Gene Mapping, Genome-Wide Transcriptome Analysis, and WGCNA Reveals the Molecular Mechanism for Triggering Programmed Cell Death in Rice Mutant pir1

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
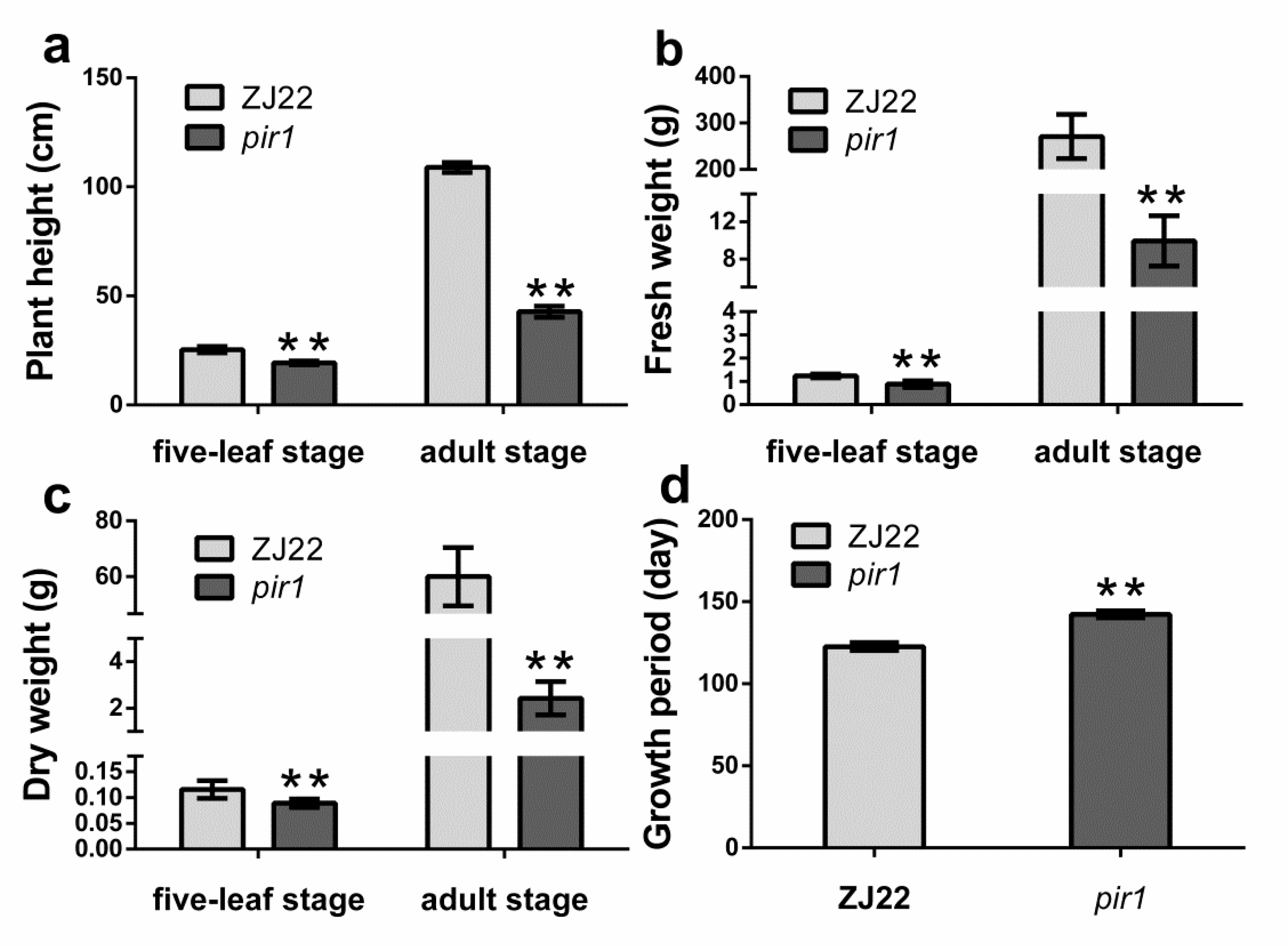
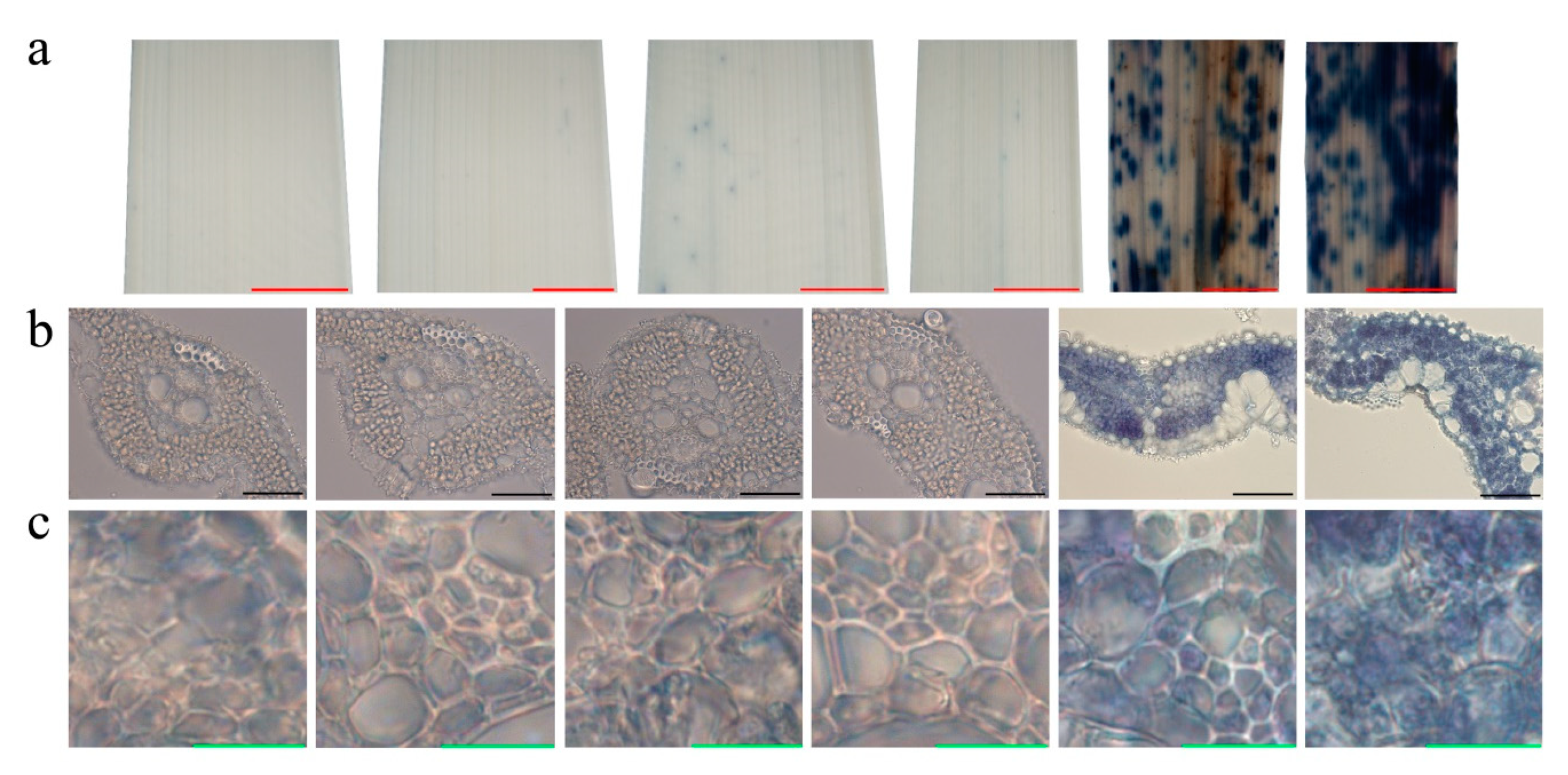
2.1. Characterization of Phenotype and Physiological Analysis
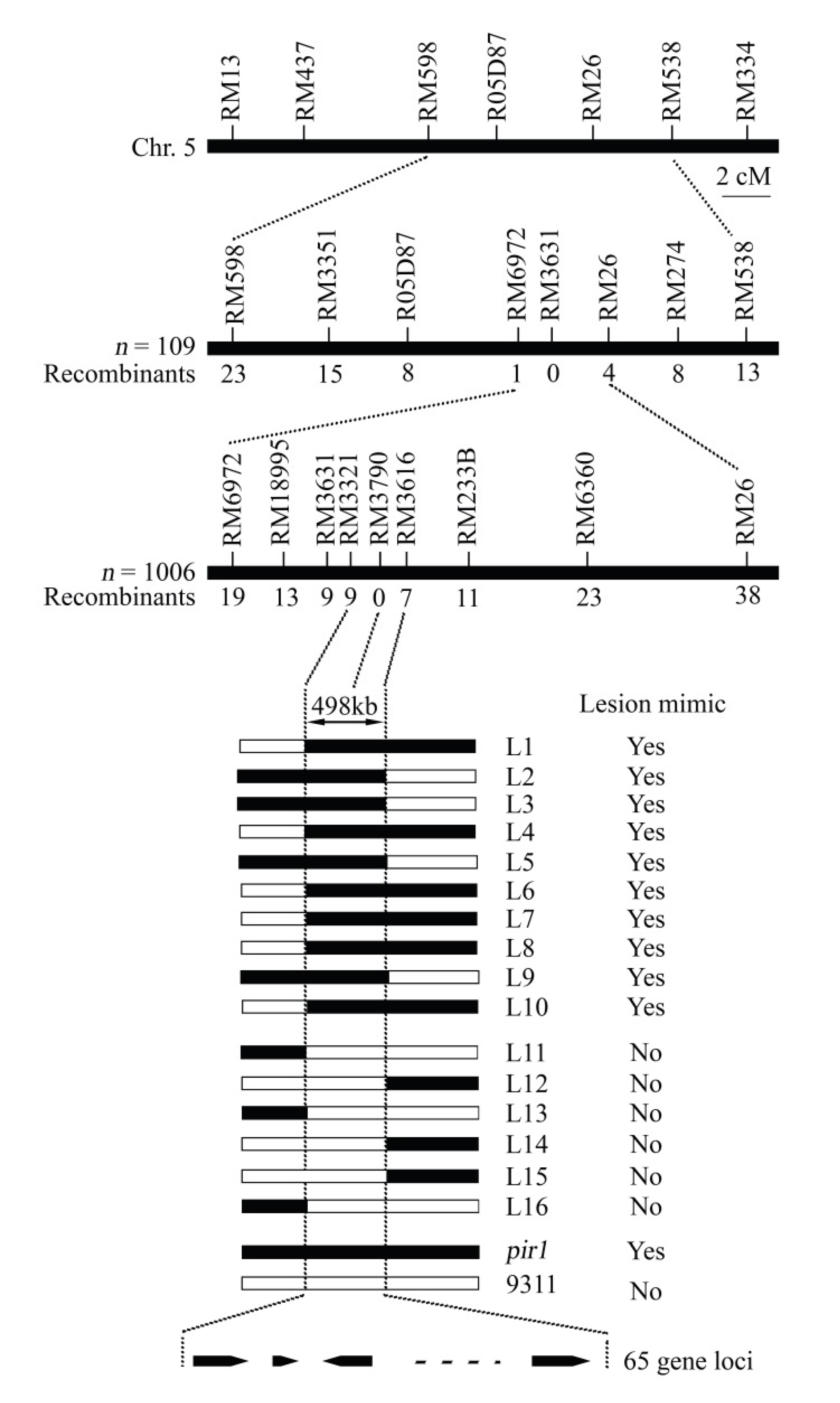
2.2. Mapping of the Gene Locus for the Rice Mutant
2.3. Evaluation of RNA-Seq Reads and Mapping Results
2.4. Sample Correlations
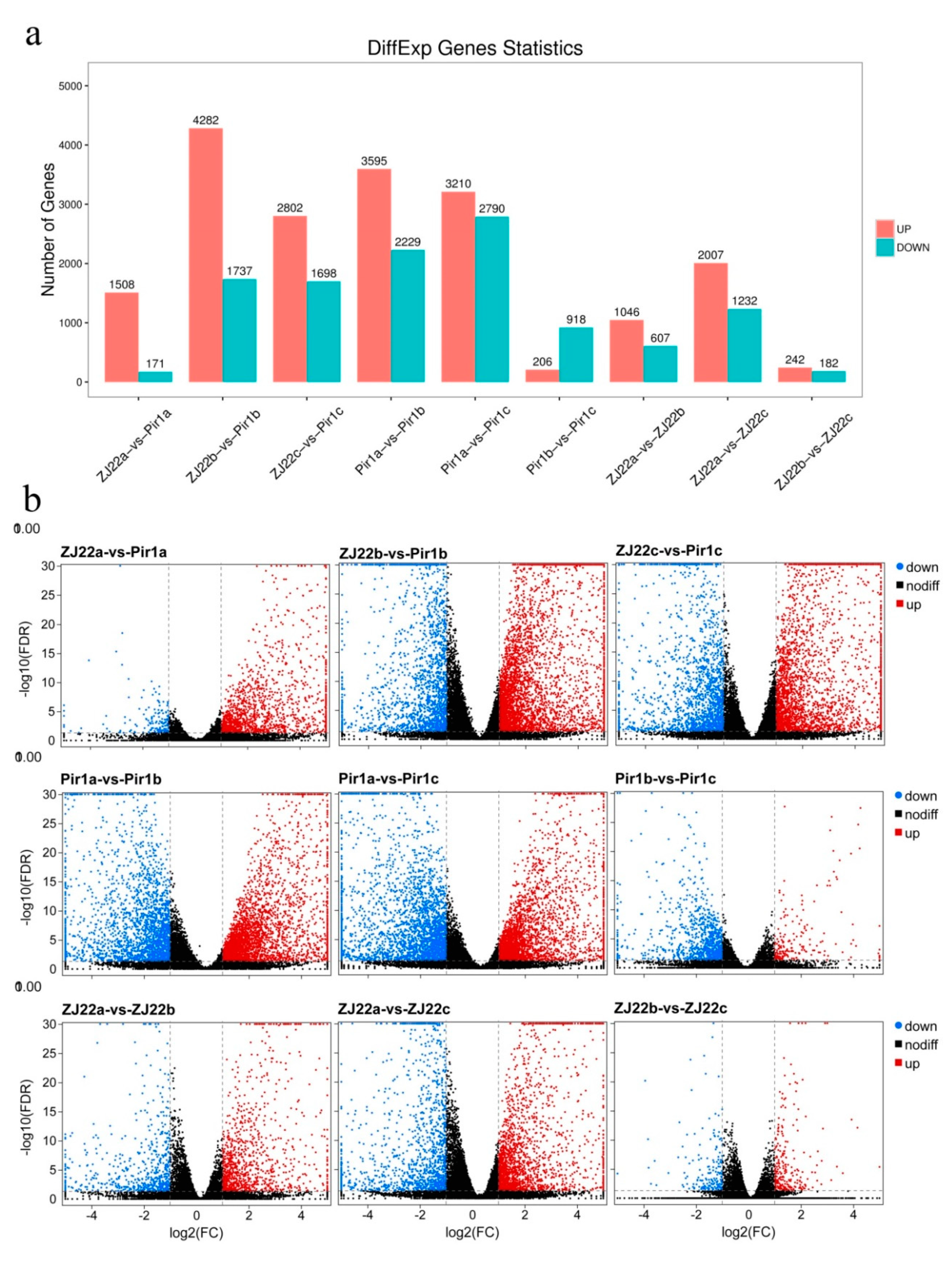
2.5. Analysis of Differentially Expressed Genes (DEGs)
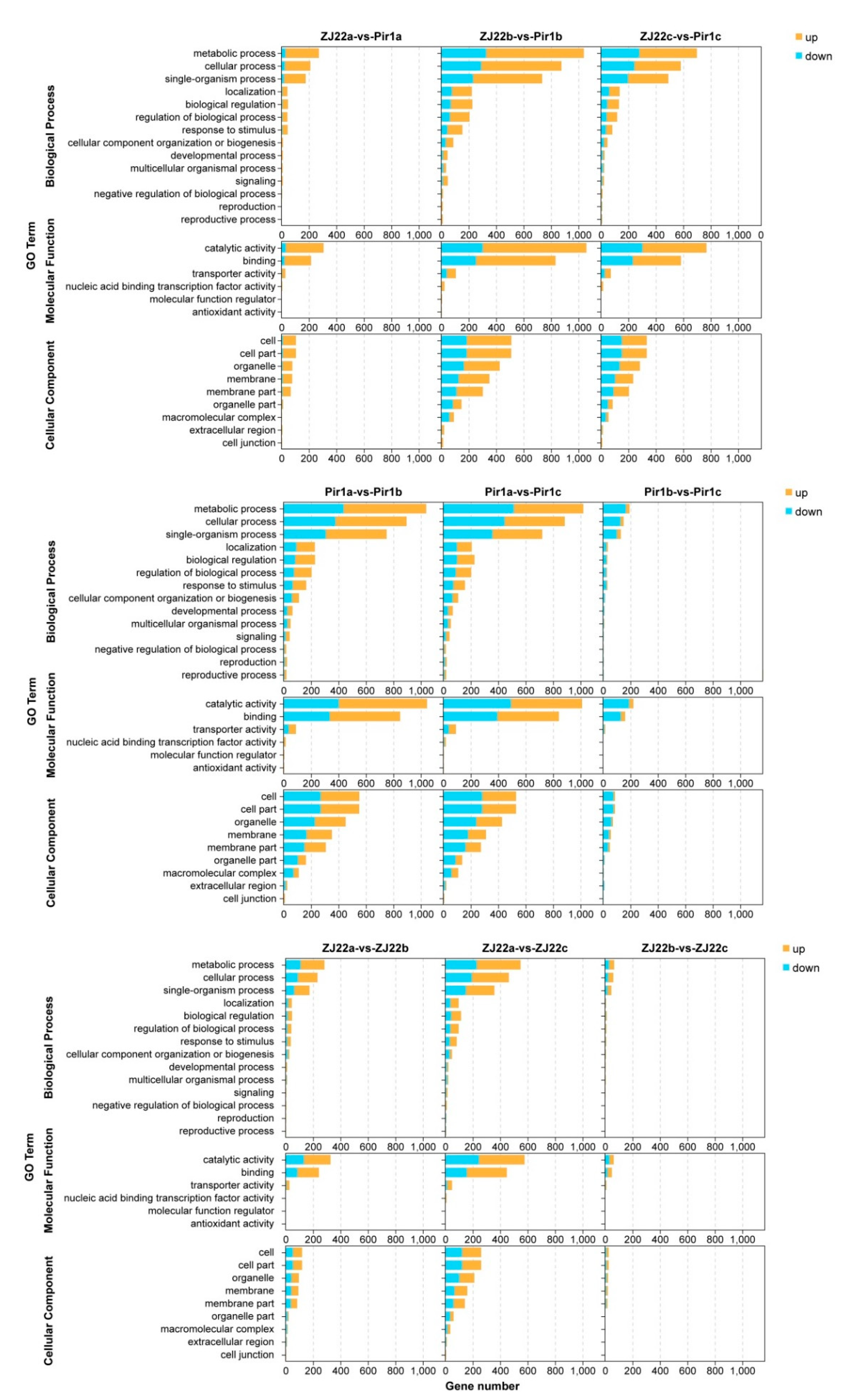
2.6. Gene Ontology (GO) Analysis of the DEGs
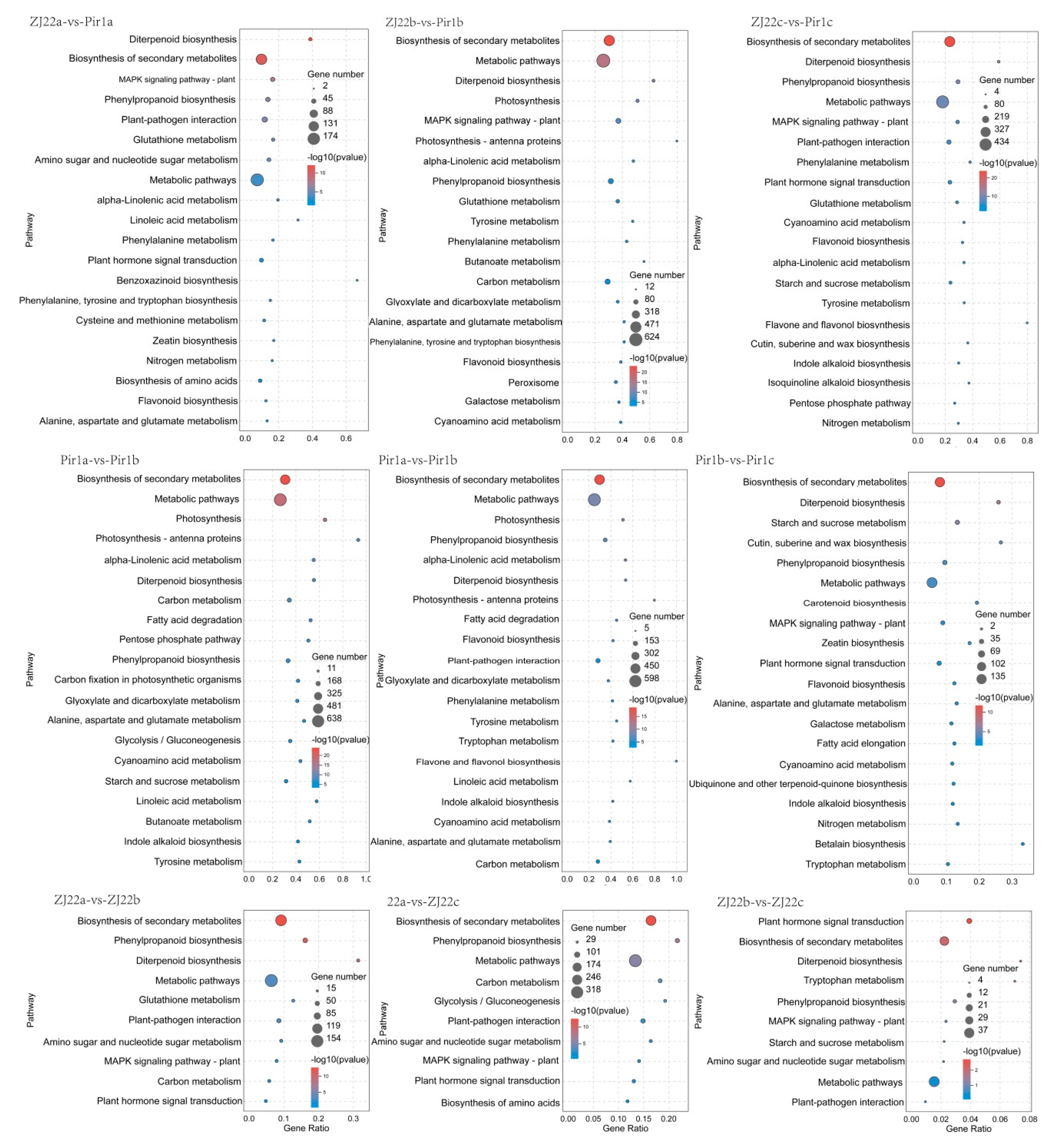
2.7. KEGG Pathway Analysis of the DEGs
2.8. Identification of Common DEGs in All Comparisons between Wild Type and Mutant Rice
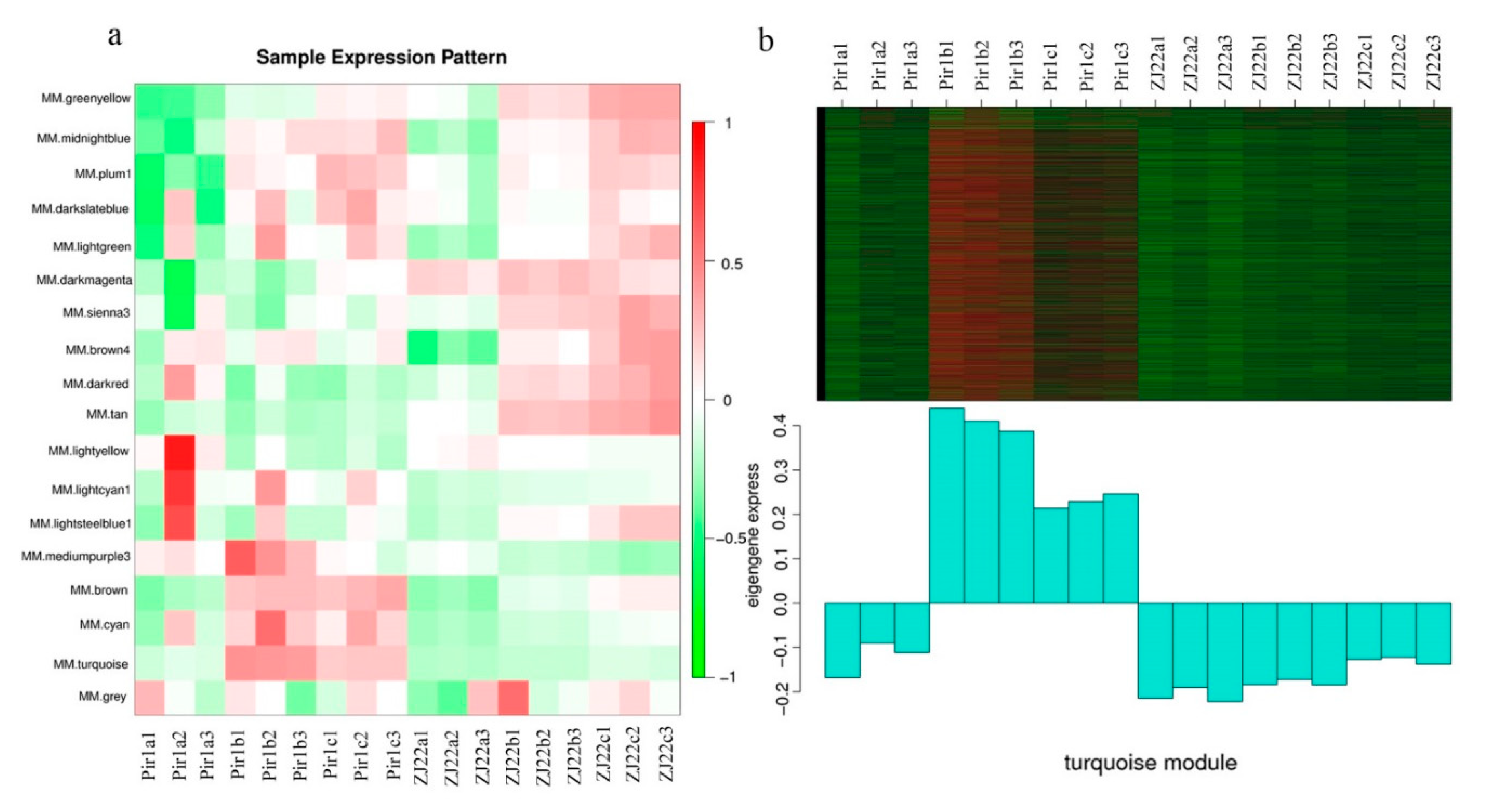
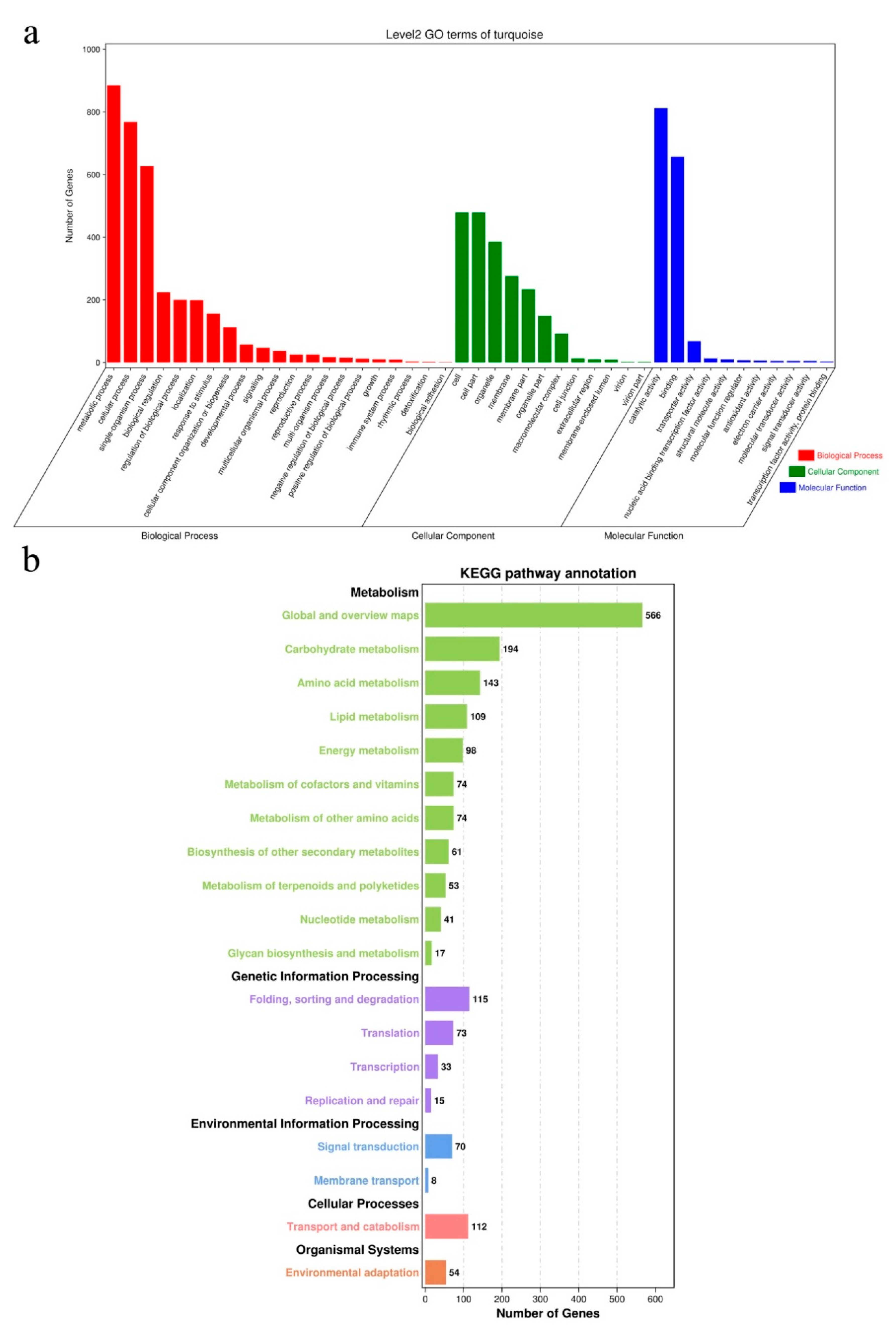
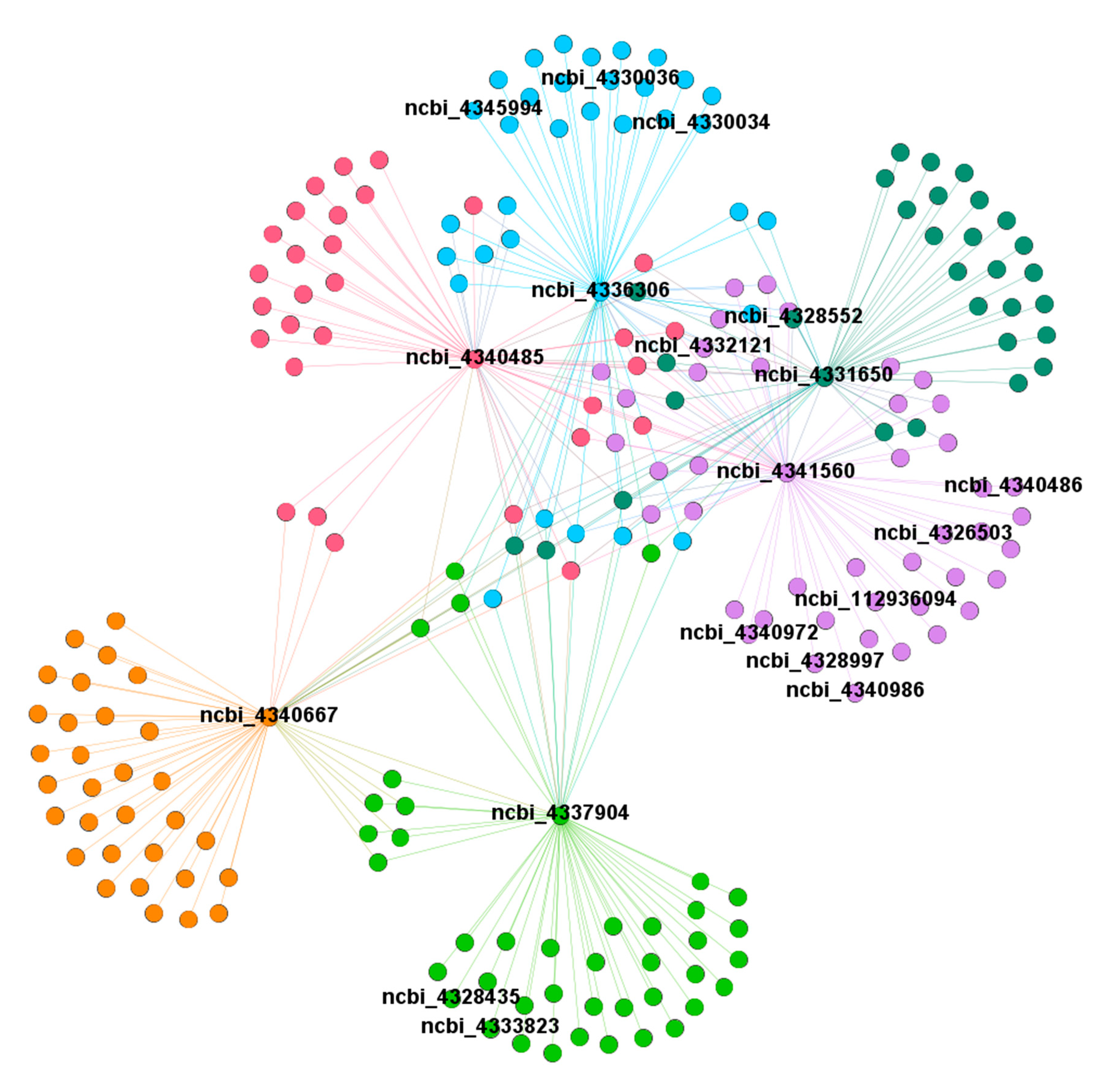
2.9. Identification of Conserved DEGs via Weighted Gene Co-Expression Network Analysis (WGCNA)
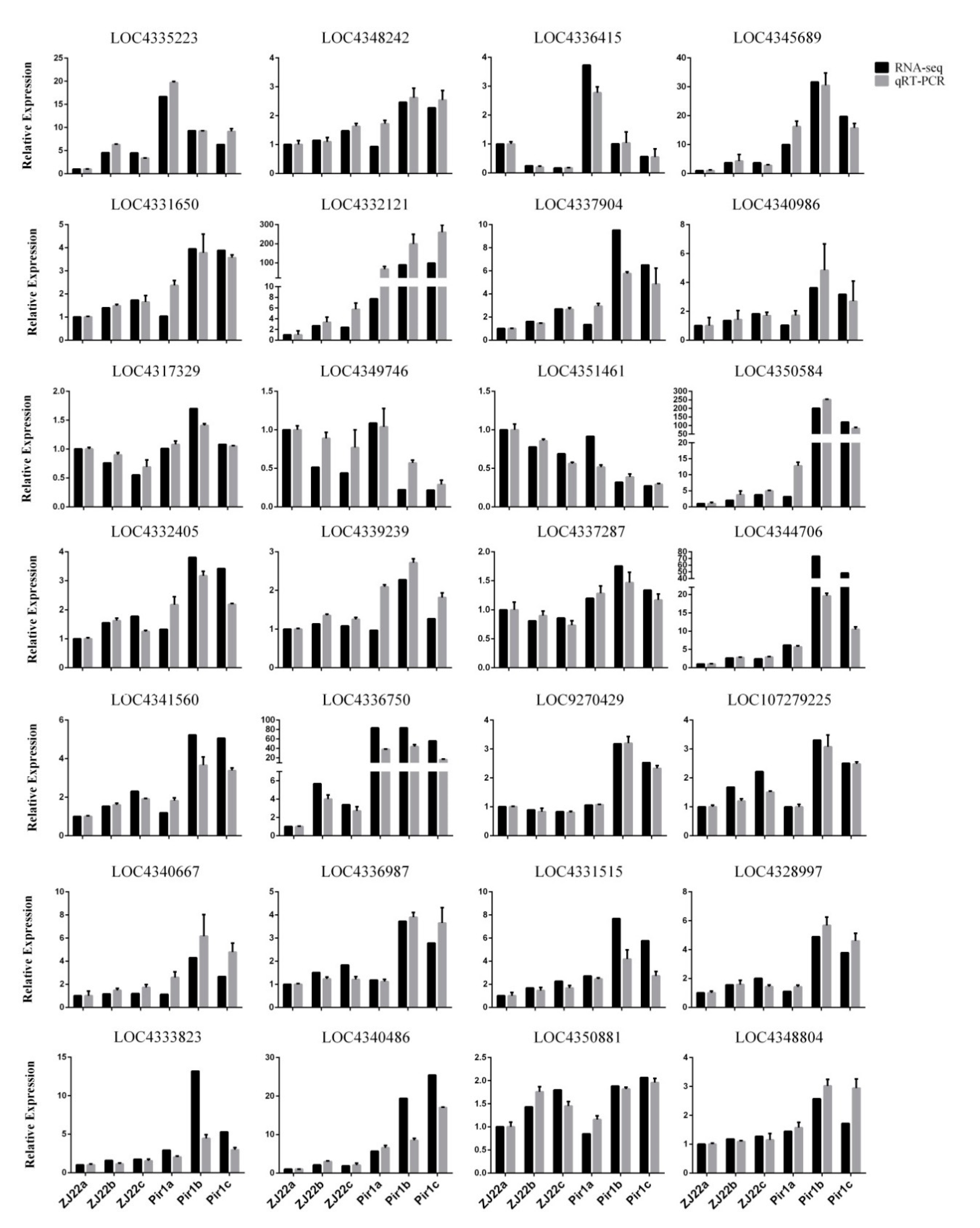
2.10. Validation of RNA-Seq Data by qRT-PCR Analysis
3. Discussion
3.1. PIR1 Gene Is a Novel Gene for Rice PCD
3.2. Co-Expression Network in Regulation of PCD
3.2.1. Involvement of Lignin Synthesis-Related Genes in Regulation of Rice PCD
3.2.2. Involvement of Plant Hormone Synthesis-Related Genes in Regulation of Rice PCD
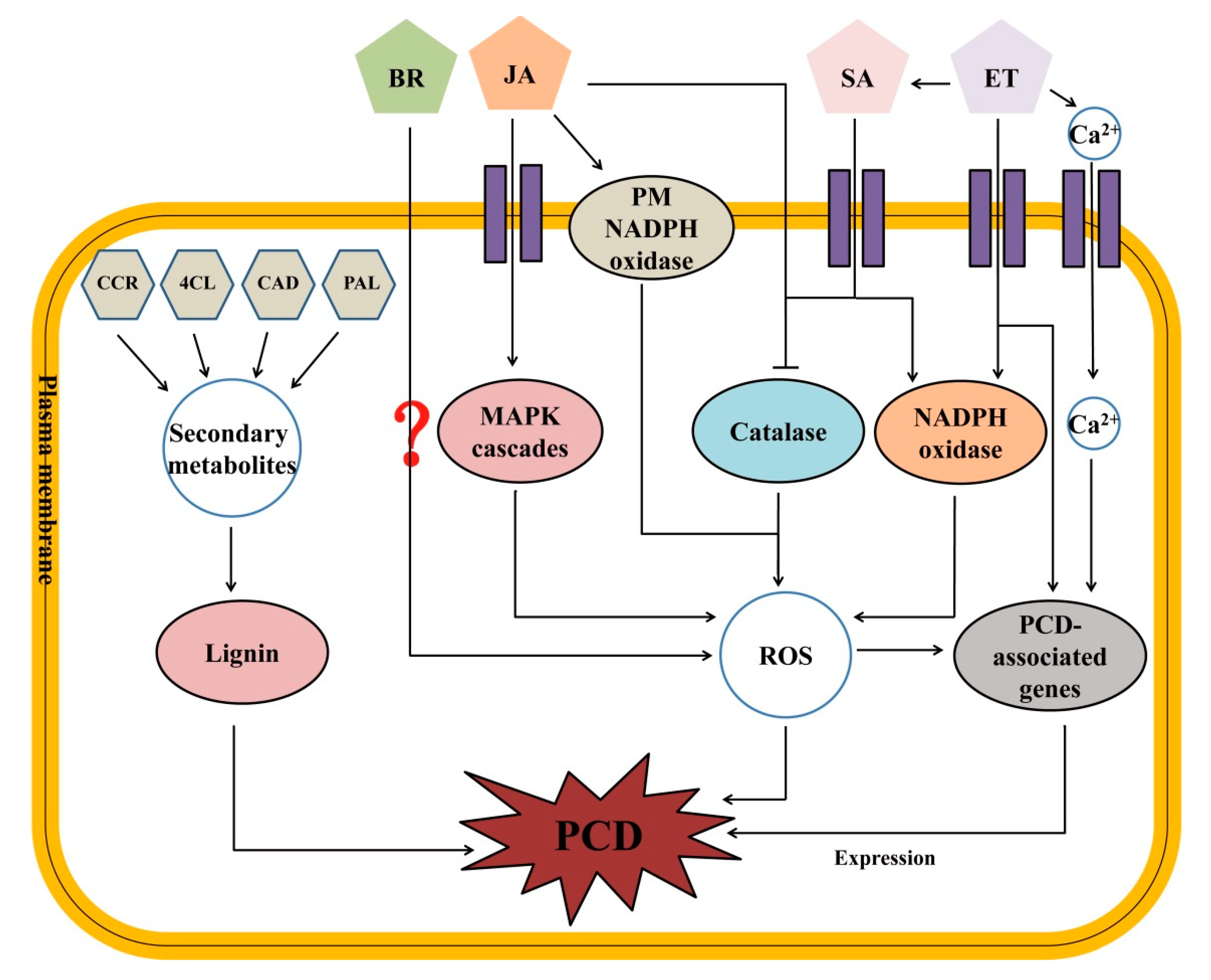
3.3. Working Model for PCD Regulation in pir1 Mutant
4. Materials and Methods
4.1. Plant Materials
4.2. Leaf Histochemical and Physiological Analyses
4.3. Mapping of the Gene Locus for the Rice Mutant
4.4. Sampling and RNA Extraction
4.5. cDNA Library Construction and Sequencing
4.6. RNA-Seq Data Analysis
4.7. Co-Expression Network Analysis for Module Construction
4.8. Validation of Gene Expression by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilchrist, D.G. Programmed cell death in plant disease: The purpose and promise of cellular suicide. Annu. Rev. Phytopathol. 1998, 36, 393–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, A.; Jf, K.; Ah, W.; Ar, C. Faculty Opinions recommendation of Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Fac. Opin. Post Publ. Peer Rev. Biomed. Lit. 2020, 26, 239–257. [Google Scholar] [CrossRef]
- Mittler, R.; Del Pozo, O.; Meisel, L.; Lam, E. Pathogen-induced programmed cell death in plants, a possible defense mechanism. Dev. Genet. 1997, 21, 279–289. [Google Scholar] [CrossRef]
- Yamamoto, R. Brassinosteroid levels increase drastically prior to morphogenesis of tracheary elements. Plant Physiol. 2001, 125, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Hirai, R.; Higaki, T.; Takenaka, Y.; Sakamoto, Y.; Hasegawa, J.; Matsunaga, S.; Demura, T.; Ohtani, M. The progression of xylem vessel cell differentiation is dependent on the activity level of VND7 in Arabidopsis thaliana. Plants 2019, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.R.; Chung, K.M.; Park, J.-H.; Oh, S.A.; Ahn, T.; Hong, S.H.; Jang, S.K.; Gil Nam, H.; Gil Nam, S.K.J. ORE9, an F-Box protein that regulates leaf senescence in arabidopsis. Plant Cell 2001, 13, 1779. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.-L.; Xue, H.-W. The MADS29 Transcription factor regulates the degradation of the nucellus and the nucellar projection during rice seed development. Plant Cell 2012, 24, 1049–1065. [Google Scholar] [CrossRef] [Green Version]
- Plackett, A.R.; Thomas, S.G.; Wilson, Z.A.; Hedden, P. Gibberellin control of stamen development: A fertile field. Trends Plant Sci. 2011, 16, 568–578. [Google Scholar] [CrossRef]
- Fendrych, M.; Van Hautegem, T.; Van Durme, M.; Olvera-Carrillo, Y.; Huysmans, M.; Karimi, M.; Lippens, S.; Guerin, C.J.; Krebs, M.; Schumacher, K.; et al. Programmed cell death controlled by ANAC033/SOMBRERO determines root cap organ size in Arabidopsis. Curr. Biol. 2014, 24, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, J.T. Programmed cell death in plant-pathogen interactions. Annu. Rev. Plant Biol. 1997, 48, 525–545. [Google Scholar] [CrossRef] [Green Version]
- Apostol, I.; Heinstein, P.F.; Low, P.S. Rapid Stimulation of an Oxidative Burst during Elicitation of Cultured Plant Cells. Plant Physiol. 1989, 90, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draper, J. Salicylate, superoxide synthesis and cell suicide in plant defence. Trends Plant Sci. 1997, 2, 162–165. [Google Scholar] [CrossRef]
- Büschges, R.; Hollricher, K.; Panstruga, R.; Simons, G.; Wolter, M.; Frijters, A.; Van Daelen, R.; Van Der Lee, T.; Diergaarde, P.; Groenendijk, J.; et al. The Barley Mlo Gene: A Novel Control Element of Plant Pathogen Resistance. Cell 1997, 88, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Rusterucci, C.; Aviv, D.H.; Holt, B.F.; Dangl, J.L.; Parker, J.E. The Disease Resistance Signaling Components EDS1 and PAD4 Are Essential Regulators of the Cell Death Pathway Controlled by LSD1 in Arabidopsis. Plant Cell 2001, 13, 2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Bordeos, A.; Madamba, M.R.S.; Baraoidan, M.; Ramos, M.; Wang, G.-L.; Leach, J.E.; Leung, H. Rice lesion mimic mutants with enhanced resistance to diseases. Mol. Genet. Genom. 2008, 279, 605–619. [Google Scholar] [CrossRef]
- Bethke, P.C.; Jones, R.L. Cell death of barley aleurone protoplasts is mediated by reactive oxygen species. Plant J. 2001, 25, 19–29. [Google Scholar]
- Nguyen, G.N.; Hailstones, D.L.; Wilkes, M.; Sutton, B.G. Drought-induced oxidative conditions in rice anthers leading to a programmed cell death and pollen abortion. J. Agron. Crop. Sci. 2009, 195, 157–164. [Google Scholar] [CrossRef]
- Monetti, E.; Kadono, T.; Tran, D.; Azzarello, E.; Arbelet-Bonnin, D.; Biligui, B.; Briand, J.; Kawano, T.; Mancuso, S.; Bouteau, F. Deciphering early events involved in hyperosmotic stress-induced programmed cell death in tobacco BY-2 cells. J. Exp. Bot. 2014, 65, 1361–1375. [Google Scholar] [CrossRef] [Green Version]
- Locato, V.; Gadaleta, C.; De Gara, L.; Romero-Puertas, M.D.C. Production of reactive species and modulation of antioxidant network in response to heat shock: A critical balance for cell fate. Plant Cell Environ. 2008, 31, 1606–1619. [Google Scholar] [CrossRef]
- Zheng, S.; Li, J.; Ma, L.; Wang, H.; Zhou, H.; Ni, E.; Jiang, D.; Liu, Z.; Zhuang, C. OsAGO2 controls ROS production and the initiation of tapetal PCD by epigenetically regulating OsHXK1 expression in rice anthers. Proc. Natl. Acad. Sci. USA 2019, 116, 7549–7558. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.H.; Jung, W.Y.; Kang, Y.H.; Kim, J.Y.; Kim, D.G.; Jeong, J.C.; Baek, D.W.; Jin, J.B.; Lee, J.Y.; O Kim, M.; et al. AtBAG6, a novel calmodulin-binding protein, induces programmed cell death in yeast and plants. Cell Death Differ. 2005, 13, 84–95. [Google Scholar] [CrossRef]
- Wang, W.; Yang, X.; Tangchaiburana, S.; Ndeh, R.; Markham, J.E.; Tsegaye, Y.; Dunn, T.M.; Wang, G.-L.; Bellizzi, M.; Parsons, J.F.; et al. An Inositolphosphorylceramide Synthase Is Involved in Regulation of Plant Programmed Cell Death Associated with Defense in Arabidopsis. Plant Cell 2008, 20, 3163–3179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajita, S.; Katayama, Y.; Omori, S. Alterations in the Biosynthesis of Lignin in Transgenic Plants with Chimeric Genes for 4-Coumarate: Coenzyme A Ligase. Plant Cell Physiol. 1996, 37, 957–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goujon, T.; Ferret, V.; Mila, I.; Pollet, B.; Ruel, K.; Burlat, V.; Joseleau, J.-P.; Barrière, Y.; Lapierre, C.; Jouanin, L. Down-regulation of the AtCCR1 gene in Arabidopsis thaliana: Effects on phenotype, lignins and cell wall degradability. Planta 2003, 217, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Aya, K.; Kondo, M.; Okuno, A.; Morinaka, Y.; Matsuoka, M. OsCAD2 is the major CAD gene responsible for monolignol biosynthesis in rice culm. Plant Cell Rep. 2012, 31, 91–101. [Google Scholar] [CrossRef]
- Zhang, K.; Qian, Q.; Huang, Z.; Wang, Y.; Li, M.; Hong, L.; Zeng, D.; Gu, M.; Chu, C.; Cheng, Z. Gold hull and internode2 encodes a primarily multifunctional cinnamyl-alcohol dehydrogenase in rice. Plant Physiol. 2006, 140, 972–983. [Google Scholar] [CrossRef] [Green Version]
- Chabannes, M.; Barakate, A.; Lapierre, C.; Marita, J.M.; Ralph, J.; Pean, M.; Danoun, S.; Halpin, C.; Grima-Pettenati, J.; Boudet, A.M. Strong decrease in lignin content without significant alteration of plant development is induced by simultaneous down-regulation of cinnamoyl CoA reductase (CCR) and cinnamyl alcohol dehydrogenase (CAD) in tobacco plants. Plant J. 2001, 28, 257–270. [Google Scholar] [CrossRef]
- Balkany, T.; Telischi, F.F. How I do it: Otology and neurotology: Fixation of the electrode cable during cochlear implantation: The split bridge technique. Laryngoscope 1995, 105, 217–218. [Google Scholar] [CrossRef]
- Liu, H.; Guo, Z.; Gu, F.; Ke, S.; Sun, D.; Dong, S.; Liu, W.; Huang, M.; Xiao, W.; Yang, G.; et al. 4-Coumarate-CoA Ligase-Like Gene OsAAE3 Negatively Mediates the Rice Blast Resistance, Floret Development and Lignin Biosynthesis. Front. Plant Sci. 2017, 7, 290. [Google Scholar] [CrossRef] [Green Version]
- De León, I.P.; Schmelz, E.A.; Gaggero, C.; Castro, A.; Álvarez, A.; Montesano, M. Physcomitrella patens activates reinforcement of the cell wall, programmed cell death and accumulation of evolutionary conserved defence signals, such as salicylic acid and 12-oxo-phytodienoic acid, but not jasmonic acid, upon Botrytis cinerea infection. Mol. Plant Pathol. 2012, 13, 960–974. [Google Scholar] [CrossRef]
- Prats, E.; Martínez, F.; Rojas-Molina, M.M.; Rubiales, D. Differential Effects of Phenylalanine Ammonia Lyase, Cinnamyl Alcohol Dehydrogenase, and Energetic Metabolism Inhibition on Resistance of Appropriate Host and Nonhost Cereal–Rust Interactions. Phytopathology 2007, 97, 1578–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Bowling, S.A.; Gordon, A.S.; Dong, X. Characterization of an Arabidopsis Mutant That Is Nonresponsive to Inducers of Systemic Acquired Resistance. Plant Cell 1994, 6, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Trobacher, C.P.; Senatore, A.; Holley, C.; Greenwood, J.S. Induction of a ricinosomal-protease and programmed cell death in tomato endosperm by gibberellic acid. Planta 2012, 237, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Mur, L.A.J.; Prats, E.; Pierre, S.; Hall, M.A.; Hebelstrup, K.H. Integrating nitric oxide into salicylic acid and jasmonic acid/ ethylene plant defense pathways. Front. Plant Sci. 2013, 4, 215. [Google Scholar] [CrossRef] [Green Version]
- Rantong, G.; Evans, R.C.; Gunawardena, A.H.L.A.N. Lace plant ethylene receptors, AmERS1a and AmERS1c, regulate ethylene-induced programmed cell death during leaf morphogenesis. Plant Mol. Biol. 2015, 89, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Grimes, H.D.; Koetje, D.S.; Franceschi, V.R. Expression, Activity, and Cellular Accumulation of Methyl Jasmonate-Responsive Lipoxygenase in Soybean Seedlings. Plant Physiol. 1992, 100, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Dhakarey, R.; Peethambaran, P.K.; Riemann, M. Functional Analysis of Jasmonates in Rice through Mutant Approaches. Plants 2016, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Song, W.C.; Funk, C.D.; Brash, A.R. Molecular cloning of an allene oxide synthase: A cytochrome P450 specialized for the metabolism of fatty acid hydroperoxides. Proc. Natl. Acad. Sci. USA 1993, 90, 8519–8523. [Google Scholar] [CrossRef] [Green Version]
- Laudert, D.; Weiler, E.W. Allene oxide synthase: A major control point in Arabidopsis thaliana octadecanoid signalling. Plant J. 1998, 15, 675–684. [Google Scholar] [CrossRef]
- Ahmad, P.; Rasool, S.; Gul, A.; Sheikh, S.A.; Akram, N.A.; Ashraf, M.; Kazi, A.M.; Gucel, S. Jasmonates: Multifunctional Roles in Stress Tolerance. Front. Plant Sci. 2016, 7, 813. [Google Scholar] [CrossRef] [Green Version]
- Sagi, M.; Davydov, O.; Orazova, S.; Yesbergenova, Z.; Ophir, R.; Stratmann, J.W.; Fluhr, R. Plant Respiratory Burst Oxidase Homologs Impinge on Wound Responsiveness and Development in Lycopersicon esculentum. Plant Cell 2004, 16, 616–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y. Systemic induction of H2O2 in pea seedlings pretreated by wounding and exogenous jasmonic acid. Sci. China Ser. C Life Sci. 2005, 48, 202. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.V.; Lee, H.-I.; Creelman, R.A.; Mullet, J.E.; Davis, K. Jasmonic Acid Signaling Modulates Ozone-Induced Hypersensitive Cell Death. Plant Cell 2000, 12, 1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, P.M.; Kazan, K.; Wilson, I.; Anderson, J.P.; Richmond, T.; Somerville, S.C.; Manners, J.M. Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proc. Natl. Acad. Sci. USA 2000, 97, 11655–11660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, D.; Yang, H.; Zhang, S. Cell Death Mediated by MAPK Is Associated with Hydrogen Peroxide Production in Arabidopsis. J. Biol. Chem. 2002, 277, 559–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Xie, D.; Cao, J.; Xia, X.; Shi, K.; Zhou, Y.; Zhou, J.; Foyer, C.H.; Yu, J. Brassinosteroid-mediated reactive oxygen species are essential for tapetum degradation and pollen fertility in tomato. Plant J. 2020, 102, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.-J.; Wang, Y.-J.; Zhou, Y.-H.; Tao, Y.; Mao, W.-H.; Shi, K.; Asami, T.; Chen, Z.; Yu, J.-Q. Reactive Oxygen Species Are Involved in Brassinosteroid-Induced Stress Tolerance in Cucumber. Plant Physiol. 2009, 150, 801–814. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.I.; Cho, H.J.; Jung, J.H.; Yoshimoto, K.; Shirasu, K.; Park, O.K. The Rab GTPase RabG3b functions in autophagy and contributes to tracheary element differentiation in Arabidopsis. Plant J. 2010, 64, 151–164. [Google Scholar] [CrossRef]
- Drew, M.C.; He, C.-J.; Morgan, P.W. Programmed cell death and aerenchyma formation in roots. Trends Plant Sci. 2000, 5, 123–127. [Google Scholar] [CrossRef]
- Huarte, H.R.; Puglia, G.D.; Prjibelski, A.D.; Raccuia, S.A. Seed Transcriptome Annotation Reveals Enhanced Expression of Genes Related to ROS Homeostasis and Ethylene Metabolism at Alternating Temperatures in Wild Cardoon. Plants 2020, 9, 1225. [Google Scholar] [CrossRef]
- De Jong, A.J.; Yakimova, E.T.; Kapchina, V.M.; Woltering, E.J. A critical role for ethylene in hydrogen peroxide release during programmed cell death in tomato suspension cells. Planta 2002, 214, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Yakimova, E.; Kapchina-Toteva, V.; Laarhoven, L.-J.; Harren, F.; Witkowska, E.W.I. Involvement of ethylene and lipid signalling in cadmium-induced programmed cell death in tomato suspension cells. Plant Physiol. Biochem. 2006, 44, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Moeder, W.; Barry, C.S.; Tauriainen, A.A.; Betz, C.; Tuomainen, J.; Utriainen, M.; Grierson, D.; Sandermann, H.; Langebartels, C.; Kangasjärvi, J. Ethylene Synthesis Regulated by Biphasic Induction of 1-Aminocyclopropane-1-Carboxylic Acid Synthase and 1-Aminocyclopropane-1-Carboxylic Acid Oxidase Genes Is Required for Hydrogen Peroxide Accumulation and Cell Death in Ozone-Exposed Tomato. Plant Physiol. 2002, 130, 1918–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trobacher, C.P. Ethylene and programmed cell death in plants. Botany Botanique 2009, 87, 757–769. [Google Scholar] [CrossRef]
- Kende, H. Ethylene Biosynthesis. Annu. Rev. Plant Biol. 1993, 44, 283–307. [Google Scholar] [CrossRef]
- Zhao, M.-G.; Tian, Q.-Y.; Zhang, W.-H. Ethylene activates a plasma membrane Ca2+-permeable channel in tobacco suspension cells. New Phytol. 2007, 174, 507–515. [Google Scholar] [CrossRef]
- Domínguez, F.; Cejudo, F.J. Identification of a nuclear-localized nuclease from wheat cells undergoing programmed cell death that is able to trigger DNA fragmentation and apoptotic morphology on nuclei from human cells. Biochem. J. 2006, 397, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Nutt, L.K.; Chandra, J.; Pataer, A.; Fang, B.; Roth, J.A.; Swisher, S.G.; O’Neil, R.G.; McConkey, D.J. Bax-mediated Ca2+Mobilization Promotes CytochromecRelease during Apoptosis. J. Biol. Chem. 2002, 277, 20301–20308. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, Y.; Hao, R.; Cao, Y.; Shan, X.; Jing, Y. Nitric Oxide Enhances Cytotoxicity of Lead by Modulating the Generation of Reactive Oxygen Species and Is Involved in the Regulation of Pb2+ and Ca2+ Fluxes in Tobacco BY-2 Cells. Plants 2019, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zheng, Z.; Huang, J.; Lai, Z.; Fan, B. Biosynthesis of salicylic acid in plants. Plant Signal. Behav. 2009, 4, 493–496. [Google Scholar] [CrossRef]
- Alvarez, M.E. Salicylic acid in the machinery of hypersensitive cell death and disease resistance. Plant Mol. Biol. 2000, 44, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Maruri-López, I.; Aviles-Baltazar, N.Y.; Buchala, A.; Serrano, M. Intra and Extracellular Journey of the Phytohormone Salicylic Acid. Front. Plant Sci. 2019, 10, 423. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.W.; Heckathorn, S.A. Mitochondrial Adaptations to NaCl. Complex I Is Protected by Anti-Oxidants and Small Heat Shock Proteins, Whereas Complex II Is Protected by Proline and Betaine. Plant Physiol. 2001, 126, 1266–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Klessig, D.F. Salicylic Acid Activates a 48-kD MAP Kinase in Tobacco. Plant Cell 1997, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.V.; Davis, K.R. Ozone-induced cell death occurs via two distinct mechanisms in Arabidopsis: The role of salicylic acid. Plant J. 1999, 17, 603–614. [Google Scholar] [CrossRef]
- Durner, J.; Klessig, D.F. Inhibition of ascorbate peroxidase by salicylic acid and 2,6-dichloroisonicotinic acid, two inducers of plant defense responses. Proc. Natl. Acad. Sci. USA 1995, 92, 11312–11316. [Google Scholar] [CrossRef] [Green Version]
- Overmyer, K.; Brosché, M.; Kangasjärvi, J. Reactive oxygen species and hormonal control of cell death. Trends Plant Sci. 2003, 8, 335–342. [Google Scholar] [CrossRef]
- Bowling, S.A.; Clarke, J.D.; Liu, Y.; Klessig, D.F.; Dong, X. The cpr5 Mutant of Arabidopsis Expresses Both NPR1-Dependent and NPR1-Independent Resistance. Plant Cell 1997, 9, 1573. [Google Scholar] [CrossRef] [Green Version]
- Thordal-Christensen, H.; Zhang, Z.; Wei, Y.; Collinge, D.B. Subcellular localization of H2O2 in plants. H2O2 accumulation in papillae and hypersensitive response during the barley-powdery mildew interaction. Plant J. 1997, 11, 1187–1194. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef] [Green Version]
- Camon, E. The Gene Ontology Annotation (GOA) Database: Sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res. 2004, 32, 262D–266D. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Singh, V.K.; Mangalam, A.; Dwivedi, S.; Naik, S. Primer Premier: Program for Design of Degenerate Primers from a Protein Sequence. BioTechniques 1998, 24, 318–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Mei, Q.; Liang, W.; Sun, J.; Wang, X.; Zhou, J.; Wang, J.; Zhou, Y.; Zheng, B.; Yang, Y.; et al. Gene Mapping, Genome-Wide Transcriptome Analysis, and WGCNA Reveals the Molecular Mechanism for Triggering Programmed Cell Death in Rice Mutant pir1. Plants 2020, 9, 1607. https://doi.org/10.3390/plants9111607
Chen X, Mei Q, Liang W, Sun J, Wang X, Zhou J, Wang J, Zhou Y, Zheng B, Yang Y, et al. Gene Mapping, Genome-Wide Transcriptome Analysis, and WGCNA Reveals the Molecular Mechanism for Triggering Programmed Cell Death in Rice Mutant pir1. Plants. 2020; 9(11):1607. https://doi.org/10.3390/plants9111607
Chicago/Turabian StyleChen, Xinyu, Qiong Mei, Weifang Liang, Jia Sun, Xuming Wang, Jie Zhou, Junmin Wang, Yuhang Zhou, Bingsong Zheng, Yong Yang, and et al. 2020. "Gene Mapping, Genome-Wide Transcriptome Analysis, and WGCNA Reveals the Molecular Mechanism for Triggering Programmed Cell Death in Rice Mutant pir1" Plants 9, no. 11: 1607. https://doi.org/10.3390/plants9111607