
Population Structure and Genetic Diversity Analyses Provide New Insight into the Endemic Species Aster spathulifolius Maxim. and Its Evolutionary History



Abstract
:1. Introduction
2. Results
2.1. Analysis of Genetic Diversity
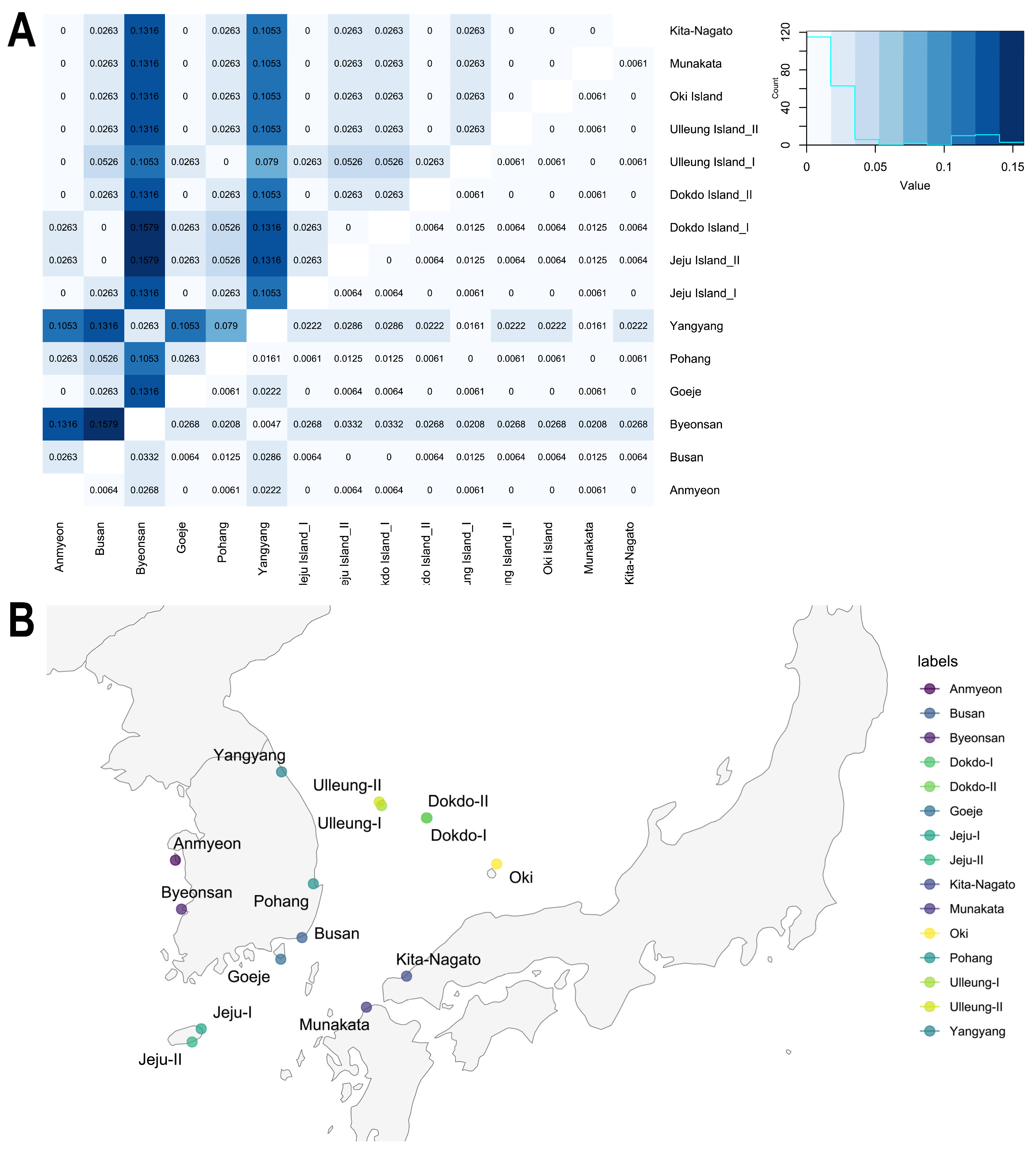
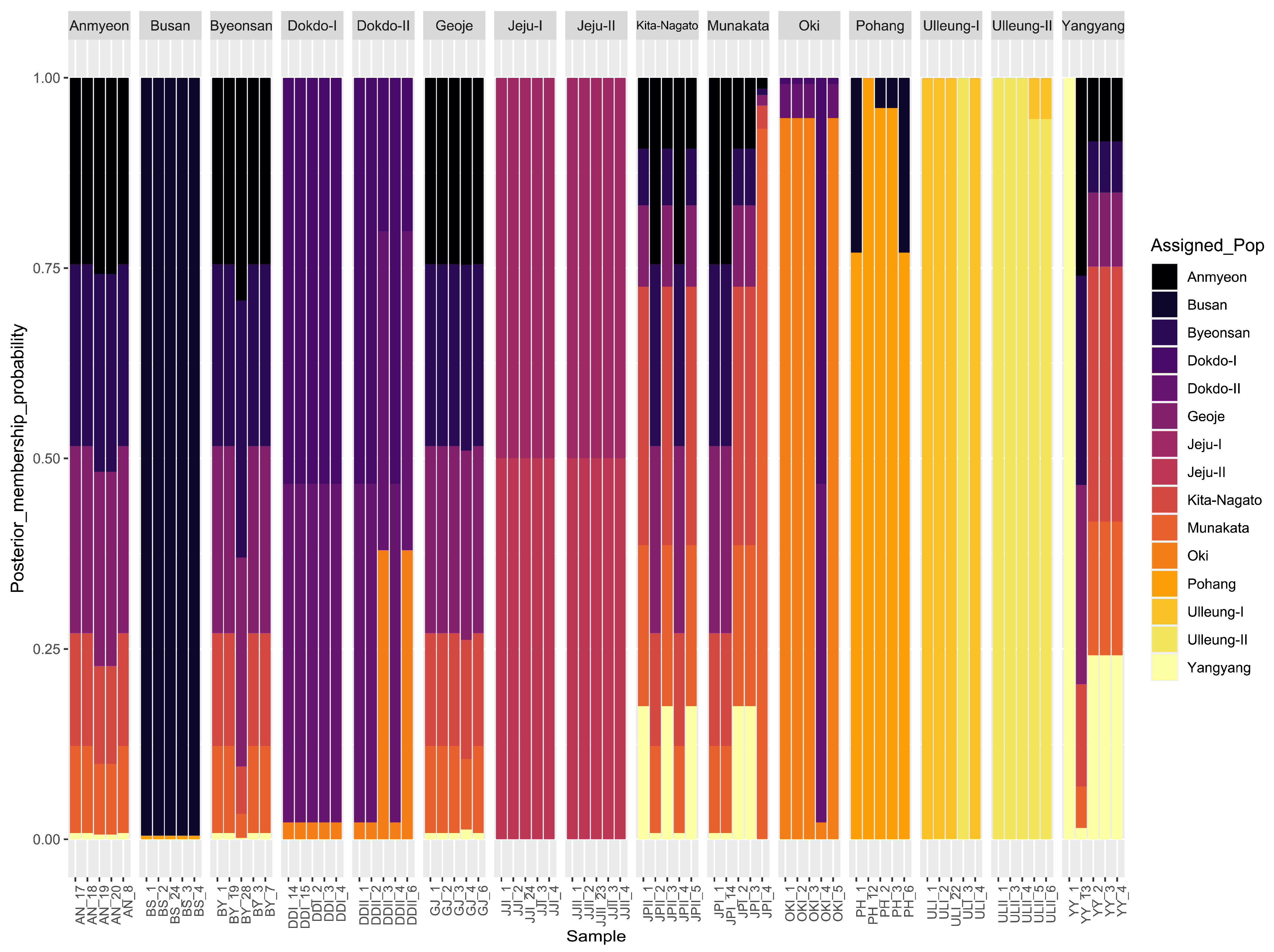
2.2. Genetic Differentiation and Genetic Flow
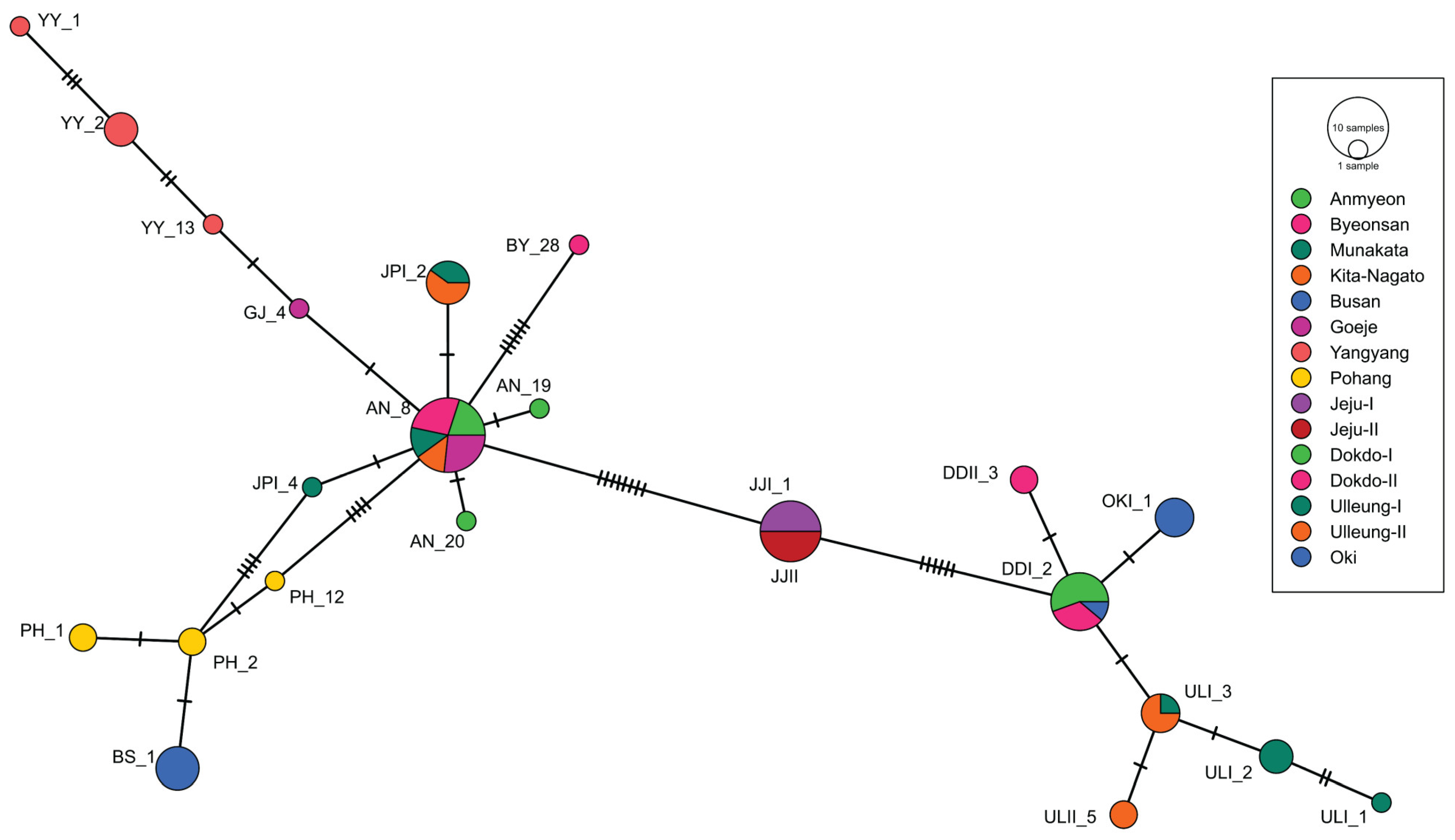
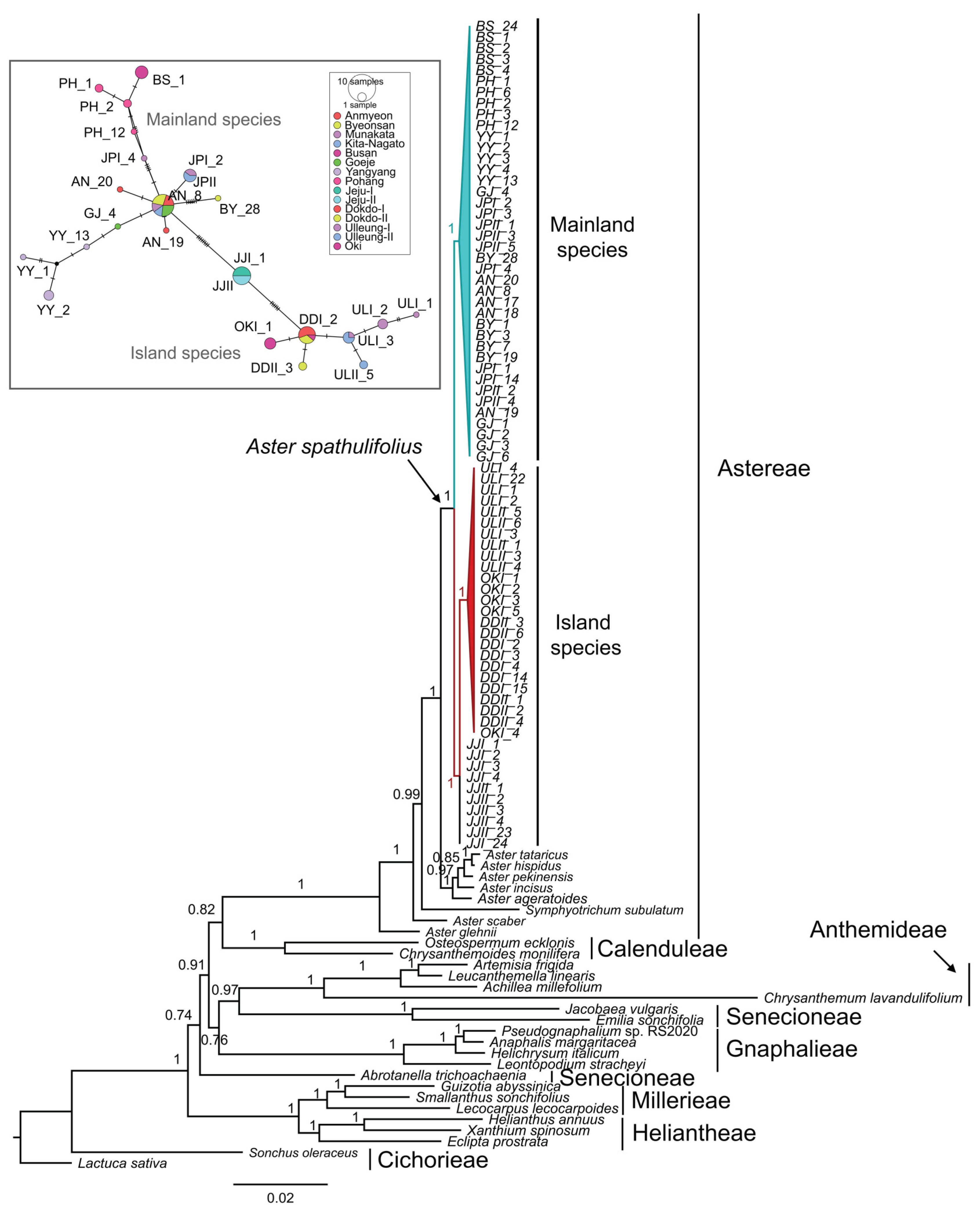
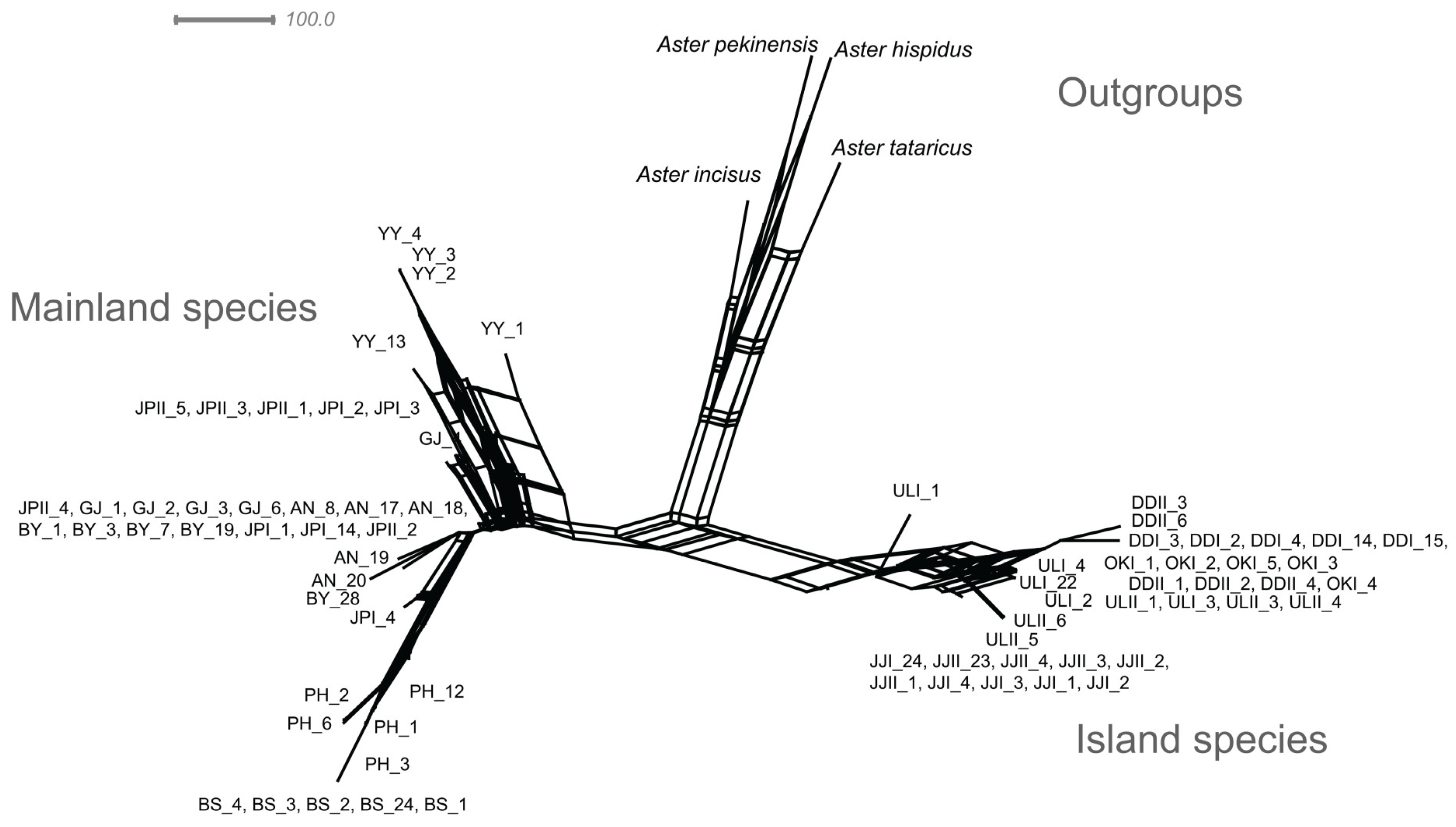
2.3. Network Analysis
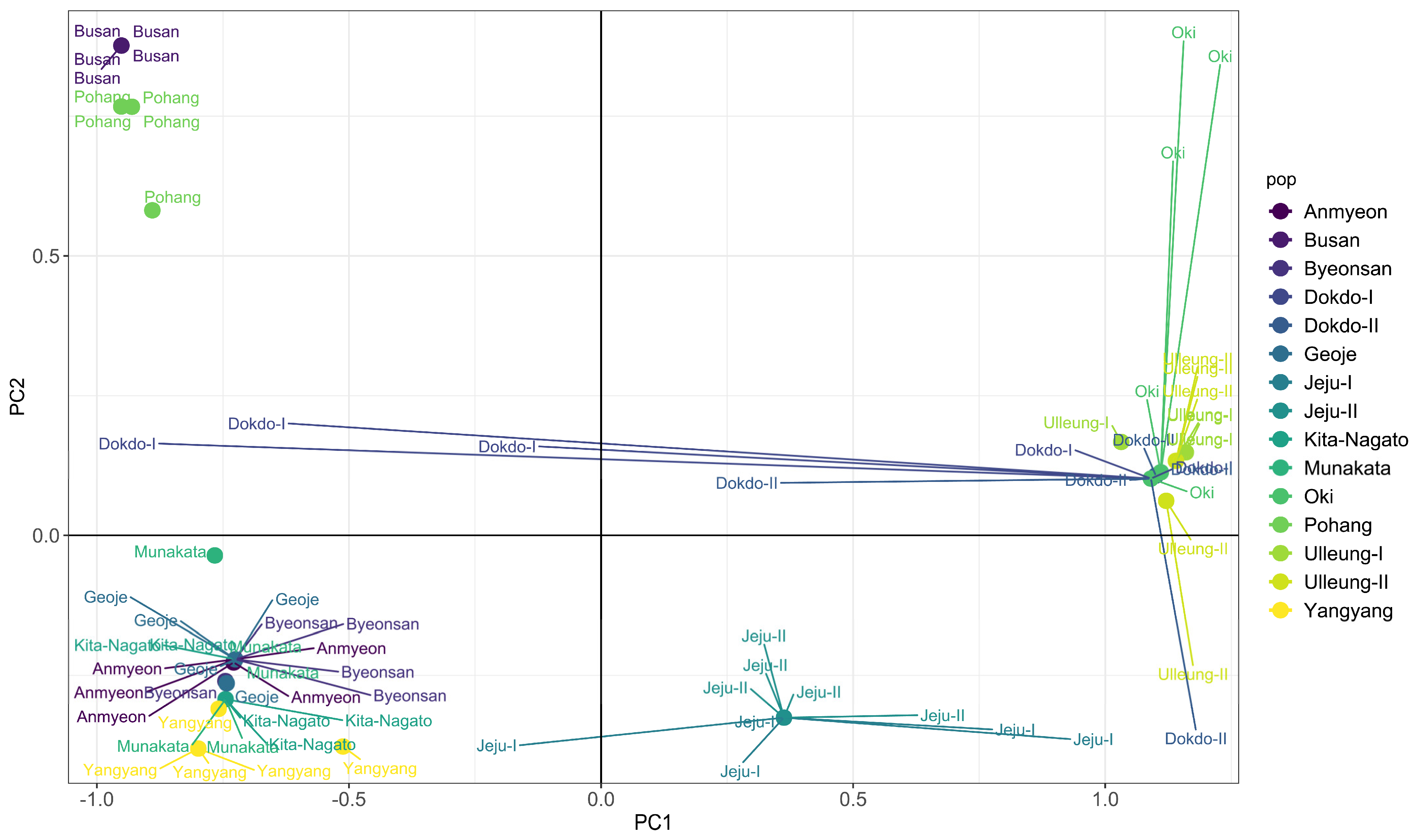
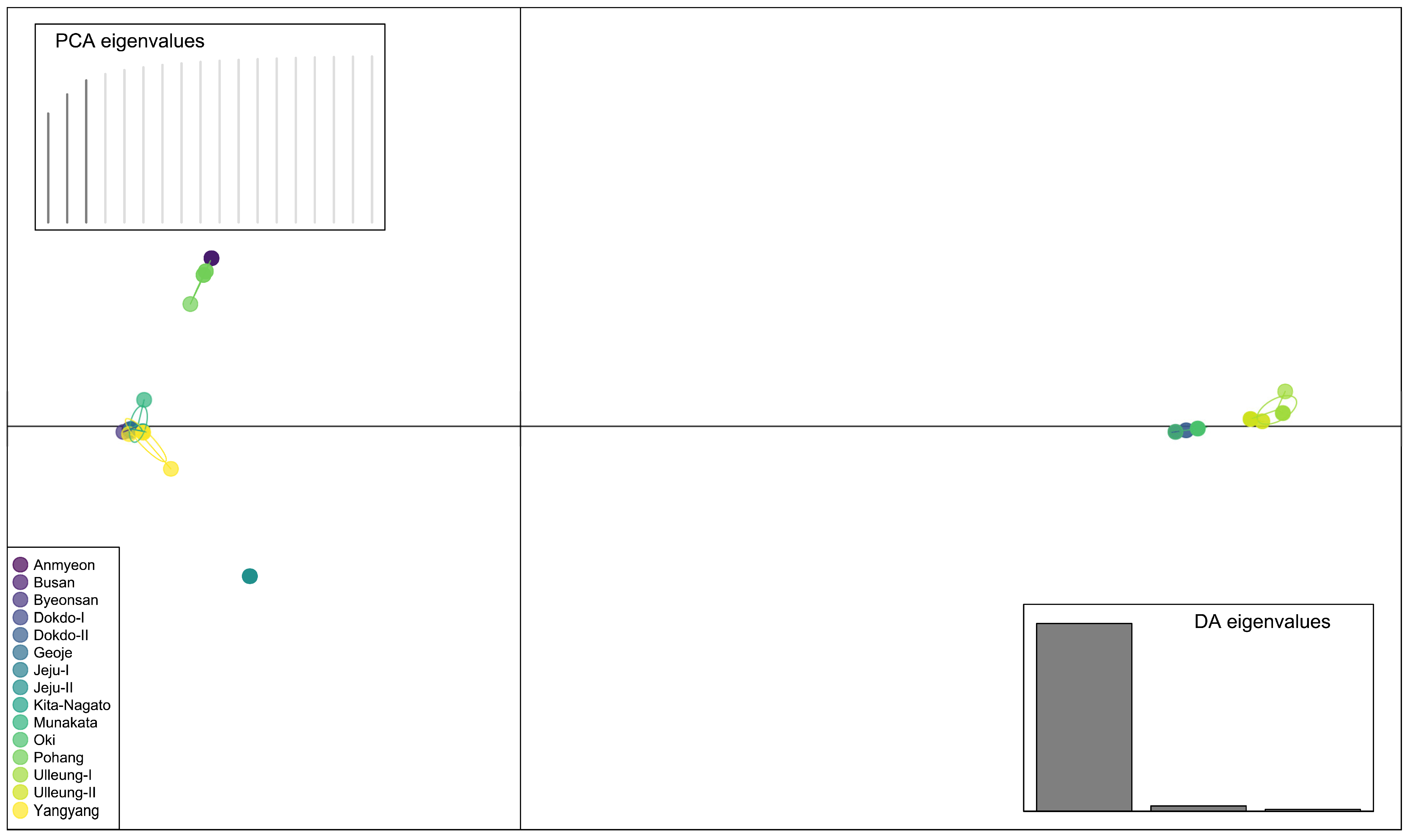
2.4. Principal Component Analysis (PCA) and Discriminant Analysis of Principal Components (DAPC)
2.5. Phylogenetic Analysis
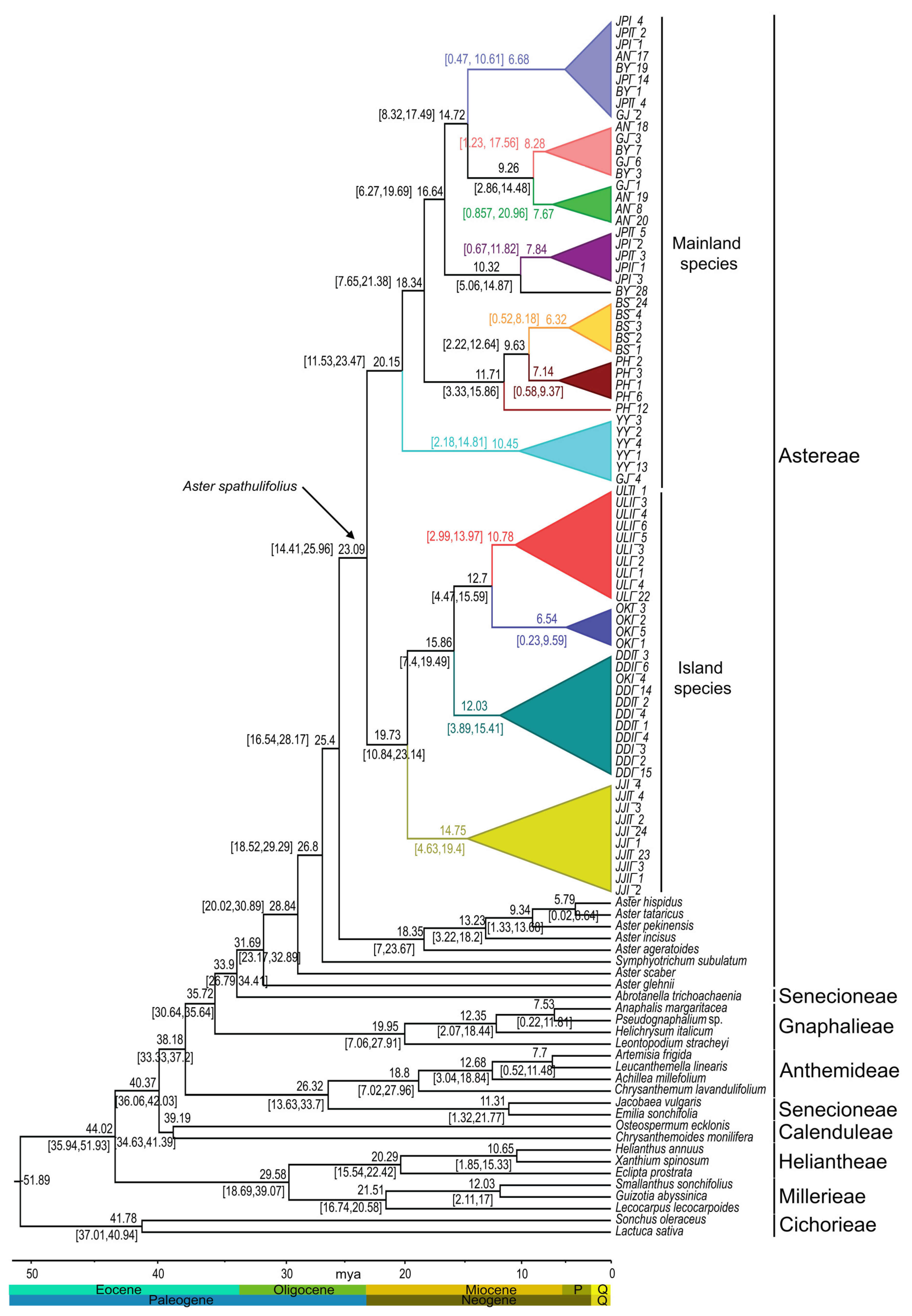
2.6. Estimation of Divergence Time
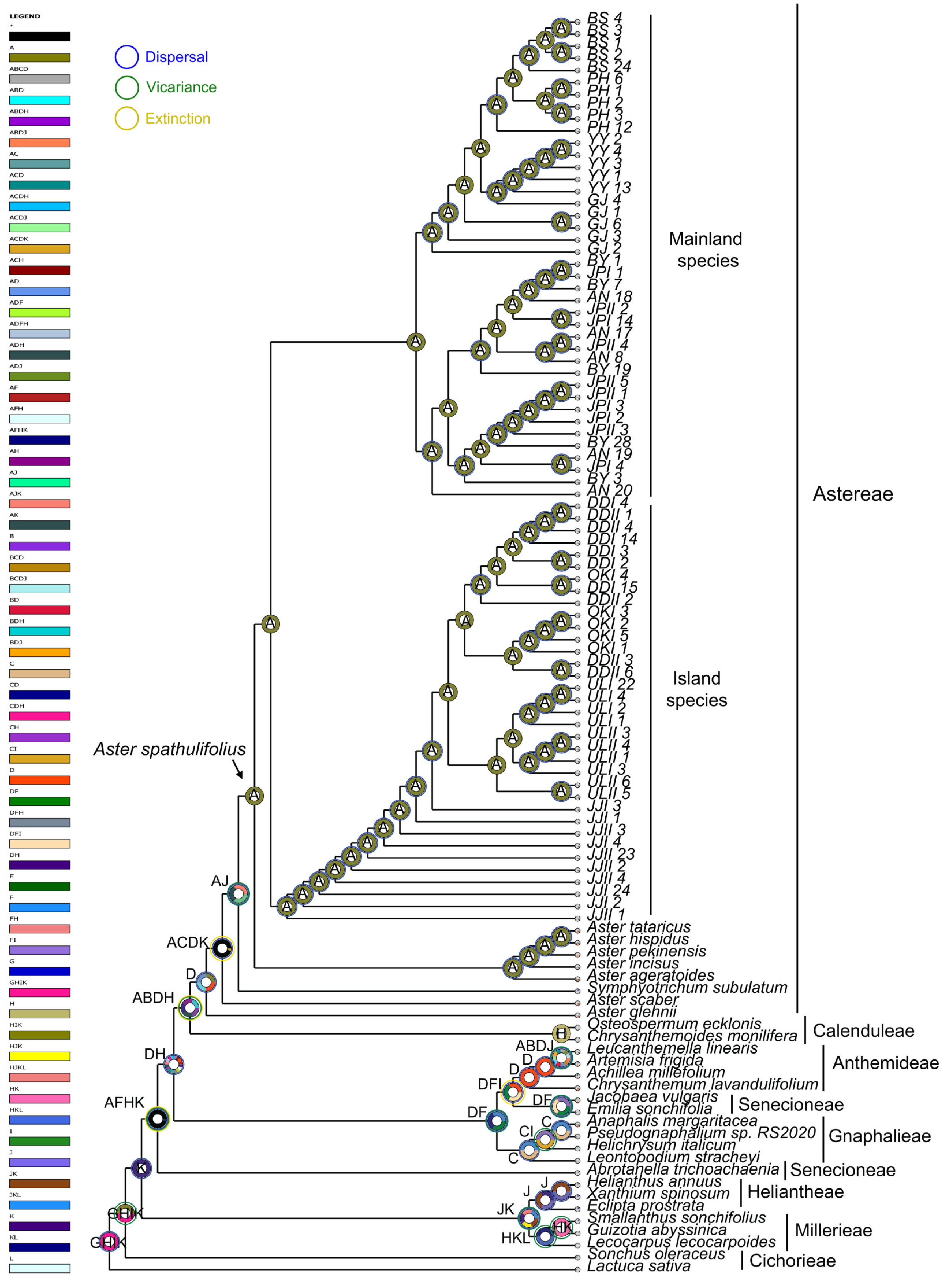
2.7. Ancestral Area Reconstruction
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. DNA Extraction, Amplification, Sequencing and Variant Calling
4.3. Genetic Diversity Analysis
4.4. Principle Component Analysis
4.5. Population Structure Analysis
4.6. Genetic Differentiation and Genetic Flow
4.7. Phylogenetic Analyses
4.8. Divergence Dating Analyses
4.9. Biogeographical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cheng, J.; Kao, H.; Dong, S. Population genetic structure and gene flow of rare and endangered Tetraena mongolica Maxim. revealed by reduced representation sequencing. BMC Plant Biol. 2020, 20, 391. [Google Scholar] [CrossRef] [PubMed]
- Robledo-Arnuncio, J.J.; Klein, E.K.; Muller-Landau, H.C.; Santamaria, L. Space, time and complexity in plant dispersal ecology. Mov. Ecol. 2014, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Vranckx, G.; Jacquemyn, H.; Muys, B.; Honnay, O. Meta-analysis of susceptibility of woody plants to loss of genetic diversity through habitat fragmentation. Conserv. Biol. 2012, 26, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Costanza, R.; dArge, R.; deGroot, R.; Farber, S.; Grasso, M.; Hannon, B.; Limburg, K.; Naeem, S.; ONeill, R.V.; Paruelo, J.; et al. The value of the world’s ecosystem services and natural capital. Nature 1997, 387, 253–260. [Google Scholar] [CrossRef]
- Park, J.S.; Takayama, K.; Suyama, Y.; Choi, B.H. Distinct phylogeographic structure of the halophyte Suaeda malacosperma (Chenopodiaceae/Amaranthaceae), endemic to Korea-Japan region, influenced by historical range shift dynamics. Plant Syst. Evol. 2019, 305, 193–203. [Google Scholar] [CrossRef]
- Clark, P.U.; Dyke, A.S.; Shakun, J.D.; Carlson, A.E.; Clark, J.; Wohlfarth, B.; Mitrovica, J.X.; Hostetler, S.W.; McCabe, A.M. The last glacial maximum. Science 2009, 325, 710–714. [Google Scholar] [CrossRef]
- Kadereit, J.W.; Arafeh, R.; Somogyi, G.; Westberg, E. Terrestrial growth and marine dispersal? Comparative phylogeography of five coastal plant species at a European scale. Taxon 2005, 54, 861–876. [Google Scholar] [CrossRef]
- Weising, K.; Freitag, H. Phylogeography of halophytes from European coastal and inland habitats. Zool. Anz. 2007, 246, 279–292. [Google Scholar] [CrossRef]
- Han, Q.; Higashi, H.; Mitsui, Y.; Setoguchi, H. Distinct Phylogeographic Structures of Wild Radish (Raphanus sativus L. var. raphanistroides Makino) in Japan. PLoS ONE 2015, 10, e0135132. [Google Scholar] [CrossRef]
- Qiu, Y.X.; Fu, C.X.; Comes, H.P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet Evol. 2011, 59, 225–244. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, J.H.; Cho, W.B.; Choi, B.H. The establishment history of alpine Leontopodium japonicum (Asteraceae) resembles that of warm-temperate plants on the Korean Peninsula. Plant Syst. Evol. 2016, 302, 1483–1494. [Google Scholar] [CrossRef]
- Jin, D.P.; Lee, J.H.; Xu, B.; Choi, B.H. Phylogeography of East Asian Lespedeza buergeri (Fabaceae) based on chloroplast and nuclear ribosomal DNA sequence variations. J. Plant Res. 2016, 129, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, D.H.; Choi, B.H. Phylogeography and genetic diversity of East Asian Neolitsea sericea (Lauraceae) based on variations in chloroplast DNA sequences. J. Plant Res. 2013, 126, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Westberg, E.; Kadereit, J.W. The influence of sea currents, past disruption of gene flow and species biology on the phylogeographical structure of coastal flowering plants. J. Biogeogr. 2009, 36, 1398–1410. [Google Scholar] [CrossRef]
- Choi, K.S.; Park, S. The complete chloroplast genome sequence of Aster spathulifolius (Asteraceae); genomic features and relationship with Asteraceae. Gene 2015, 572, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Itow, S.; Nakanishi, H. Natural Vegetation of Tsushima; Biogeography of the Tsushima island; Nagasaki Prefecture Office, Ed.; Nagasaki Prefecture Office: Nagasaki, Japan, 1987. [Google Scholar]
- Maki, M.; Morita, H. Genetic diversity in island and mainland populations of Aster spathulifolius (Asteraceae). Int. J. Plant Sci. 1998, 159, 148–152. [Google Scholar] [CrossRef]
- Lee, T.B. Illustrated Flora of Korea; Hyangmunsa Press: Seoul, Republic of Korea, 1993. [Google Scholar]
- Jean Claude, S.; Park, S. Aster spathulifolius Maxim. a leaf transcriptome provides an overall functional characterization, discovery of SSR marker and phylogeny analysis. PLoS ONE 2020, 15, e0244132. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Ng, K.K.; Lee, S.L.; Koh, C.L. Spatial structure and genetic diversity of two tropical tree species with contrasting breeding systems and different ploidy levels. Mol. Ecol. 2004, 13, 657–669. [Google Scholar] [CrossRef]
- Karron, J.D. A comparison of levels of genetic polymorphism and self-compatibility in geographically restricted and widespread plant congeners. Evol. Ecol. 1987, 1, 47–58. [Google Scholar] [CrossRef]
- Nguyen, H.T.T.; Choi, K.S.; Park, S. Genetic diversity and differentiation of a narrowly distributed and endemic species, Aster spathulifolius Maxim (Asteraceae), revealed with inter simple sequence repeat markers. J. Korean Soc. Appl. Biol. Chem. 2013, 56, 255–262. [Google Scholar] [CrossRef]
- Tyagi, S.; Jung, J.A.; Kim, J.S.; Won, S.Y. Comparative analysis of the complete chloroplast genome of mainland Aster spathulifolius and other Aster species. Plants 2020, 9, 568. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.M.; Itaki, T.; Tada, R.; Kamikuri, S.-i. Paleoceanographic history of the Japan Sea over the last 9.5 million years inferred from radiolarian assemblages (IODP Expedition 346 Sites U1425 and U1430). Prog. Earth Planet. Sci. 2018, 5, 54. [Google Scholar] [CrossRef]
- Chung, J.-M.; Shin, J.-K.; Kim, H.-M. Diversity of vascular plants native to the Ulleungdo and Dokdo islands in Korea. J. Asia-Pac. Biodivers. 2020, 13, 701–708. [Google Scholar] [CrossRef]
- Seo, H.S.; Kim, S.H.; Kim, S.C. Chloroplast DNA insights into the phylogenetic position and anagenetic speciation of Phedimus takesimensis (Crassulaceae) on Ulleung and Dokdo islands, Korea. PLoS ONE 2020, 15, e0239734. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Iida, K.; Nunome, M.; Kim, Y.-K.; Han, S.-H.; Sohn, J.-H.; Ohmori, Y.; Oh, H.-S.; Kimura, J.; Hondo, E. Phylogeography of Miniopterus fuliginosus (Chiroptera) with special reference to Jeju Island, South Korea. Mammalia 2019, 83, 610–614. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Raman, G.; Park, S.; Lee, E.M.; Park, S. Evidence of mitochondrial DNA in the chloroplast genome of Convallaria keiskei and its subsequent evolution in the Asparagales. Sci. Rep. 2019, 9, 5028. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Chu, R.; Xu, X.; Lu, Z.; Ma, Y.; Cheng, H.; Zhu, S.; Bakker, F.T.; Schranz, M.E.; Wei, Z. Plastome-based phylogeny and biogeography of Lactuca L. (Asteraceae) support revised lettuce gene pool categories. Front. Plant Sci. 2022, 13, 978417. [Google Scholar] [CrossRef]
- Yu, Y.; Blair, C.; He, X. RASP 4: Ancestral state reconstruction tool for multiple genes and characters. Mol. Biol. Evol. 2020, 37, 604–606. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Populations | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mainland Populations | Island Populations | Average/ Population | Mainland Populations | Island Populations | ||||||||||||||
| AN | BS | BY | GC | PH | YY | JPI | JPII | JJI | JJII | DDI | DDII | ULI | ULII | OKI | ||||
| S | 2 | 0 | 6 | 1 | 1 | 3 | 1 | 0 | 0 | 0 | 0 | 3 | 1 | 0 | 0 | 1.2 | 1.75 | 0.571429 |
| h | 3 | 1 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 3 | 2 | 1 | 1 | 1.666 | 1.875 | 1.428571 |
| Hd | 0.700 | 0 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0 | 0 | 0 | 0 | 0.7 | 0.4 | 0 | 0 | 0.25333 | 0.3375 | 0.157143 |
| π | 0.00019 | 0 | 0.00057 | 0.0001 | 0.0001 | 0.00029 | 0.0001 | 0 | 0 | 0 | 0 | 0.00029 | 0.0001 | 0 | 0 | 0.000116 | 0.000169 | 0.000056 |
| κ | 0.8 | 0 | 2.4 | 0.4 | 0.4 | 1.2 | 0.4 | 0 | 0 | 0 | 0 | 1.2 | 0.4 | 0 | 0 | 0.48 | 0.7 | 0.228571 |
| Tajima’s D | 0.96 | 0 | 2.880 | 0.48 | 1.44 | 0.48 | 0 | 0 | 0 | 0 | 1.44 | 0.48 | 0 | 0 | 0.582857 | 0.891429 | 0.274286 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raman, G.; Choi, K.S.; Park, S. Population Structure and Genetic Diversity Analyses Provide New Insight into the Endemic Species Aster spathulifolius Maxim. and Its Evolutionary History. Plants 2024, 13, 88. https://doi.org/10.3390/plants13010088
Raman G, Choi KS, Park S. Population Structure and Genetic Diversity Analyses Provide New Insight into the Endemic Species Aster spathulifolius Maxim. and Its Evolutionary History. Plants. 2024; 13(1):88. https://doi.org/10.3390/plants13010088
Chicago/Turabian StyleRaman, Gurusamy, Kyoung Su Choi, and SeonJoo Park. 2024. "Population Structure and Genetic Diversity Analyses Provide New Insight into the Endemic Species Aster spathulifolius Maxim. and Its Evolutionary History" Plants 13, no. 1: 88. https://doi.org/10.3390/plants13010088