Li, T. Ripk3 mediates cardiomyocyte necrosis through targeting mitochondria and the JNK-Bnip3 pathway under hypoxia-reoxygenation injury. J. Recept. Signal Transduct. 2019, 39, 331–340. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [Green Version]
- Rivera, J.; Sobey, C.G.; Walduck, A.K.; Drummond, G.R. Nox isoforms in vascular pathophysiology: Insights from transgenic and knockout mouse models. Redox Rep. Commun. Free Radic. Res. 2010, 15, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress-A concise review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar] [PubMed]
- Adameova, A.; Dhalla, N.S. Role of microangiopathy in diabetic cardiomyopathy. Heart Fail. Rev. 2014, 19, 25–33. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Ganguly, P.K.; Bhullar, S.K.; Tappia, P.S. Role of catecholamines in the pathogenesis of diabetic cardiomyopathy (1). Can. J. Physiol. Pharmacol. 2019, 97, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Shah, A.K.; Tappia, P.S. Role of Oxidative Stress in Metabolic and Subcellular Abnormalities in Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 2413. [Google Scholar] [CrossRef] [Green Version]
- Tappia, P.S.; Adameova, A.; Dhalla, N.S. Attenuation of Diabetes-induced Cardiac and Subcellular Defects by Sulphur-containing Amino Acids. Curr. Med. Chem. 2018, 25, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.; Bornstein, S.; Linkermann, A.; Shapiro, A.M.J. Regulated Cell Death Seen through the Lens of Islet Transplantation. Cell Transplant. 2018, 27, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, J.; Bermudez, V.; Palmar, J.; Martínez, M.S.; Olivar, L.C.; Nava, M.; Tomey, D.; Rojas, M.; Salazar, J.; Garicano, C.; et al. Pancreatic Beta Cell Death: Novel Potential Mechanisms in Diabetes Therapy. J. Diabetes Res. 2018, 2018, 9601801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinawong, K.; Apaijai, N.; Wongsuchai, S.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Necrostatin-1 Mitigates Cognitive Dysfunction in Prediabetic Rats With No Alteration in Insulin Sensitivity. Diabetes 2020, 69, 1411–1423. [Google Scholar] [CrossRef]
- Xu, H.; Du, X.; Liu, G.; Huang, S.; Du, W.; Zou, S.; Tang, D.; Fan, C.; Xie, Y.; Wei, Y.; et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol. Metab. 2019, 23, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, Y.; Luo, H.; Xu, M.; Meng, G.; Zhang, W. Ca2+/calmodulin-dependent protein kinase II regulation by inhibitor 1 of protein phosphatase 1 alleviates necroptosis in high glucose-induced cardiomyocytes injury. Biochem. Pharmacol. 2019, 163, 194–205. [Google Scholar] [CrossRef]
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.; Zhang, Z.; et al. A Role of RIP3-Mediated Macrophage Necrosis in Atherosclerosis Development. Cell Rep. 2013, 3, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Chen, H.; Li, Y.; Huang, D.; Huang, B.; Sun, D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol. Int. 2020, 44, 1481–1490. [Google Scholar] [CrossRef]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Chen, M.; Zheng, D.; He, J.; Song, M.; Mo, L.; Feng, J.; Lan, J. A novel damage mechanism: Contribution of the interaction between necroptosis and ROS to high glucose-induced injury and inflammation in H9c2 cardiac cells. Int. J. Mol. Med. 2017, 40, 201–208. [Google Scholar] [CrossRef]
- Liang, W.; Chen, M.; Zheng, D.; Li, J.; Song, M.; Zhang, W.; Feng, J.; Lan, J. The Opening of ATP-Sensitive K+ Channels Protects H9c2 Cardiac Cells Against the High Glucose-Induced Injury and Inflammation by Inhibiting the ROS-TLR4-Necroptosis Pathway. Cell. Physiol. Biochem. 2017, 41, 1020–1034. [Google Scholar] [CrossRef]
- Yin, W.; Wang, C.; Peng, Y.; Yuan, W.; Zhang, Z.; Liu, H.; Xia, Z.; Ren, C.; Qian, J. Dexmedetomidine alleviates H2O2-induced oxidative stress and cell necroptosis through activating of α2-adrenoceptor in H9C2 cells. Mol. Biol. Rep. 2020, 47, 3629–3639. [Google Scholar] [CrossRef]
- Yu, S.; Yang, H.; Guo, X.; Sun, Y. Klotho attenuates angiotensin II-induced cardiotoxicity through suppression of necroptosis and oxidative stress. Mol. Med. Rep. 2021, 23, 11705. [Google Scholar] [CrossRef]
- Wang, C.; Hu, L.; Guo, S.; Yao, Q.; Liu, X.; Zhang, B.; Meng, X.; Yang, X. Phosphocreatine attenuates doxorubicin-induced cardiotoxicity by inhibiting oxidative stress and activating TAK1 to promote myocardial survival in vivo and in vitro. Toxicology 2021, 460, 152881. [Google Scholar] [CrossRef]
- Chi, R.F.; Li, L.; Wang, A.L.; Yang, H.; Xi, J.; Zhu, Z.F.; Wang, K.; Li, B.; Yang, L.G.; Qin, F.Z.; et al. Enhanced oxidative stress mediates pathological autophagy and necroptosis in cardiac myocytes in pressure overload induced heart failure in rats. Clin. Exp. Pharmacol. Physiol. 2022, 49, 60–69. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Zhuang, W.; Yuan, G.; Sun, T.; Jiang, X.; Zhou, Z.; Yuan, H.; Zhang, Z.; Dong, H. PEDF and PEDF-derived peptide 44mer protect cardiomyocytes against hypoxia-induced apoptosis and necroptosis via anti-oxidative effect. Sci. Rep. 2014, 4, 5637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Ding, Y.; Dai, G.L.; Zhang, Y.; Xu, M.T.; Shen, J.R.; Meng, G.L. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 2021, 42, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, M.I.; Liu, J.; Arslan, F.; den Ouden, K.; van Middelaar, B.J.; Doevendans, P.A.; Sluijter, J.P. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res. Cardiol. 2012, 107, 270. [Google Scholar] [CrossRef] [PubMed]
- Koudstaal, S.; Oerlemans, M.I.; Van der Spoel, T.I.; Janssen, A.W.; Hoefer, I.E.; Doevendans, P.A.; Sluijter, J.P.; Chamuleau, S.A. Necrostatin-1 alleviates reperfusion injury following acute myocardial infarction in pigs. Eur. J. Clin. Investig. 2015, 45, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feng, Q.; Wang, T. Necrostatin-1 Protects Against Paraquat-Induced Cardiac Contractile Dysfunction via RIP1-RIP3-MLKL-Dependent Necroptosis Pathway. Cardiovasc. Toxicol. 2018, 18, 346–355. [Google Scholar] [CrossRef]
- Horvath, C.; Szobi, A.; Kindernay, L.; Ravingerova, T.; Adameova, A. Pleiotropic, non-cell death-associated effects of inhibitors of receptor-interacting protein kinase 1 in the heart. Mol. Cell. Biochem. 2021, 476, 3079–3087. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.C.; Davidson, S.M.; Lim, S.Y.; Simpkin, J.C.; Hothersall, J.S.; Yellon, D.M. Necrostatin: A potentially novel cardioprotective agent? Cardiovasc. Drugs Ther. 2007, 21, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Davidson, S.M.; Mocanu, M.M.; Yellon, D.M.; Smith, C.C. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc. Drugs Ther. 2007, 21, 467–469. [Google Scholar] [CrossRef] [Green Version]
- Vandenabeele, P.; Grootjans, S.; Callewaert, N.; Takahashi, N. Necrostatin-1 blocks both RIPK1 and IDO: Consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013, 20, 185–187. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Metz, R.; Muller, A.J. Towards a genetic definition of cancer-associated inflammation: Role of the IDO pathway. Am. J. Pathol. 2010, 176, 2082–2087. [Google Scholar] [CrossRef]
- Wang, X.F.; Wang, H.S.; Wang, H.; Zhang, F.; Wang, K.F.; Guo, Q.; Zhang, G.; Cai, S.H.; Du, J. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell. Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef]
- Szobi, A.; Rajtik, T.; Adameova, A. Effects of necrostatin-1, an inhibitor of necroptosis, and its inactive analogue Nec-1i on basal cardiovascular function. Physiol. Res. 2016, 65, 861–865. [Google Scholar] [CrossRef]
- Takahashi, N.; Duprez, L.; Grootjans, S.; Cauwels, A.; Nerinckx, W.; DuHadaway, J.B.; Goossens, V.; Roelandt, R.; Van Hauwermeiren, F.; Libert, C.; et al. Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012, 3, e437. [Google Scholar] [CrossRef] [Green Version]
- Teng, X.; Degterev, A.; Jagtap, P.; Xing, X.; Choi, S.; Denu, R.; Yuan, J.; Cuny, G.D. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5039–5044. [Google Scholar] [CrossRef]
- Degterev, A.; Maki, J.L.; Yuan, J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013, 20, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Weisel, K.; Berger, S.; Papp, K.; Maari, C.; Krueger, J.G.; Scott, N.; Tompson, D.; Wang, S.; Simeoni, M.; Bertin, J.; et al. Response to Inhibition of Receptor-Interacting Protein Kinase 1 (RIPK1) in Active Plaque Psoriasis: A Randomized Placebo-Controlled Study. Clin. Pharmacol. Ther. 2020, 108, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.H.; Park, S.Y.; Mah, S.; Park, J.H.; Hong, S.S.; Hong, S.; Kim, Y.S. HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Exp. Mol. Med. 2018, 50, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Wang, Q.; Phan, N.; Ren, J.; Yang, H.; Feldman, C.C.; Feltenberger, J.B.; Ye, Z.; Wildman, S.A.; Tang, W.; et al. Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Yang, C.H.; et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef] [Green Version]
- Cruz, S.A.; Qin, Z.; Stewart, A.F.R.; Chen, H.H. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen. Res. 2018, 13, 252–256. [Google Scholar] [CrossRef]
- Fauster, A.; Rebsamen, M.; Huber, K.V.; Bigenzahn, J.W.; Stukalov, A.; Lardeau, C.H.; Scorzoni, S.; Bruckner, M.; Gridling, M.; Parapatics, K.; et al. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 2015, 6, e1767. [Google Scholar] [CrossRef]
- Zhang, A.; Mao, X.; Li, L.; Tong, Y.; Huang, Y.; Lan, Y.; Jiang, H. Necrostatin-1 inhibits Hmgb1-IL-23/IL-17 pathway and attenuates cardiac ischemia reperfusion injury. Transpl. Int. 2014, 27, 1077–1085. [Google Scholar] [CrossRef]
- Rubbelke, M.; Fiegen, D.; Bauer, M.; Binder, F.; Hamilton, J.; King, J.; Thamm, S.; Nar, H.; Zeeb, M. Locking mixed-lineage kinase domain-like protein in its auto-inhibited state prevents necroptosis. Proc. Natl. Acad. Sci. USA 2020, 117, 33272–33281. [Google Scholar] [CrossRef]
- Münzel, T.; Gori, T.; Keaney, J.F., Jr.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Pol, A.; Van Gilst, W.H.; Voors, A.A.; Van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]


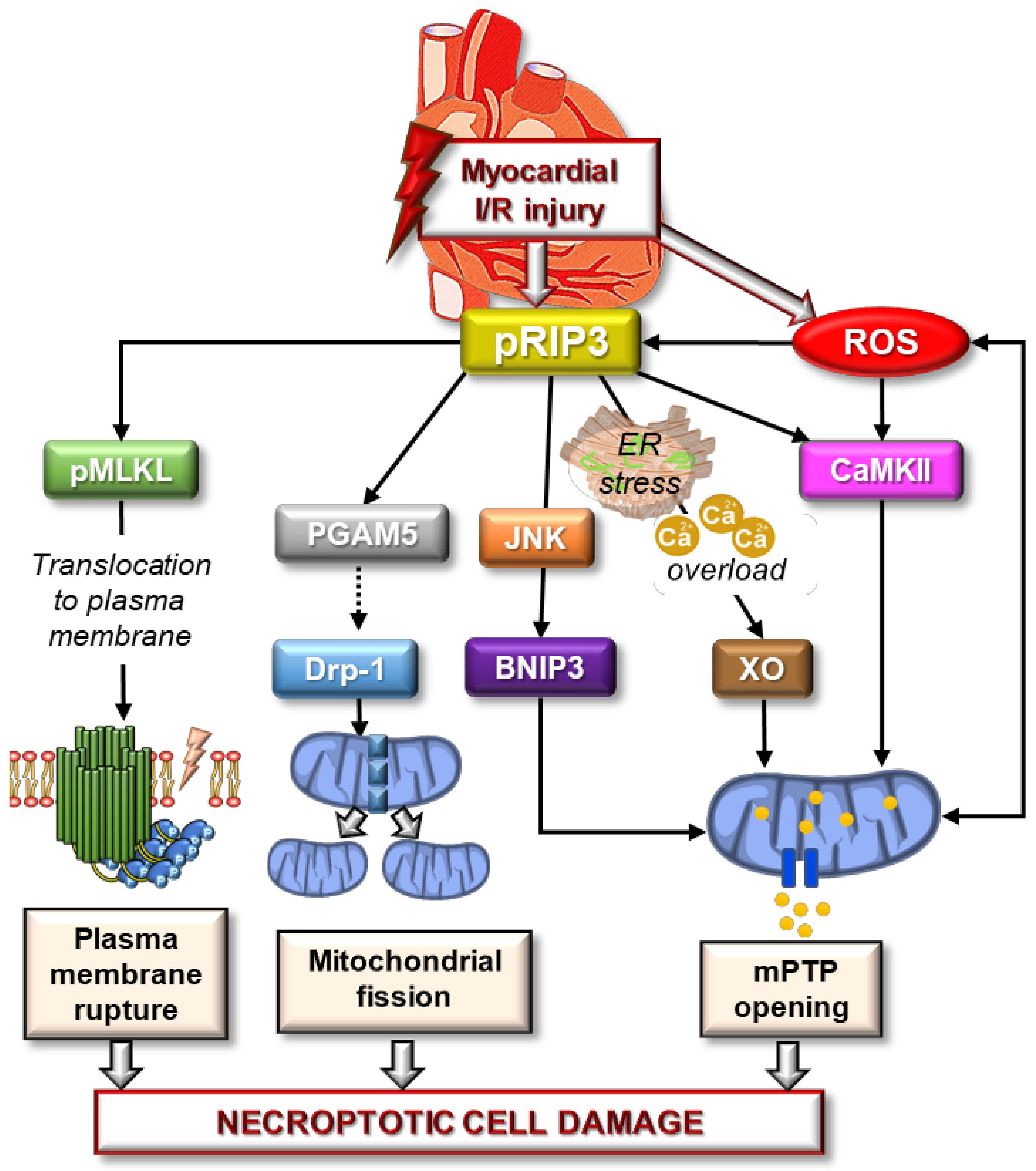{kind=link}
| Signaling Type | Proteins Involved | Reference | |
|---|---|---|---|
| Canonical | RIP3–MLKL | [73,74,90,91] | |
| Non-canonical | Associated with the mitochondria | RIP3–CaMKII | [35] |
| RIP3–PGAM5–Drp-1 | [85,86] | ||
| RIP3–JNK–BNIP3 | [89] | ||
| Associated with the sarcoplasmic reticulum | RIP3–XO | [36] |
| Model of OS-Induced Injury | Main Findings | Reference | |
|---|---|---|---|
| Protocol | Cell/Animals | ||
| 30-min ischemia in vivo | Isolated adult mouse cardiomyocytes | ROS scavenger (Tiron) and knockdown of Nox2 mitigated RIP3-induced necroptosis | [35] |
| High glucose (25.5 mM) | Neonatal rat ventricular myocytes | I1PP1 overexpression decreased oxCaMKII and ROS levels and limited necroptosis | [106] |
| High glucose (35 mM) | H9c2 | KATP channel opening was protective against high glucose-induced injury by inhibiting ROS-TLR4-necroptosis pathway | [111] |
| 12-/24-/48-h hypoxia | H9c2 | Pigment epithelium-derived factor ameliorated hypoxia-induced necroptosis and apoptosis activation via its antioxidant effect | [116] |
| H2O2 (500 μM) | H9c2 | Dexmedetomidine prevents OS-induced necroptosis | [112] |
| Doxorubicin (1 μM for 24 h) | H9c2 | NAC pre-treatment attenuated necroptosis by downregulating RIP3 and CaMKII expression | [114] |
| 45-min LAD ligation | C57BL/6 mice | RIP3 mediates I/R injury via SR stress-Ca2+ overload-XO-ROS-mPTP pathway | [36] |
| Streptozocin-induced diabetes mellitus | C57BL/6 mice | Sirtuin 3 deficiency increased ROS production and promoted necroptosis | [117] |
| Abdominal aortic constriction | Rats | NAC (500 mg/kg) treatment prevented the increase in OS and necroptosis and improved LV systolic function | [115] |
| Conditions | Experimental Protocol | Findings | Reference | |
|---|---|---|---|---|
| with ischemic insult | 30-min LAD ligation in vivo | Mice | Nec-1 (3.3 mg/kg) altered gene expression of NOS2, COX-2, GAB1 GPX1, CYBA and TXNIP | [118] |
| 30-min global ischemia ex vivo | Rat | GSK’872 (250 nM) decreased the expression of XO and MnSOD | [37] | |
| 75-min LCx ligation in vivo | Pigs | Nec-1 (1.0 mg/kg; 3.3 mg/kg) decreased nuclear ROS levels | [119] | |
| without ischemic insult | Angiotensin II (10 nM) | H9c2 | Nec-1 (10 mM) reduced angiotensin II-induced ROS production | [113] |
| High glucose (35 mM) | H9c2 | Nec-1 (100 µM) reduced high glucose-induced ROS production | [110] | |
| Paraquat (45 mg/kg) administration in vivo | Mice | Nec-1 (3.5 mg/kg) reversed paraquat-induced ROS production in the heart | [120] | |
| Ex vivo perfusion | Rat | Nec-1 (1.2 µM), Nec-1i (1.2 µM) and Nec-1s (0.5 µM), but not GSK’772 increased protein tyrosine nitration | [121] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adameova, A.; Horvath, C.; Abdul-Ghani, S.; Varga, Z.V.; Suleiman, M.S.; Dhalla, N.S. Interplay of Oxidative Stress and Necrosis-like Cell Death in Cardiac Ischemia/Reperfusion Injury: A Focus on Necroptosis. Biomedicines 2022, 10, 127. https://doi.org/10.3390/biomedicines10010127
Adameova A, Horvath C, Abdul-Ghani S, Varga ZV, Suleiman MS, Dhalla NS. Interplay of Oxidative Stress and Necrosis-like Cell Death in Cardiac Ischemia/Reperfusion Injury: A Focus on Necroptosis. Biomedicines. 2022; 10(1):127. https://doi.org/10.3390/biomedicines10010127
Chicago/Turabian StyleAdameova, Adriana, Csaba Horvath, Safa Abdul-Ghani, Zoltan V. Varga, M. Saadeh Suleiman, and Naranjan S. Dhalla. 2022. "Interplay of Oxidative Stress and Necrosis-like Cell Death in Cardiac Ischemia/Reperfusion Injury: A Focus on Necroptosis" Biomedicines 10, no. 1: 127. https://doi.org/10.3390/biomedicines10010127